Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Allopurinol in Patients with Pulmonary Hypertension Associated with Chronic Lung Disease
Authors Liu-Shiu-Cheong PSK, Lipworth BJ ![]() , Weir-McCall JR
, Weir-McCall JR ![]() , Houston JG
, Houston JG ![]() , Struthers AD
, Struthers AD
Received 1 May 2020
Accepted for publication 15 July 2020
Published 25 August 2020 Volume 2020:15 Pages 2015—2024
DOI https://doi.org/10.2147/COPD.S260917
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Patrick SK Liu-Shiu-Cheong,1,2 Brian J Lipworth,3 Jonathan R Weir-McCall,1,4 J Graeme Houston,5 Allan D Struthers1
1Division of Molecular and Clinical Medicine, University of Dundee, Dundee DD1 9SY, UK; 2Department of Respiratory Medicine, Victoria Hospital, NHS Fife, Kirkcaldy KY2 5AH, UK; 3Scottish Centre for Respiratory Research, Medical Research Institute, University of Dundee, Dundee DD1 9SY, UK; 4Department of Radiology, University of Cambridge, Cambridge CB2 0QQ, UK; 5Imaging Science and Technology, University of Dundee, Dundee DD1 9SY, UK
Correspondence: Brian J Lipworth
Scottish Centre for Respiratory Research, Medical Research Institute, University of Dundee, Ninewells Hospital, Dundee DD1 9SY, United Kingdom
Tel +44 1382 383188
Email [email protected]
Background: Oxidative stress (OS) has been implicated in the development of pulmonary hypertension (PH) and ventricular hypertrophy. Xanthine oxidase is a well-recognised source of reactive oxygen species, which lead to OS. The aim of this proof of concept study was to assess whether allopurinol (xanthine oxidase inhibitor) would reduce right ventricular mass (RVM) in patients with PH-associated chronic lung disease (PH-CLD).
Methods: We conducted a randomised, double-blind, parallel-group, placebo-controlled trial in patients with PH-CLD (93% COPD, 7% IPF) who were randomly assigned to receive allopurinol or placebo for 12 months. The primary outcome was the mean change in RVM, as assessed by cardiac magnetic resonance imaging (CMRI). Secondary outcomes included quality of life (QOL), spirometry and six-minute walk test (6MWT).
Results: Seventy-one patients were recruited: mean age 71 years, mean pulmonary arterial pressure 30 mm Hg, FEV1 60% and resting SpO2 96%. After 12 months, there was no significant difference in the change in RVM from baseline (allopurinol 1.85g vs placebo 0.97g with mean difference 0.88g, CI − 4.77 to 3.01, p =0.7). There were also no significant changes in other cardiac parameters measured on MRI, in QOL, spirometry and 6MWT. Subgroup analysis showed that allopurinol significantly reduced RVM compared to placebo with -6.16g vs 0.75g and mean difference 6.92g (CI 1.14 to 12.69, p = 0.02) in COPD patients with more severe airflow limitation.
Conclusion: Allopurinol had no overall impact on patients with PH-CLD but had potential benefit in COPD patients with more severe airflow limitation.
Keywords: pulmonary hypertension, right ventricle, allopurinol, chronic lung disease
Introduction
The development of pulmonary hypertension (PH) is a well-recognised complication of many chronic lung diseases and is generally mild to moderate in severity. The most common chronic lung diseases associated with PH are chronic obstructive pulmonary disease (COPD) and interstitial lung disease (ILD).1 Irrespective of the underlying pulmonary aetiology, the presence of even mild PH is associated with deterioration in exercise capacity, worsening of hypoxaemia and shorter survival.2,3
Pulmonary hypertension in hypoxaemic lung diseases is attributed to multiple factors including pulmonary vasoconstriction caused by alveolar hypoxia and the distortion of pulmonary vessels by parenchymal changes.4 Right ventricular hypertrophy (RVH) is believed to be the consequence of chronic pulmonary hypertension.5 RVH is the initiating step in the progression to right ventricular (RV) failure in lung disease.
At present, there is no specific therapy recommended for patients with group 3 PH associated with lung disease.1 The European Respiratory Society advocates optimising the treatment of the underlying lung disease and advises that patients should receive long-term oxygen therapy (LTOT) if they are hypoxaemic as LTOT has been shown to partially reduce the progression of PH in COPD.1
Allopurinol is a xanthine oxidase inhibitor which decreases both uric acid (UA) and oxidative stress (OS). Five experimental studies have shown that allopurinol inhibits hypoxia-induced pulmonary vasoconstriction, pulmonary hypertension, endothelial dysfunction and vascular remodelling.6–10 Hypoxia is known from many studies to up-regulate xanthine oxidase and therefore to increase its production of UA and OS.6 Hypoxia-induced oxidative stress has also been shown to have a direct effect in the development of ventricular hypertrophy.11 Allopurinol has also been shown in clinical studies to reduce left ventricular mass in patients with chronic kidney disease, optimally treated ischaemic heart disease and type 2 diabetes mellitus.12–14
The aim of this proof of concept study was to investigate whether high-dose allopurinol would reduce right ventricular mass in patients with pulmonary hypertension associated with chronic lung disease (PH-CLD).
The hypothesis is that allopurinol would reduce the production of reactive oxygen species, thereby decreasing oxidative stress, which would lead to reduction in right ventricular mass in patients with PH-CLD.
Methods
Study Design and Patients
This is a randomised, placebo-controlled, double-blind, parallel-group study conducted over a 12-month treatment period at a single centre in Dundee, Scotland between April 2015 and July 2017. The trial was approved by the East of Scotland Research Ethics Committee (reference: 14/ES/1035) and was carried out in accordance with the Declaration of Helsinki. This study has been registered with the International Standard Randomized Controlled Trial Number register (ISRCTN11081180) and European Clinical Trials Database (EudraCT no: 2014–002305-38).
We recruited patients from the chest clinics at the local hospital, from local respiratory databases, from general practices covering two UK National Health Service boards (Tayside and Fife) via the Scottish Primary Care Research Network, and from the Scottish Health Research Register. Box 1 shows the study eligibility criteria. All patients provided written informed consent. All their usual medications were continued throughout the study.
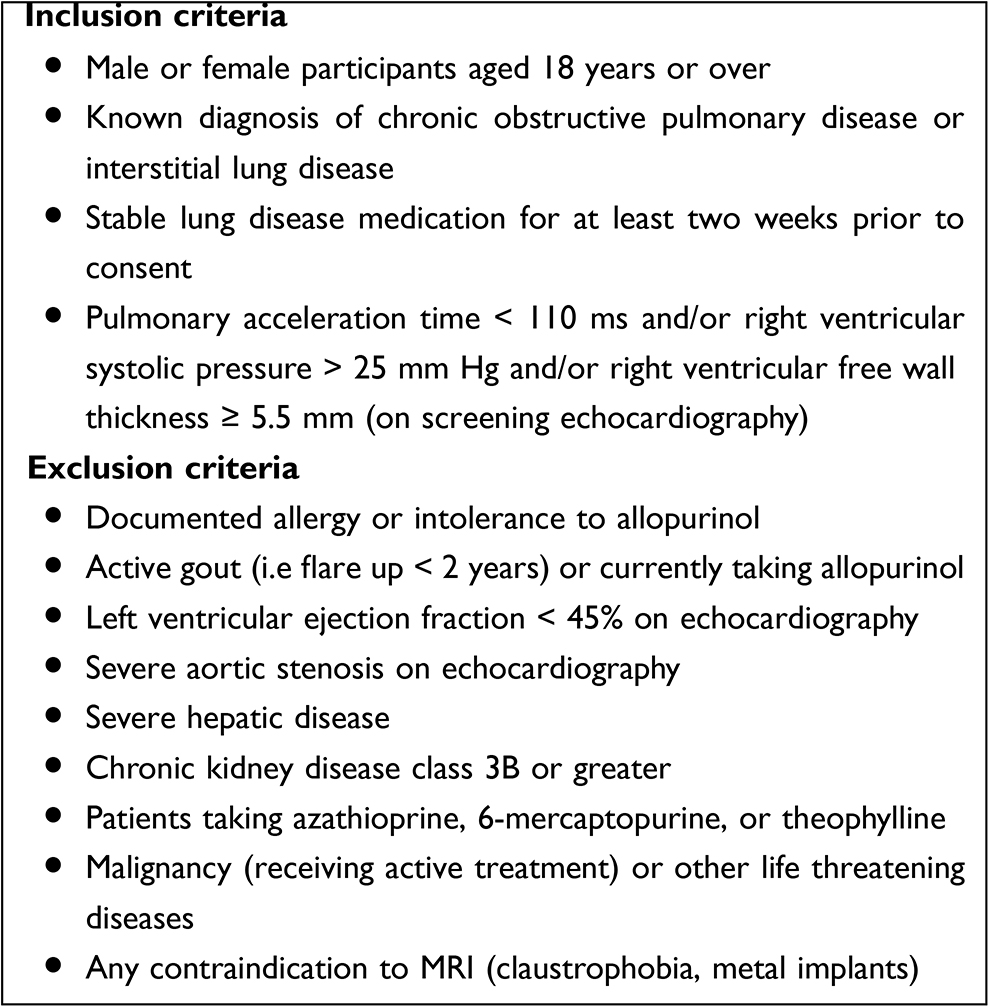 |
Box 1 Inclusion and Exclusion Criteria |
Randomisation and Masking
Eligible patients were randomly assigned by a validated computer-generated random allocation sequence to a study number. The patients were given the trial medication, which was either allopurinol or matched placebo, carrying their unique study number. At baseline visit, they would receive either allopurinol 100 mg/day or matched placebo orally for two weeks. If this was tolerated, the trial medication was increased to 300mg of allopurinol or placebo for four weeks. The dosage was further increased after this four week period to the target dose of 600mg (given as 300 mg twice daily) for the remaining duration of the trial. The intervention and placebo capsules and bottles looked identical and double blinding was maintained throughout. Patients, study investigators and outcome assessors were masked to group allocation. Unmasking was done only after the end of the trial.
Procedures
All recruited participants underwent screening echocardiography to check if they were eligible. PLSC undertook echocardiography with either Philips iE33 system (Philips, Netherlands) or Philips Epiq7 ultrasound system (Philips Ultrasound, USA). If the measurements were difficult to obtain using standard acoustic windows, the oblique subcostal windows were used to assess the pulmonary acceleration time with pulse-wave Doppler.15 The estimated mean pulmonary arterial pressure (MPAP) was then calculated with Dabestani equation, and/or the right ventricular systolic pressure with the Bernoulli equation.16
All participants underwent spirometry, six-minute walk test (6MWT) and cardiac MRI scan at randomisation visit and these measures were repeated at final visit. Spirometry was performed using MicroLoop spirometer (Micro Medical, Rochester, UK) as per ATS/ERS guidelines and 6MWT was performed as per ATS guidelines.17,18 Patients were allowed to use their usual walking aids but participants on LTOT did their 6MWT off oxygen. Oxygen saturation during the 6MWT was measured using portable pulse oximeter.
Quality of life (QOL) at baseline and final visits was assessed using the St George’s Respiratory Questionnaire (SGRQ) and the King’s Brief Interstitial Lung Disease (K-BILD) questionnaire. The 36-Item Short Form Survey (SF-36) was also used to assess general QOL measure.
Blood samples were taken at baseline and final visits for N-terminal prohormone of brain natriuretic peptide (NT-proBNP) and high-sensitive troponin I (hs-Trop I).
Cardiac MRI
Cardiac magnetic resonance imaging (CMRI) was performed at baseline and at final visit only. The images were acquired on a 32 RF cardiac receiver channel, 3 Tesla MRI scanner (Prisma, Siemens, Erlangen, Germany) using dedicated cardiac coils as previously described.19 The images were exported, and analysis performed offline by an independent, blinded radiologist (JWM), with five years experience in CMRI, using CVI 42 (Circle Cardiovascular Imaging software, Calgary, Canada).
The epicardial and endocardial contours were drawn around the right ventricle at end systole and end diastole. Trabeculae were included in the mass measurement and excluded from the volume calculation. The septum was treated as belonging to the left ventricle and was excluded from the right ventricular mass (RVM). Ventricular mass and volumes were indexed to body surface area, which was calculated using the Mosteller formula.20
Outcomes
The primary outcome was the mean difference between the baseline and final right ventricular mass index after 12 months of allopurinol compared to placebo. Secondary outcomes included change in other cardiac MRI measurements, lung function (spirometry), exercise capacity (6MWT), quality of life measures (SGRQ, K-BILD, SF-36), and blood markers (NT-proBNP, hs-Trop I).
Statistical Analysis
Bradlow et al,21 advised that 34 patients are required to have 80% power at p < 0·05 to detect changes in the manually measured outcomes of RV mass of 10g. However, a 10g change in ventricular mass was over ambitious. 66 patients were required to achieve 80% power to detect a 5g change in RVM at a significance level of p < 0·05, based on previous studies investigating the allopurinol’s effect on left ventricular mass using CMRI.12–14 In order to allow for 10% drop-outs, 72 eligible patients were recruited.
The primary analysis was based on the intention-to-treat principle. The extent of missing data was examined and the reason for drop-out was ascertained. Multiple imputations have been used to impute missing values and where assumptions for missing at random data were met. Complete case analysis where missing patients are excluded was carried out as a secondary analysis.
The data for continuous outcome measures were assessed for normality of distribution prior to analysis. The descriptive statistics was reported in the form of mean ± standard deviation (SD) for normally distributed continuous variables, median and interquartile range (IQR) for non-normally distributed continuous variables, and percentages and denominators for categorical variables are tabulated at the baseline visit. The comparison between continuous variables is analysed using paired t-tests (to test within group differences), independent t-test (to test between group differences), or Mann–Whitney U-test (for non-normally distributed data) whilst categorical variables were analysed using Chi-squared test.
All statistical analyses were undertaken blinded using IBM SPSS Statistics v22·0 (IBM, United States). A two-sided p value < 0·05 was considered statistically significant.
Results
The flow of participants is depicted in the CONSORT diagram (Figure 1). A total of 72 participants were recruited for the study. One patient was randomised but excluded from analysis as he was unable to undergo baseline CMRI due to claustrophobia. 36 received allopurinol and 35 received placebo. 63 (89%) participants completed the study per protocol and the average follow-up period was 11·3 months for completers. The compliance from tablet counting was high at 96%. All the participants tolerated a daily dosage of 600 mg of allopurinol except 1 participant who was left on 300 mg as he developed fatigue and lethargy while taking 600 mg.
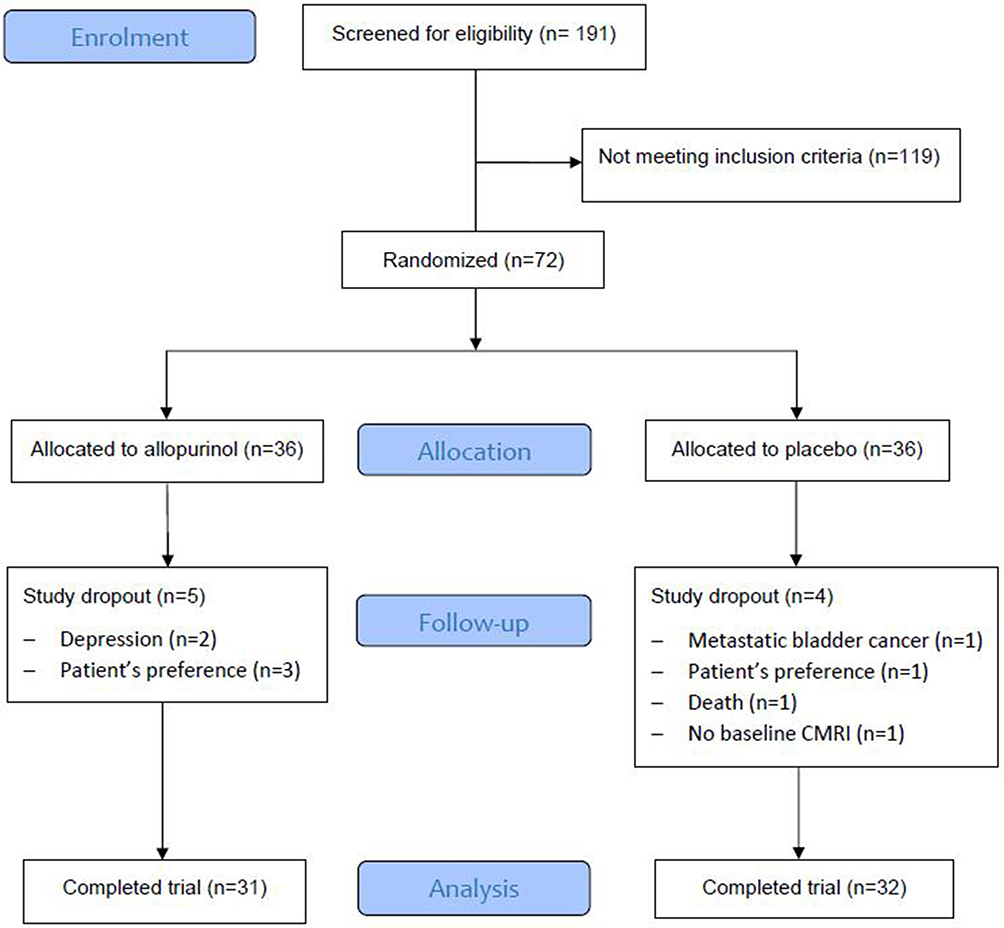 |
Figure 1 CONSORT diagram showing participant flow through the study. |
The baseline characteristics were similar between the two groups (Table 1): mean age 71 years, estimated MPAP 30 mm Hg, mean FEV1 60% predicted, resting oxygen saturation (SaO2) 96% and predominantly male (62%). 66 (93%) participants had COPD and five (7%) had IPF. Five patients were on LTOT. There were eight study dropouts (allopurinol n = 5, placebo = 3): participant’s preference (n = 4), depression (n = 2), metastatic bladder cancer (n = 1) and death (n = 1). No patient receiving LTOT needed an increase in oxygen dose. One patient in the placebo who was already on ambulatory oxygen was prescribed LTOT during the trial period.
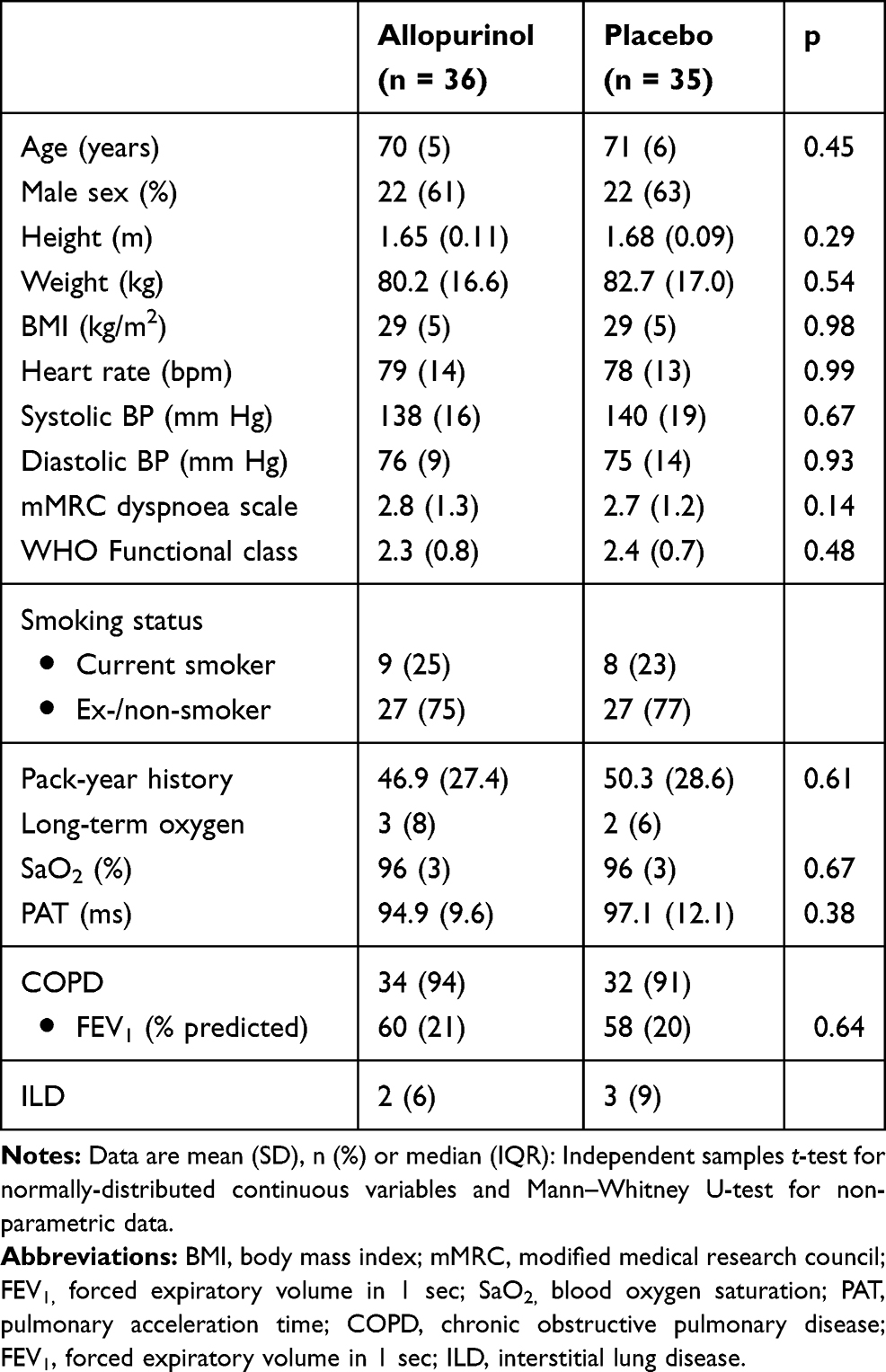 |
Table 1 Baseline Characteristics of Study Participants |
At baseline, RVM and RVMI were similar in both groups (Table 2). In the intention-to-treat analysis (Table 3), there was no significant difference between allopurinol group and placebo group in the change in RVM (allopurinol group 1.85 ± 1.56 g vs placebo group 0.97 ± 1.20 g; p = 0.66) and in the change in RVMI (allopurinol group 0.70 ± 0.75 g/m2 vs placebo group 0.50 ± 0.60 g/m2; p = 0.83). There was also no significant difference in the change in RVM and RVMI when per-protocol analysis was performed. In the post-hoc exploratory subgroup analysis according to severity of airflow limitation (Table 4), participants with more severe airflow limitation (GOLD 3 and 4) showed significant reduction in RVM (allopurinol group −6.16 ± 1.97 g vs placebo group 0.75 ± 1.71 g; p = 0.02) and in RVMI (allopurinol group −3.13 ± 1.00 g/m2 vs placebo group 0.54 ± 0.85 g/m2; p = 0.02). (Figure 2)
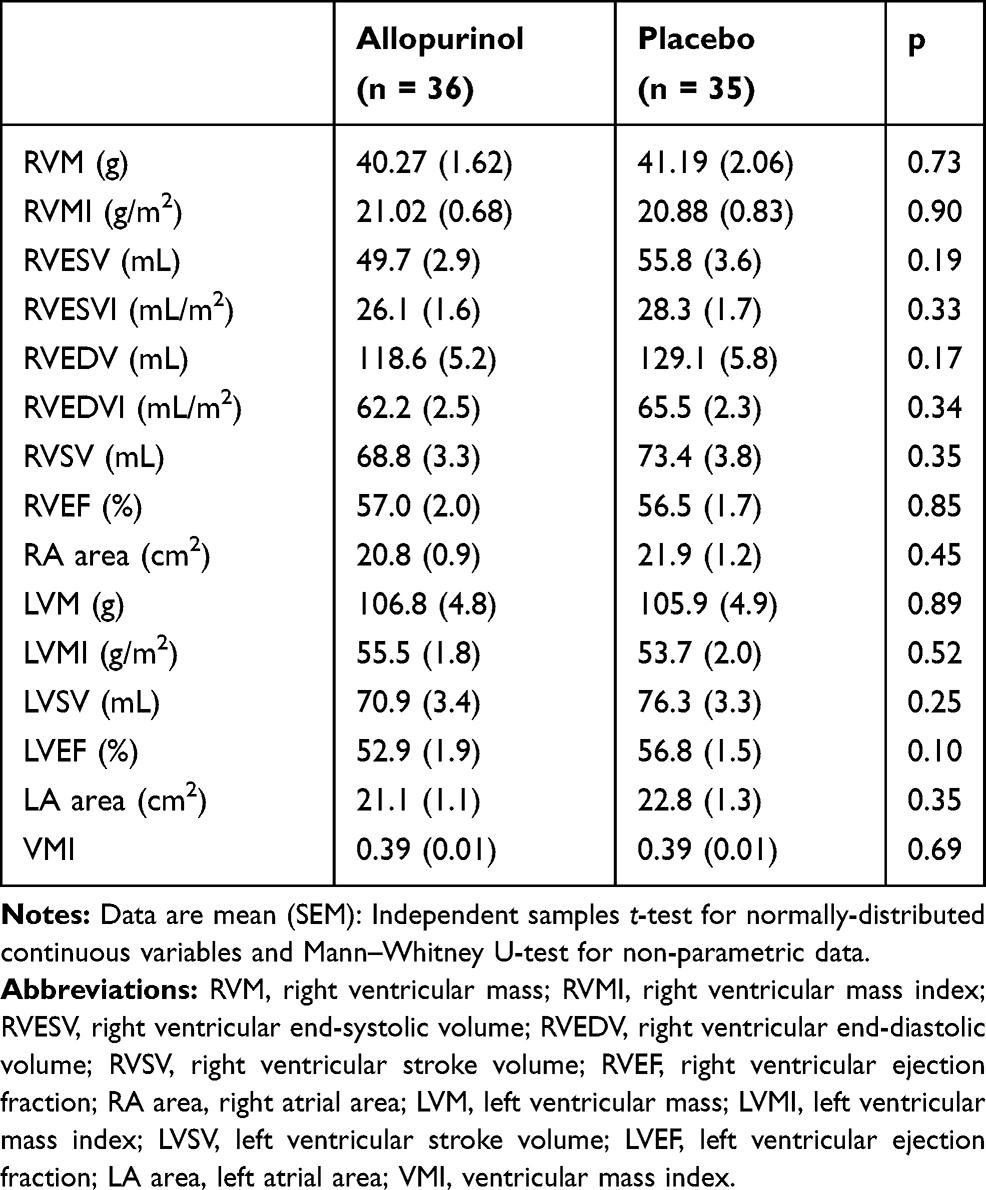 |
Table 2 Baseline Cardiac MRI Measurements |
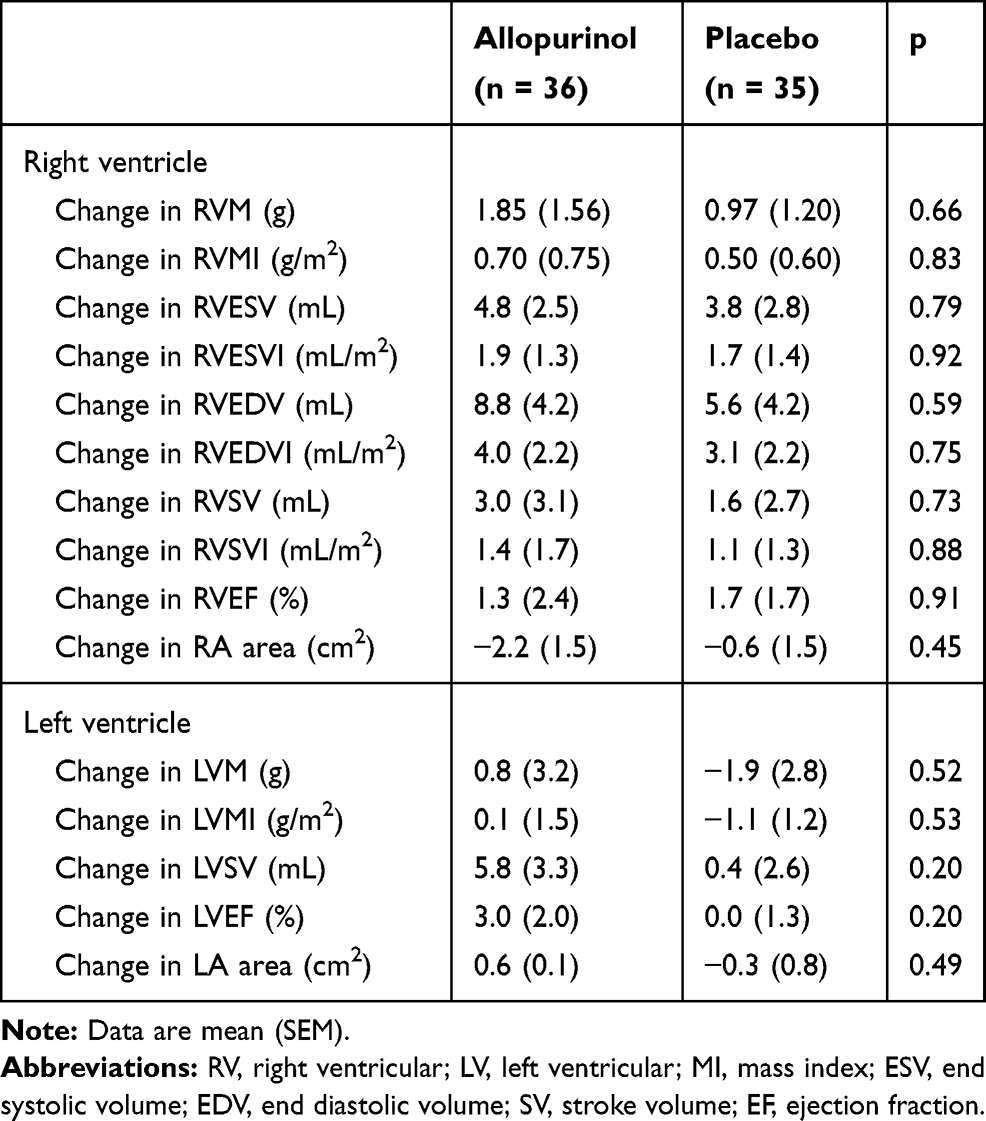 |
Table 3 CMRI Changes After Allopurinol Treatment (Intention-to-Treat Analysis) |
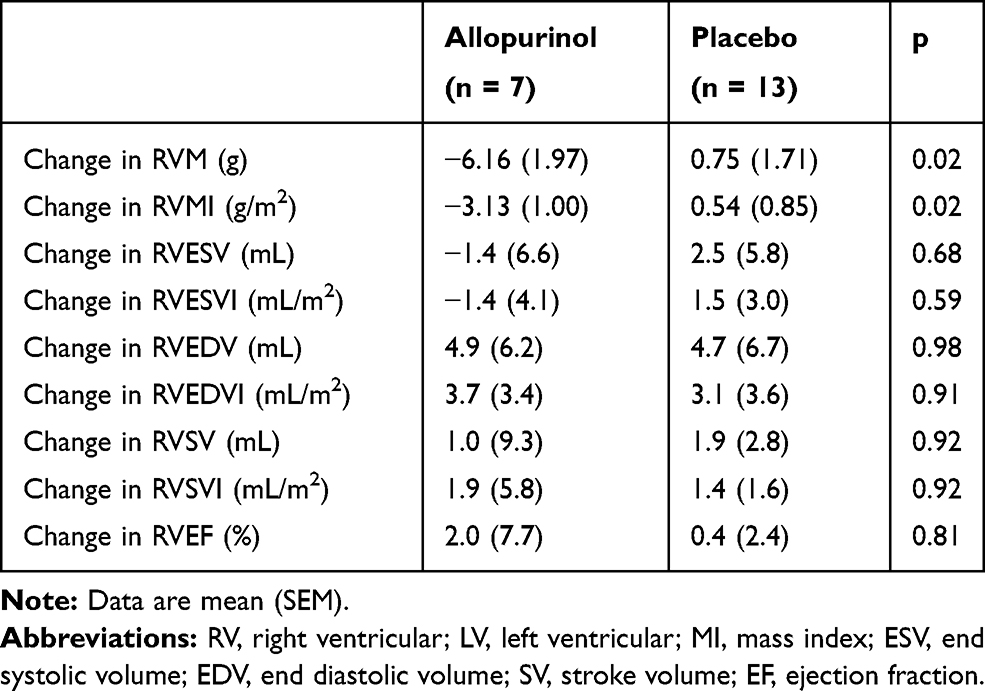 |
Table 4 CMRI Changes for RV After Allopurinol Treatment in Subgroup GOLD 3 and 4 |
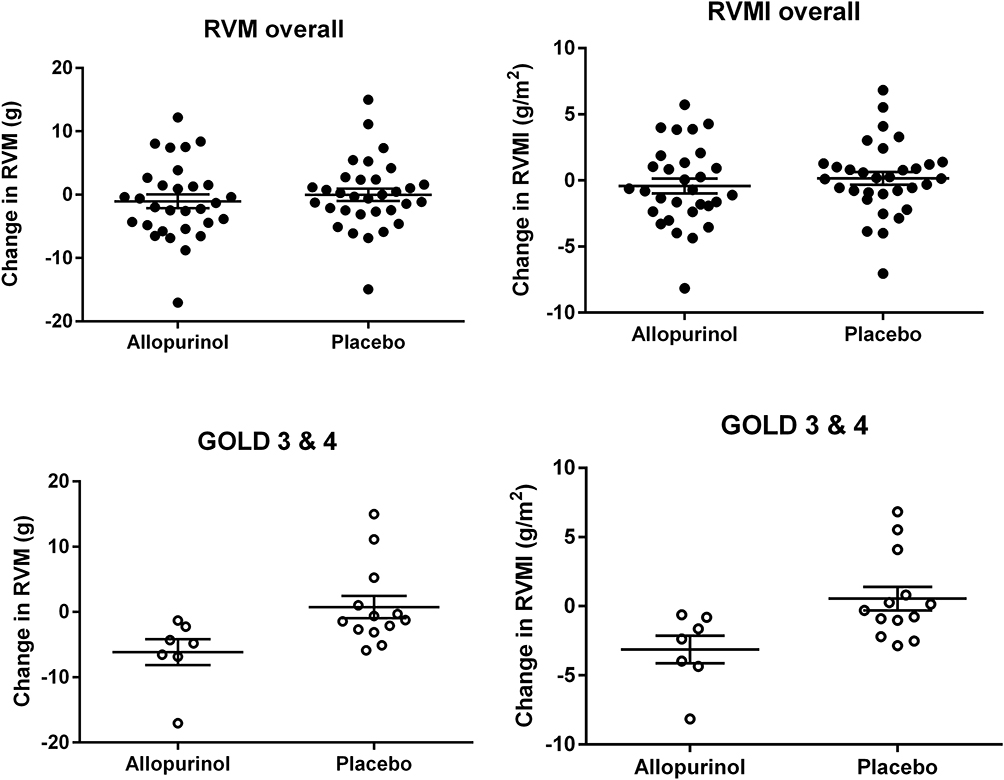 |
Figure 2 Scatter plots of RVM and RVMI (mean and SEM) for overall population (allopurinol n=31, placebo n=32) and subgroup GOLD 3 and 4 (allopurinol n=7, placebo n=13). There were significant (p = 0·02) differences in RVM and RVMI in the subgroup of GOLD 3/4 patients. |
For the other parameters measured on CMRI for both right and left ventricles, there were no significant changes in end-systolic volume (ESV), end-diastolic volume (EDV), ejection fraction (EF), stroke volume (SV) and their indexed values (Table 3). There were also no statistically significant changes in any of the quality-of-life, 6MWT and spirometric measurements (Table 5). There was no significant change in NT-proBNP and hs-Trop I levels over the study period (Table 6). There was a significant reduction in uric acid level by 59% with allopurinol treatment.
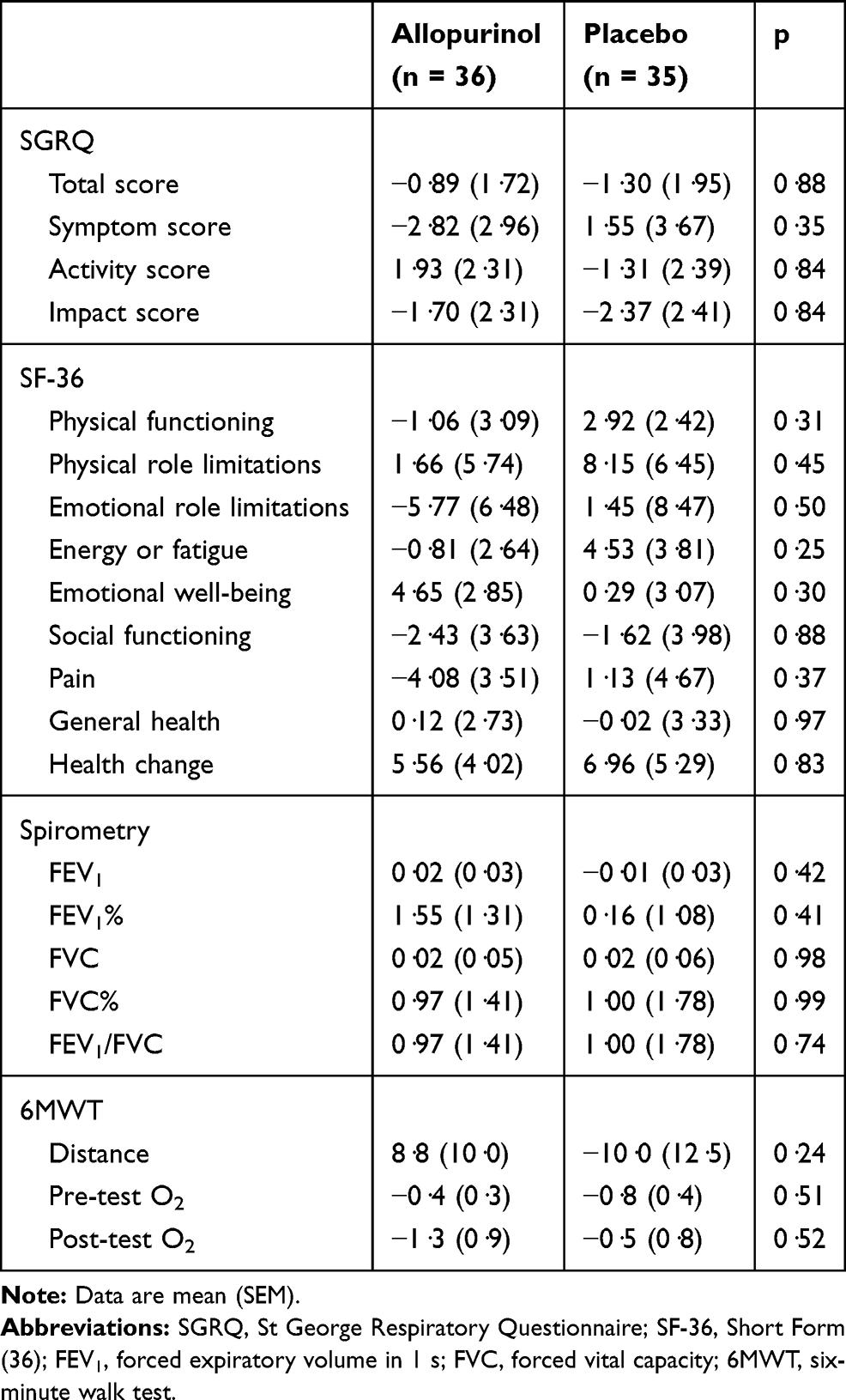 |
Table 5 Mean Change in Quality-of-Life Scores, Spirometry and Six-Minute Walk Measurements at 52 Weeks Compared to Baseline (Intention-to-Treat Analysis) |
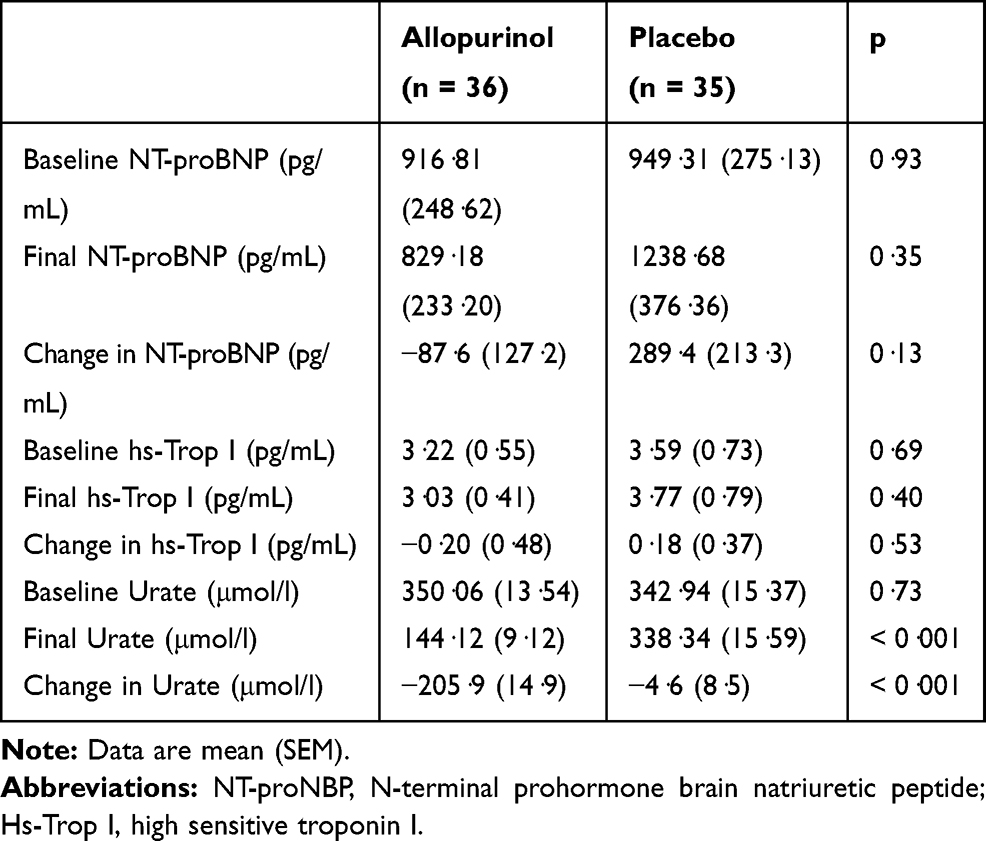 |
Table 6 Mean Change in Blood Markers at 52 Weeks Compared to Baseline (Intention-to-Treat Analysis) |
Another post-hoc exploratory subgroup analysis when stratifying by NT-proBNP level showed that the patients (n = 31) with higher NT-proBNP level (>489 pg/mL) had a greater improvement in LVEF with allopurinol 5.12 ± 2.36% vs placebo −1.62 ± 1.51% (p = 0.02). There was no significant difference in the change in RVM (allopurinol group 1.04 ± 1.33 g vs placebo −0.56 ± 4.42 g; p = 0.44) and in RVMI (allopurinol group 0.63 ± 0.72 g/m2 vs placebo group −0.05 ± 0.67 g/m2; p = 0.50) in this subgroup.
There was a total of 179 adverse events (AE) recorded with 80 (45%) in the allopurinol group and 99 (55%) in the placebo group. There were 7 serious adverse events (SAE) in the allopurinol group compared to 15 SAE in the placebo group.
Discussion
High dose allopurinol exhibited no overall effect on the RVM, as measured by cardiac MRI. This could be due to the heterogeneity of severity in chronic lung disease. Recent studies have demonstrated the wide range of phenotypes and heterogeneity of patients suffering from COPD.22 The overall effect of allopurinol could have been different if we had considered only patients with specific phenotypes of the disease. As this was a proof of concept study, it was not possible to restrict to specific phenotypes as there were no previous studies that had already evaluated this.
The right ventricular mass of our study population was relatively not too high compared to studies in patients with idiopathic pulmonary arterial hypertension23,28 This is probably because pulmonary hypertension in COPD and ILD patients tends to be milder in severity compared to group 1 patients.1 One could say that because RVM was not high, then our population did not have pulmonary hypertension. However, there was evidence of pulmonary hypertension on CMRI as demonstrated by a higher ventricular mass index (VMI) in our population compared to similar population of COPD and no PH studied by Johns et al.29 VMI correlates with invasive measurement of pulmonary artery pressure and has been shown to be a prognostic marker in pulmonary hypertension.30,31
Another possible explanation for the negative result for the primary outcome is that the study population consisted mostly of patients with COPD (93%) and a few patients suffering from ILD (7%). It was agreed at the time when the study was designed to include patients suffering from group 3 pulmonary hypertension. However, it is still not completely clear whether the pathophysiology underlying the development of PH and RVH is the same for all the chronic lung diseases.
The heterogeneous study population may have masked a possible effect of allopurinol. This is supported by the finding that there was a statistically significant difference in the allopurinol group amounting to a mean difference of 6.92g in a subgroup of patients with more severe airflow obstruction corresponding to GOLD groups 3 and 4. This in turn suggests that the more severe the degree of airflow limitation, the more likely allopurinol may reduce right ventricular mass. A potential explanation is that such patients are more likely to be more hypoxic on exertion (despite having satisfactory resting oxygen saturation), causing increasing oxidative stress, leading to cardiac hypertrophy and increased pulmonary arterial pressures on exercise. The observed trend towards a reduction in RVESV conferred by allopurinol suggests that offloading the RV may have been the mechanism for the RVH regression, further supported by similar observations of left ventricular mass reduction and improved systolic function reported by Rekhraj et al study13 of left ventricular hypertrophy.
The overall negative results in our study may be because the current study population had too mild chronic lung disease. The positive signal that was found in our subgroup analysis suggests that the study needs to be repeated in patients with more severe chronic lung disease to ascertain the benefits of allopurinol in that population who have the greatest need of new treatments.
It is difficult to compare the results of the current study with other randomised clinical trials (RCT) as to date there is no other RCT looking at the cardiac mass, volume and function in patients with group 3 pulmonary hypertension. The closest studies that could be compared with are longitudinal ones in patients with group 1 pulmonary hypertension who received drug therapies already licensed to treat pulmonary hypertension in this group. The results from these studies were mixed: the studies by Michelakis et al,26 van Wolferen et al,23 and van de Veerdonk,28 and the SERAPH study24 have reported a reduction in RVM from baseline while the study by Roeleveld et al,27 and the EURO-MR study25 reported no change in RVM.
Two recent studies32,33 have reported improvements in ventricular volumes and function in COPD population following treatment with inhaled therapies. However, these studies were looking at a particular phenotype of COPD, namely those with lung hyperinflation which induces cardiac under-filling. Lung hyperinflation or gas trapping was not specifically evaluated in the current study.
We have also found that allopurinol improved left ventricular ejection fraction in a subgroup of patients who had higher NT-proBNP level. A possible mechanism for this finding could be the ability of allopurinol to improve cardiac energetics by improving relative and absolute concentrations of myocardial high-energy phosphates and ATP flux through creatinine kinase, which has been observed in left heart failure.34
One of the limitations of this study is that right heart catheterisation (RHC), which is the gold standard for assessing pulmonary haemodynamics, was not performed. RHC is invasive and it would introduce unnecessary risk and reduce the number of patients willing to be recruited, hence potentially making recruits atypical. It is also not routine practice in patients with COPD and suspected borderline or mild pulmonary hypertension. Pulmonary acceleration time is a good alternative and non-invasive echo measurement that has a correlation coefficient of r = 0.88 with pulmonary pressures.35
Another limitation is the lack of data on transfer factor of the lung for carbon monoxide (TLCO). Reduction in TLCO is common in COPD, due to emphysema, while pulmonary hypertension causes a further reduction in the TLCO which would have helped to further characterise the study population. However, as it was not expected that allopurinol would have any beneficial effect on transfer factor and to limit testing burden on the trial participants, it was not included in the current study.
Conclusion
In conclusion, there was no overall effect of allopurinol on right ventricular mass in patients with pulmonary hypertension associated with chronic lung disease. There was also no overall effect of allopurinol on LV and RV measurements, quality of life, spirometry, six-minute walk test and blood markers.
There was a potential benefit of allopurinol in COPD patients with more severe airflow limitation (FEV1 < 50% predicted) and in patients with higher NT-proBNP levels. Further studies are warranted to assess the longer-term impact of allopurinol in this population.
Abbreviations
6MWT, six minute walk test; AE, adverse event; CMRI, cardiac magnetic resonance imaging; COPD, chronic obstructive pulmonary disease; EDV, end diastolic volume; EF, ejection fraction; ESV, end systolic volume; FEV1, forced expiratory volume in 1 second; Hs-Trop I, high-sensitive troponin I; ILD, interstitial lung disease; IPF, idiopathic pulmonary fibrosis; K-BILD, King’s brief interstitial lung disease; LTOT, long term oxygen therapy; LV, left ventricle; MPAP, mean pulmonary arterial pressure; NT-proBNP, N-terminal prohormone of brain natriuretic peptide; OS, oxidative stress; PH, pulmonary hypertension; PH-CLD, pulmonary hypertension associated with chronic lung disease; QOL, quality of life; RCT, randomised controlled trial; RHC, right heart catheterisation; RV, right ventricle; RVH, right ventricular hypertrophy; RVM, right ventricular mass; RVMI, right ventricular mass index; SAE, serious adverse event; SF-36, 36-Item Short Form Survey; SGRQ, St George’s Respiratory Questionnaire; SpO2, oxygen saturation; SV, stroke volume; TLCO, transfer factor of the lung for carbon monoxide; UA, uric acid; VMI; ventricular mass index.
Data Sharing Statement
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
The trial was approved by the East of Scotland Research Ethics Committee (reference: 14/ES/1035) and was carried out in accordance with the Declaration of Helsinki. All patients provided written informed consent.
Acknowledgments
The authors would like to thank the Scottish Primary Care Research Network (SPCRN) and the Scottish Health Research Register (SHARE) for helping in recruiting patients. This trial was supported by the Tayside Clinical Trials Unit.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Dr Liu-Shiu-Cheong reports personal fees from Chiesi, outside the submitted work. Dr Lipworth reports grants, personal fees and non-financial support from Chiesi, Boerhinger Ingeheim, personal fees and non-financial support from AstraZeneca and Thorasys, non-financial support from GSK, and personal fees from Novartis, Sanofi Genzyme, Lupin, Glenmark, Vectura, and Circassia, outside the submitted work. Dr Weir-McCall and Dr Houston have nothing to disclose. Dr Struthers reports grants from British Heart Foundation, during the conduct of the study; and in addition, has a patent issued for the use of xanthine oxidase inhibitors to treat chest pain in angina pectoris. The authors report no other potential conflicts of interest for this work.
References
1. Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46(4):903–975. doi:10.1183/13993003.01032-2015
2. Kessler R, Faller M, Weitzenblum E, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164(2):219–224. doi:10.1164/ajrccm.164.2.2006129
3. Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129(3):746–752. doi:10.1378/chest.129.3.746
4. Han MK, McLaughlin VV, Criner GJ, Martinez FJ. Pulmonary diseases and the heart. Circulation. 2007;116(25):2992–3005. doi:10.1161/CIRCULATIONAHA.106.685206
5. Pinsky MR. The right ventricle: interaction with the pulmonary circulation. Crit Care. 2016;20:266. doi:10.1186/s13054-016-1440-0
6. Terada LS, Guidot DM, Leff JA, et al. Hypoxia injures endothelial cells by increasing endogenous xanthine oxidase activity. Proc Natl Acad Sci U S A. 1992;89(8):3362–3366. doi:10.1073/pnas.89.8.3362
7. Kjæve J, Veel T, Bjertnies L. Allopurinol inhibits hypoxic pulmonary vasoconstriction. Role of toxic oxygen metabolites. Acta Anaesthesiol Scand. 1990;34(5):384–388. doi:10.1111/j.1399-6576.1990.tb03107.x
8. Williams A, Chen L, Scharf S. Effects of allopurinol on cardiac function and oxidant stress in chronic intermittent hypoxia. Sleep Breath. 2010;14(1):51–57. doi:10.1007/s11325-009-0279-x
9. Dopp JM, Philippi NR, Marcus NJ, et al. Xanthine oxidase inhibition attenuates endothelial dysfunction caused by chronic intermittent hypoxia in rats. Respiration. 2011;82(5):458–467. doi:10.1159/000329341
10. El Solh AA, Saliba R, Bosinski T, Grant BJB, Berbary E, Miller N. Allopurinol improves endothelial function in sleep apnoea: a randomised controlled study. Eur Resp J. 2006;27(5):997–1002. doi:10.1183/09031936.06.00101005
11. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. 2007;49(2):241–248. doi:10.1161/01.HYP.0000254415.31362.a7
12. Kao MP, Ang DS, Gandy SJ, et al. Allopurinol benefits left ventricular mass and endothelial dysfunction in chronic kidney disease. J Am Soc Nephrol. 2011;22(7):1382–1389. doi:10.1681/ASN.2010111185
13. Rekhraj S, Gandy SJ, Szwejkowski BR, et al. High-dose allopurinol reduces left ventricular mass in patients with ischemic heart disease. J Am Coll Cardiol. 2013;61(9):926–932. doi:10.1016/j.jacc.2012.09.066
14. Szwejkowski BR, Gandy SJ, Rekhraj S, et al. Allopurinol reduces left ventricular mass in patients with type 2 diabetes and left ventricular hypertrophy. J Am Coll Cardiol. 2013;62(24):2284–2293. doi:10.1016/j.jacc.2013.07.074
15. Ferrazza A, Marino B, Giusti V, Affinito V, Ragonese P. Usefulness of left and right oblique subcostal view in the echo-Doppler investigation of pulmonary arterial blood flow in patients with chronic obstructive pulmonary disease. The subxiphoid view in the echo-Doppler evaluation of pulmonary blood flow. Chest. 1990;98(2):286–289. doi:10.1378/chest.98.2.286
16. Dabestani A, Mahan G, Gardin JM, et al. Evaluation of pulmonary artery pressure and resistance by pulsed Doppler echocardiography. Am J Cardiol. 1987;59(6):662–668. doi:10.1016/0002-9149(87)91189-1
17. Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. doi:10.1183/09031936.05.00034805
18. ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166(1):111–117. doi:10.1164/ajrccm.166.1.at1102
19. Weir-McCall JR, Liu-Shiu-Cheong PS, Struthers AD, Lipworth BJ, Houston JG. Pulmonary arterial stiffening in COPD and its implications for right ventricular remodelling. Eur Radiol. 2018;28:3464–3472. doi:10.1007/s00330-018-5346-x
20. M RD. Simplified calculation of body-surface area. N Engl J Med. 1987;317(17):1098.
21. Bradlow WM, Hughes ML, Keenan NG, et al. Measuring the heart in pulmonary arterial hypertension (PAH): implications for trial study size. J Magnetic Resonance Imag. 2010;31(1):117–124. doi:10.1002/jmri.22011
22. Kovacs G, Agusti A, Barbera JA, et al. Pulmonary vascular involvement in chronic obstructive pulmonary disease. is there a pulmonary vascular phenotype? Am J Respir Crit Care Med. 2018;198(8):1000–1011. doi:10.1164/rccm.201801-0095PP
23. van Wolferen SA, Boonstra A, Marcus JT, et al. Right ventricular reverse remodelling after sildenafil in pulmonary arterial hypertension. Heart. 2006;92(12):1860–1861. doi:10.1136/hrt.2005.085118
24. Wilkins MR, Paul GA, Strange JW, et al. Sildenafil versus Endothelin Receptor Antagonist for Pulmonary Hypertension (SERAPH) study. Am J Respir Crit Care Med. 2005;171(11):1292–1297. doi:10.1164/rccm.200410-1411OC
25. Peacock AJ, Crawley S, McLure L, et al. Changes in right ventricular function measured by cardiac magnetic resonance imaging in patients receiving pulmonary arterial hypertension-targeted therapy: the EURO-MR study. Circ Cardiovasc Imaging. 2014;7(1):107–114. doi:10.1161/CIRCIMAGING.113.000629
26. Michelakis ED, Tymchak W, Noga M, et al. Long-term treatment with oral sildenafil is safe and improves functional capacity and hemodynamics in patients with pulmonary arterial hypertension. Circulation. 2003;108(17):2066–2069. doi:10.1161/01.CIR.0000099502.17776.C2
27. Roeleveld RJ, Vonk-Noordegraaf A, Marcus JT, et al. Effects of epoprostenol on right ventricular hypertrophy and dilatation in pulmonary hypertension. Chest. 2004;125(2):572–579. doi:10.1378/chest.125.2.572
28. van de Veerdonk MC, In TVAE H, Marcus JT, et al. Upfront combination therapy reduces right ventricular volumes in pulmonary arterial hypertension. Eur Respir J. 2017;49:6. doi:10.1183/13993003.00007-2017
29. Johns CS, Rajaram S, Capener DA, et al. Non-invasive methods for estimating mPAP in COPD using cardiovascular magnetic resonance imaging. Eur Radiol. 2018;28(4):1438–1448. doi:10.1007/s00330-017-5143-y
30. Saba TS, Foster J, Cockburn M, Cowan M, Peacock AJ. Ventricular mass index using magnetic resonance imaging accurately estimates pulmonary artery pressure. Eur Respir J. 2002;20(6):1519–1524. doi:10.1183/09031936.02.00014602
31. Simpson CE, Damico RL, Kolb TM, et al. Ventricular mass as a prognostic imaging biomarker in incident pulmonary arterial hypertension. Eur Respir J. 2019;53:4. doi:10.1183/13993003.02067-2018
32. Hohlfeld JM, Vogel-Claussen J, Biller H, et al. Effect of lung deflation with indacaterol plus glycopyrronium on ventricular filling in patients with hyperinflation and COPD (CLAIM): a double-blind, randomised, crossover, placebo-controlled, single-centre trial. Lancet Respir Med. 2018;6(5):368–378. doi:10.1016/S2213-2600(18)30054-7
33. Stone IS, Barnes NC, James WY, et al. Lung deflation and cardiovascular structure and function in chronic obstructive pulmonary disease. a randomized controlled trial. Am J Respir Crit Care Med. 2016;193(7):717–726. doi:10.1164/rccm.201508-1647OC
34. Hirsch GA, Bottomley PA, Gerstenblith G, Weiss RG. Allopurinol acutely increases adenosine triphospate energy delivery in failing human hearts. J Am Coll Cardiol. 2012;59(9):802–808. doi:10.1016/j.jacc.2011.10.895
35. Kiely DG, Cargill RI, Wheeldon NM, Coutie WJ, Lipworth BJ. Haemodynamic and endocrine effects of type 1 angiotensin II receptor blockade in patients with hypoxaemic cor pulmonale. Cardiovasc Res. 1997;33(1):201–208. doi:10.1016/S0008-6363(96)00180-0
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.