Back to Journals » Breast Cancer: Targets and Therapy » Volume 17
ALKBH5 Promotes Breast Cancer Stemness Through Regulating Wnt/β-Catenin Signaling
Received 3 February 2025
Accepted for publication 4 June 2025
Published 6 June 2025 Volume 2025:17 Pages 471—482
DOI https://doi.org/10.2147/BCTT.S520532
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Robert Clarke
Kailin Wang,1,2 Kaiting Wang1
1Institute for Hepatology, National Clinical Research Center for Infectious Disease, Shenzhen Third People’s Hospital, The Second Affiliated Hospital, School of Medicine, Southern University of Science and Technology, Shenzhen, 518112, People’s Republic of China; 2Breast Cancer Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, 510289, People’s Republic of China
Correspondence: Kaiting Wang, Institute for Hepatology, National Clinical Research Center for Infectious Disease, Shenzhen Third People’s Hospital, The Second Affiliated Hospital, School of Medicine, Southern University of Science and Technology, Shenzhen, 518112, People’s Republic of China, Tel +86-0755-61238989, Email [email protected]
Background: Breast cancer is the most prevalent disease and the fourth cause of cancer death among female globally. The N6-methyladenylate methylation (m6A) demethylase alpha-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) decreases modification of RNA, while its role in regulating breast cancer development remains unclear.
Methods: ALKBH5-silenced breast cancer cell-line MCF-7 was constructed to investigate its functional impact. Cell proliferation, migration and invasion ability were evaluated by CCK8 and transwell assays under ALKBH5 inhibition. Spheroid formation and in vitro extreme limiting dilution analysis (ELDA) were performed to elucidate the effect of ALKBH5 deficiency on stemness of MCF-7 cells. The m6A modification level of CTNNB1 and the interaction of ALKBH5 and CTNNB1 were investigated by Methylated RNA immunoprecipitation (MeRIP) and RIP assay respectively.
Results: Silencing ALKBH5 significantly suppressed MCF-7 cell proliferation, migration, and invasion abilities. Moreover, ALKBH5 depletion also diminished the stemness of breast cancer cells in vitro. Further investigation illustrated that ALKBH5 may regulate Wnt/β-catenin signaling via an m6A-dependant manner. Clinical data analysis demonstrated a strong positive relationship between ALKBH5 and β-catenin expression.
Conclusion: This study establishes a link between ALKBH5 and cancer stemness in breast cancer, providing insights into the functional role of demethylase ALKBH5 in breast cancer progression.
Keywords: N6-methyladenosine, (m6A), breast cancer, ALKBH5, stemness, CTNNB1
Introduction
Breast cancer is the second leading cause of tumor-related mortality among women worldwide. Early detection and screening are crucial for reducing morbidity and mortality, and improving survival rate.1,2 However, due to the complexity and heterogeneity of breast invasive carcinoma (BRCA), it is challenging to select the appropriate treatment options and conduct accurate clinical prognostic analysis.3–5 A deeper understanding of the underlying molecular mechanisms driving breast cancer development is essential to address these challenges.6
RNA N6-methyladenosine (m6A) modification is the most abundant mRNA modification, playing a critical role in regulating gene expression.7,8 The m6A RNA modification is a dynamic and reversible process coordinated by methyltransferase (m6A writers), m6A-binding proteins (m6A readers), and m6A demethylase (m6A erasers).9–12 The m6A modification determines the fate of mRNA, including splicing, translation, or degradation.13–15 Accumulating evidence demonstrated that alteration of m6A levels participated in tumor pathogenesis and progression by regulating expression of tumor-related genes.16,17
In breast cancer, it was found that m6A writer METTL3 induced m6A modification of LATS1 mRNA, which was further recognized by m6A reader YTHDF2, promoting both tumorigenesis and glycolysis.18 IGF2BP1, as an m6A reader, could induce breast cancer metastasis by enhancing CPT1A mRNA stability in an m6A-dependent manner.19 Fat mass and obesity associated protein (FTO) and a-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) were two major m6A erasers, reversing the RNA methylation process.20–22 Niu et al demonstrated that FTO promoted proliferation, colony formation and metastasis of breast cancer cell in vitro and in vivo through demethylation of m6A marks from BNIP3.23 In addition, several studies have also indicated the role of FTO in facilitating chemoresistance and epithelial–mesenchymal transition (EMT) in breast tumor cells.24–26 However, the contribution of ALKBH5 to breast cancer remain poorly understood. It was found that ALKBH5-mediated m6A demethylation not only induced glycolysis and resistance to HER2-targeted therapy, but also contributed to cancer stem cells (CSCs) characteristics and doxorubicin-resistance in breast cancer.27,28 There are yet few studies on the involvement of ALKBH5 in breast cancer progression.
This study aimed to investigate the potential role of ALKBH5 in breast tumor development and progression. It is expected to provide new clues and insights into breast cancer management.
Methods
Cell Culture
Breast cancer cell-line MCF-7 was obtained from American Type Culture Collection (ATCC). Cells were cultured in DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in a humidified atmosphere with 5% CO2 at 37°C.
Lentiviral Transfection
We performed cell infection experiments using lentiviral vectors encoding shRNA sequences targeting the gene of ALKBH5 (shALKBH5 sequence: GATCCTGGAAATGGACAAAGA). Breast cancer cells were seeded in a 6-well plate and infected with the lentiviral particles at a multiplicity of infection (MOI) of 20. After 48 h of infection, the cells were subjected to 4 μg/mL puromycin selection to establish stable ALKBH5-knockdown cell line.
Western Blot Analysis
Total protein was extracted from MCF-7 breast cancer cell line using RIPA buffer supplemented with protease inhibitors. Protein concentrations were determined using a BCA protein assay kit (ThermoFisher Scientific). Equal amounts of protein were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. Membranes were blocked in 5% non-fat milk and then incubated with primary antibodies targeting ALKBH5 (Novus, NBP1-82188), β-catenin (Abcam, ab32572), AFP (Abcam, ab284388), CK19 (ABclonal, A19040), C-Myc (SANTA CRUZ, sc-40), Cyclin D1 (Cell Signaling Technology, 2922), GAPDH (ABclonal, A19056), N6-methyladenosine (ABclonal, A22411) overnight at 4°C. Following incubation with secondary antibodies in room temperature for 1 h, protein bands were visualized using the LI-COR Odyssey imaging system.
RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted from cells using Trizol reagent following the manufacturer’s instructions. Complementary DNA was synthesized using a reverse transcription kit (Promega). Quantitative PCR was carried out with SYBR® Green Master Mix (Roche) and a Roche LightCycler 96. All primers used in this study are listed in Additional file Table S1.
Methylated RNA Immunoprecipitation-qPCR (MeRIP-qPCR)
The MeRIP assay was performed with the RNA Immunoprecipitation Kit. MCF-7 cells were used to extract RNA. RNA samples were then immunoprecipitated with m6A antibody or IgG. The isolated m6A-RIP RNA was quantified by qRT-PCR.
RNA Immunoprecipitation (RIP)
RIP was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit. Cells were lysed, and the RNA-binding proteins were immunoprecipitated using specific antibody against ALKBH5. RNA was extracted from immunoprecipitated complexes and subjected to qRT-PCR.
M6A Dot Blot Analysis
The levels of N6-methyladenosine modification in RNA were determined using a dot blot assay. Briefly, RNA samples were spotted onto a nitrocellulose membrane and UV-crosslinked (254nm). The membrane was then blocked and probed with an m6A-specific antibody overnight at 4°C. The membrane was incubated with fluorescent secondary antibody and the dot blots were visualized by Odyssey® CLx Infrared Imaging System. Total input RNA was spotted by staining with 0.02% Methylene blue.
Cell Viability Assay
Cell viability was assessed using the Cell Counting Kit-8 (CCK-8) assay according to the manufacturer’s instructions. Briefly, cells were seeded into 96-well plates at a density of 1000 cells per well. CCK-8 solution was added to each well and incubated for 1 hours at 37°C. Absorbance at 450 nm was measured and the cell viability was normalized to day 0.
Colony Formation Assay and Sphere Formation Assay
Cells were plated at a density of 1000 cells per well in a 6-well plate and allowed to grow for 7–10 days. At the end of the incubation period, colonies were fixed with 10% formalin and stained with crystal violet. Sphere formation assay was performed using ultra-low attachment 6-well plates and the cells were incubated for 7–14 days to allow for spheroid formation. Spheroid formation was assessed using an inverted microscope, and the number was measured.
Migration and Invasion Assay
For the migration assay, a total of 1x10^5 cells were seeded in the upper chamber of a 24-well Transwell plate with a pore size of 8 μm. The lower chamber contained complete growth medium with 10% FBS as a chemoattractant. After 48 hours of incubation, the cells that had migrated through the membrane were fixed with 10% paraformaldehyde, stained with crystal violet, and then photographed under a light microscope. For the invasion assay, a similar setup was used as in the migration assay, but the Transwell membrane was pre-coated with Matrigel (BD Biosciences) to mimic the extracellular matrix. After 48 hours of incubation, the invaded cells were fixed, stained, and photographed as described for the migration assay.
Extreme Limiting Dilution Assay (ELDA)
To assess the stemness of breast cancer cells, ELDA was performed. MCF-7 cells were sorted into ultra-low attachment 96-well plates at a range of dilutions, starting from 500 cells down to 1 cell per well. The plates were incubated for 1–2 weeks to allow for the formation of mammospheres. The frequency of tumorigenic cells was calculated using the ELDA web tool.
Statistical Analysis
Data were analyzed using GraphPad Prism 9.0 software. Results were presented as means ± standard deviation of at least three independent experiments. Statistical significance was determined using two-tailed T-test, with p<0.05 considered significant.
Results
ALKBH5 Promotes Breast Cancer Cell Proliferation, Colony Formation, Migration and Invasion Abilities
To investigate the role of demethylase ALKBH5 in breast cancer cell, we generated stable ALKBH5-knockdown (shALKBH5) and control (shNC) MCF-7 cell line. The transfection efficiency was verified by qRT-PCR and Western blot (Figure 1A). Next, we performed CCK8 and colony formation assay to evaluate cell growth under ALKBH5 knockdown and found that depletion of ALKBH5 significantly inhibited MCF-7 cell proliferation and colony formation abilities (Figure 1B and C). Transwell assay further demonstrated that ALKBH5 silencing dramatically reduced cell migration and invasion abilities (Figure 1D). These functional experiments collectively revealed silencing ALKBH5 dampened malignant behaviors of MCF-7 cells, including proliferation, colony formation, migration and invasion abilities in vitro.
 |
Figure 1 ALKBH5 promotes MCF-7 cells proliferation, migration, and invasion. (A) The knockdown efficacy of shALKBH5 in MCF-7 cells was verified by qRT-PCR and Western blot. (B). Cell growth was measured using CCK8 assay. (C). Representative images of colony formation assay and the number of colonies was counted in ALKBH5-knockdown cells. (D). Transwell assay was conducted to determine cell migration and invasion abilities after silencing ALKBH5. Statistical significance was determined using two-tailed T-test, with p<0.05 considered significant. *P<0.05, **P<0.01, ****P<0.0001. |
Depleting ALKBH5 Decreases CSCs Characteristics of MCF-7 Cells
Methyltransferase WTAP was proved to be affected by NRP1 to enhance stem cell properties and induce radioresistance of breast cancer cells.29 METTL3 regulated m6A level of SOX2 mRNA to promote the stemness and malignant progression of MCF-7 cell.30 YTHDF1 could target E2F8 and modulate its mRNA stability, leading to breast cancer cell growth, DNA damage and chemoresistance.31 Given the established roles of m6A regulators in stem cell properties and breast cancer progression, we investigated whether ALKBH5 was involved in breast cancer stemness. The correlation of m6A modification enzymes (ALKBH5, FTO, WTAP, METTL14, YTHDF2, and IGF2BP1) and stemness-related genes (EPCAM, KRT19, CD44, NANOG, MYC and AFP) was analyzed by the Breast Cancer Gene-Expression Miner v5.1 database (bc-GenExMiner v5.1) (Figure 2 and Supplementary Figure 1).32 The result showed ALKBH5 but not other enzymes was positively correlated with most stemness marker genes, indicating ALKBH5 may contribute to CSCs feature regulation. Sphere formation assay is commonly used to determine cell-line stemness.33 As shown in Figure 3A and B, knockdown of ALKBH5 suppressed sphere formation in MCF-7 cells (Figure 3A and B). Furthermore, in vitro limiting dilution assays verified significant decrease of stem cell frequency in shALKBH5 MCF-7 cells compared with counterpart control cells (p=0.0381) (Figure 3C and D). Stem cell frequency was estimated using public ELDA software (http://bioinf.wehi.edu.au/software/elda/).34 We also assessed the mRNA level of stemness marker genes following ALKBH5 knockdown using qRT-PCR (Figure 4E and Supplementary Figure 2). AFP and CK19 represented the degree of differentiation. Western blot assay suggested ALKBH5 silence notably decreased the protein level of AFP and CK19, further confirmed the impact of ALKBH5 on CSCs characteristics of MCF-7 cells (Figure 4F). Taken together, our results strongly suggested that ALKBH5 plays a critical role in enhancing stemness in breast tumor cell.
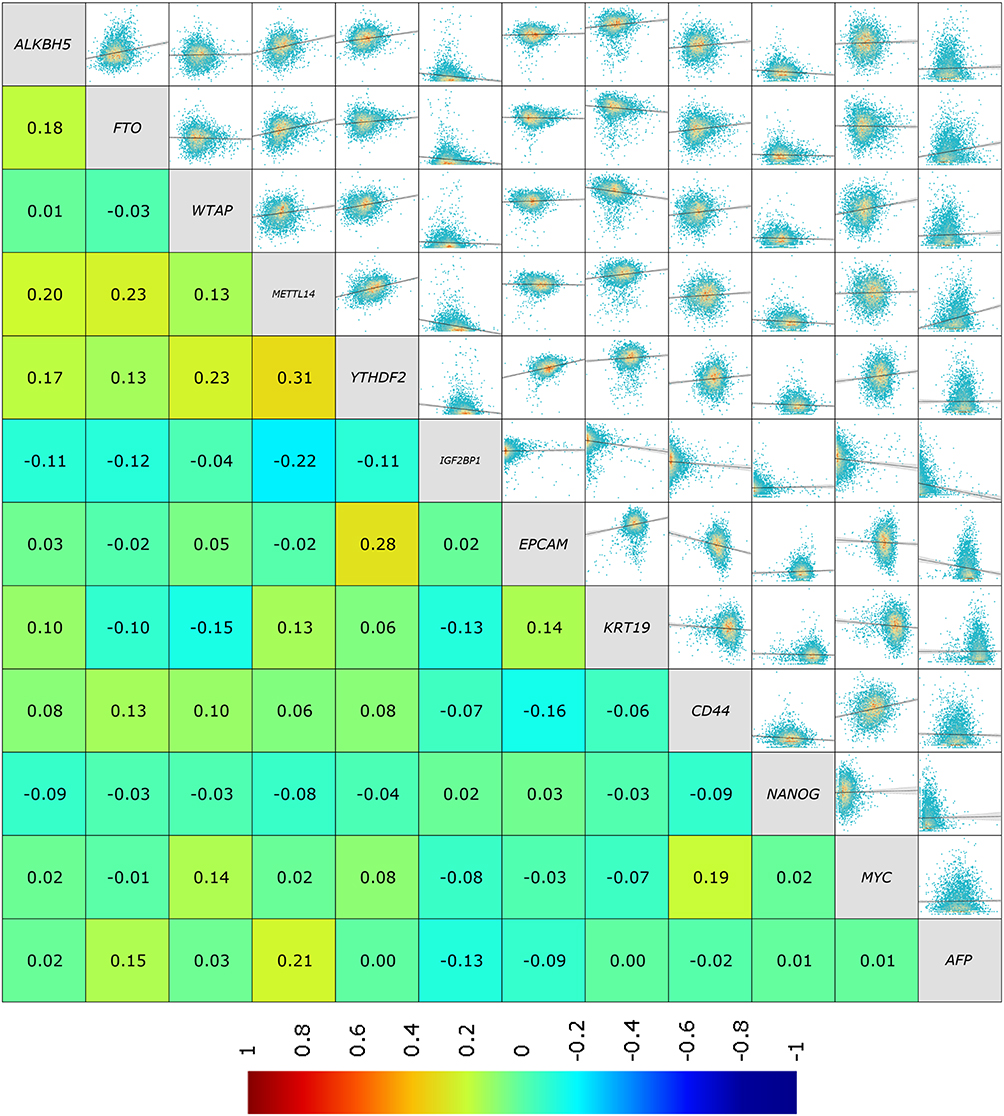 |
Figure 2 Correlation between m6A modification enzymes and stemness-associated genes. The relationship between m6A modification enzymes gene and stemness-associated genes was analyzed by the Breast Cancer Gene-Expression Miner v5.1 database (bc-GenExMiner v5.1). |
 |
Figure 3 Silencing ALKBH5 inhibits sphere formation capability of MCF-7 cells. (A) Mammosphere-forming ability of the stable cell lines was measured in ultra-low attachment plates. (B) The number of spheroids formed was quantified. (C) Representative images of spheroids formed in ELDA assay. (D) ELDA expressing median values from ALKBH5-knockdown MCF-7 cells (Control, black curves; ALKBH5-knockdown, red curves). The table shows the stem cell frequency and p value calculated by ELDA. (E) The mRNA level of stemness-related genes (NANOG, EPCAM, CK19) was measured by qRT-PCR in ALKBH5-knockdown MCF-7 spheroids. (F) The protein levels of AFP and CK19 in nonsphere cell line. Statistical significance was determined using two-tailed T-test, with p<0.05 considered significant. *P<0.05, **P<0.01, ***P<0.001. |
 |
Figure 4 ALKBH5 regulates β-catenin expression via an m6A-dependent manner. (A) Dot blot analyses of m6A levels in total RNA. (B) The mRNA level of CTNNB1 in adherent MCF-7 cells and spheroids under ALKBH5 inhibition. (C) The protein expression level of β-catenin, C-MYC, and Cyclin D1 in ALKBH5-silenced MCF-7 cells. (D) The protein level of β-catenin in MCF-7 forming sphere after suppressing ALKBH5. (E) Enrichment of m6A in CTNNB1 mRNA by Me-RIP qPCR assay. (F) RIP-qPCR confirmed ALKBH5 binding to CTNNB1 mRNA. Statistical significance was determined using two-tailed T-test, with p<0.05 considered significant. *P<0.05, **P<0.01. |
ALKBH5 Regulated m6A Modification of CTNNB1
M6A dot blot assay was employed to examine m6A level of cells. The result showed that ALKBH5 knockdown significantly increased the global RNA m6A modification levels (Figure 4A). Wnt/β-catenin signaling is known to be involved in maintaining stemness and cell proliferation.35–38 Interestingly, among ALKBH5-associated stemness genes analyzed by public dataset, CD44 and MYC were canonical Wnt target genes (Figure 2).39,40 This prompted us to investigate the expression of CTNNB1 in MCF-7 sphere and nonsphere cells by qRT-PCR. A consistent decline of CTNNB1 mRNA was observed after ALKBH5 loss, indicating Wnt/β-catenin pathway may be potential target of ALKBH5 in breast tumor (Figure 4B). Western blot analysis further demonstrated this signaling was markedly suppressed in ALKBH5-silenced MCF-7 cells, as evidenced by reduced levels of β-catenin and its downstream target, C-MYC and Cyclin D1, in adherent cells (Figure 4C).41 Moreover, this downregulation of β-catenin protein was also observed in ALKBH5-silenced MCF-7 spheroid cells (Figure 4D). According to the above results, we preliminarily speculated that CTNNB1 might be a direct target of ALKBH5. To substantiate this conjecture, the m6A abundance of CTNNB1 was measured by methylated RNA immune-precipitation qPCR (Me-RIP qPCR), which revealed that CTNNB1 mRNA was affected by m6A modification (Figure 4E). In addition, the direct binding of ALKBH5 and CTNNB1 mRNA was observed by RIP-qPCR using anti-ALKBH5 antibody (Figure 4F). The m6A sites of CTNNB1 were predicted using two databases, RMBase v3.0 (https://rna.sysu.edu.cn/rmbase3/) and RM2Target (http://rm2target.canceromics.org/).42–46 A total of 98 and 4 potential m6A sites were identified by these two tools, respectively (Figure 5A and B). Collectively, these initial analyses have suggested that ALKBH5 depletion may regulate Wnt/β-catenin signaling pathway via an m6A-dependent manner, contributing to impaired cell proliferation, migration, invasion, and stemness characteristics. CTNNB1 appeared to be a potential target of ALKBH5, warranting further exploration to elucidate this interaction.
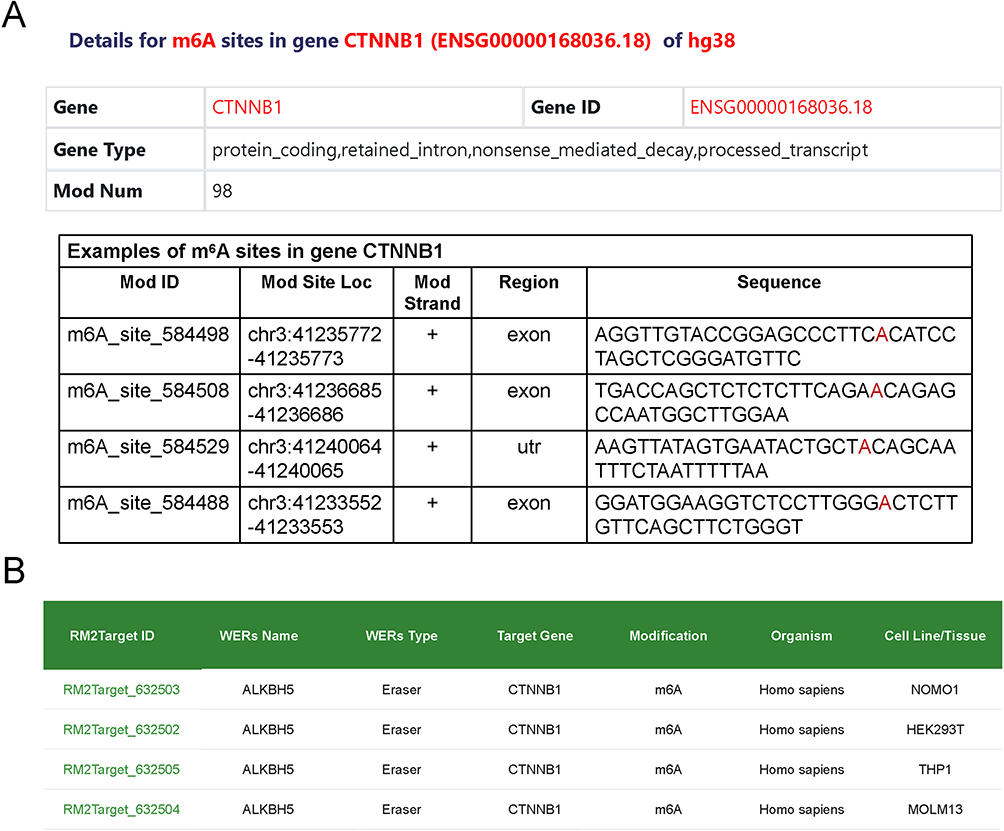 |
Figure 5 The predicted m6A modification sites. (A and B) 98 and 4 predicted m6A sites generated by RMBase v3.0 (A) and RM2Target (B) respectively. |
The Positive Correlation of β-Catenin and ALKBH5 in Clinical Data
To discuss the clinical significance of ALKBH5 in breast cancer, we first utilized the Cancer Proteogenomic Data Analysis Site database (cProSite) (https://cprosite.ccr.cancer.gov) to explore the relative protein abundance of ALKBH5 across various cancers compared to their corresponding normal tissues (Figure 6A). Specifically, relative ALKBH5 expression was the most significantly elevated in breast tumor, followed by lung adenocarcinoma (Figure 6B). This implied a potential pro-carcinoma function of ALKBH5 in breast cancer. Kaplan–Meier survival analysis of overall survival (OS) and disease-free survival (DFS) was generated with Breast Cancer Gene-Expression Miner v5.1 platform (bc-GenExMiner v5.1). Contrary to our expectations, ALKBH5 was not significantly correlated with overall survival (OS) of breast cancer patients. Moreover, higher expression of ALKBH5 even predicted a better disease-free survival (DFS), suggesting the involvement of other mechanisms regulated by ALKBH5 and highlighting its complex role in breast cancer (Figure 6C). Moreover, we analyzed the correlation between ALKBH5 expression and prognosis across different subtypes, including ER, PR, nodal status, and molecular subtype HER2-E classification. Consistent with prior observations, ALKBH5 expression did not demonstrate a significant prognostic impact (Supplementary Figure 3). We further analyze the association between ALKBH5 and β-catenin expression. The result revealed that patients with high ALKBH5 mRNA levels tended to have high CTNNB1 expression (Figure 6D). This positive correlation was also observed by the Gene Expression Profiling Interactive Analysis (GEPIA) database (Figure 6E).47 Additionally, according to analysis from cBioportal (http://www.cbioportal.org/),48 the protein levels of ALKBH5 correlated positively with β-catenin expression (Figure 6F). Altogether, although ALKBH5 may not be an ideal prognostic factor on its own, its significant association with β-catenin was evident in multiple clinical datasets. These findings suggest that combining ALKBH5 and β-catenin expression levels may offer a more robust prognostic marker for breast cancer.
 |
Figure 6 ALKBH5 is positively correlated with CTNNB1 expression in breast cancer. (A and B) Using cProSite database to analyse the relative ALKBH5 protein abundance in various tumor types (A) and the relative protein expression level of ALKBH5 in breast tumor and adjacent normal tissue (B). (C) Kaplan-Meier survival analysis of overall survival (OS) and disease-free survival (DFS) by Breast Cancer Gene-Expression Miner v5.1 platform. (D) Patients with high ALKBH5 expression demonstrated high CTNNB1 mRNA levels. (E) Gene Expression Profiling Interactive Analysis (GEPIA) database showed the positive correlation of ALKBH5 and CTNNB1 mRNA. (F) The protein level of ALKBH5 was significantly positively associated with β-catenin protein level performed with cBioportal database. |
Conclusion
The involvement of m6A epigenetic modifications in breast cancer has been extensively studied in recent years. Previous research has found that high expression of METTL14, WTAP and FTO, but not ALKBH5 was correlated with good metastasis relapse-free survival in breast cancer. Induction of m6A by overexpression of METTL14 or knockdown of ALKBH5 inhibited cell viability, colony formation, and migration.49 Zhang et al proved that knockdown of ALKBH5 not only decreased expression of NANOG, which is a pluripotency factor promoting CSCs phenotype, but also confirmed ALKBH5 was required for tumorigenesis and lung metastasis in breast cancer within the hypoxia condition.50,51 These researches highlighted the significant role of ALKBH5 in breast tumor development and progression. In our study, we suggested ALKBH5 mediated demethylation of CTNNB1 mRNA, thus affecting Wnt/β-catenin signaling pathway and stemness associated genes including NANOG. Whether NANOG was regulated directly by ALKBH5 in this study warranted further verification. In addition to Wnt/β-catenin pathway, it is important to note that other stemness-related pathways, such as Hippo, Hedgehog may also be involved.52–54 Further research is needed to explore the potential interplay of ALKBH5 with these pathways in the context of breast cancer stemness.
While our study focuses on ALKBH5, it is essential to acknowledge the complex interplay between different m6A regulators in breast cancer. Previous studies highlight that other m6A erasers like FTO also contribute to tumorigenesis and metastasis.25 Further exploration of which m6A reader recognizing m6A modification on CTNNB1 induced by ALKBH5 in MCF-7 cells was still needed. Understanding how ALKBH5 interacts with other regulators would provide a more comprehensive picture of the m6A landscape in breast cancer and its implications for targeted treatment. Despite we revealed that ALKBH5 could promote stemness of breast cancer cell in vitro, several avenues like the role of ALKBH5 in different breast cancer subtypes and its potential as a biomarker for patient stratification are worthy of future studies. Additionally, in vivo models would be invaluable for assessing the therapeutic potential of targeting ALKBH5 in breast cancer treatment.
In conclusion, we found depletion of ALKBH5 significantly inhibited the proliferation, colony formation, and migration, invasion abilities of MCF-7 cells in vitro. We also observed that the CSCs characteristics in MCF-7 cells were suppressed under ALKBH5 knockdown. These findings establish a crucial link between ALKBH5 and breast cancer progression, particularly through its impact on cancer stemness and the Wnt/β-catenin signaling pathway. These insights pave the way for potential therapeutic interventions aimed at modulating ALKBH5 activity in breast cancer management.
Data Sharing Statement
All data analyzed during this study are included in this published article and its supplementary information files.
Ethical Approval
No ethical approval is required in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca a Cancer J Clinicians. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Jokar N, Velikyan I, Ahmadzadehfar H, et al. Theranostic approach in breast cancer: a treasured tailor for future oncology. Clin Nucl Med. 2021;46:e410–e20. doi:10.1097/rlu.0000000000003678
3. Kawiak A. Molecular research and treatment of breast cancer. Int J Mol Sci. 2022. doi:10.3390/ijms23179617
4. Veronesi U, Boyle P, Goldhirsch A, Orecchia R, Viale G. Breast cancer. Lancet. 2005;365:1727–1741. doi:10.1016/s0140-6736(05)66546-4
5. Trapani D, Ginsburg O, Fadelu T, et al. Global challenges and policy solutions in breast cancer control. Cancer Treat Rev. 2022;104:102339. doi:10.1016/j.ctrv.2022.102339
6. Lüönd F, Tiede S, Christofori G. Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during malignant progression. Br J Cancer. 2021;125:164–175. doi:10.1038/s41416-021-01328-7
7. Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacothe. 2019;112:108613. doi:10.1016/j.biopha.2019.108613
8. Qiu Y, Man C, Zhu L, et al. R-loops’ m6A modification and its roles in cancers. Mol Cancer. 2024;23:232. doi:10.1186/s12943-024-02148-y
9. Zhang H, Shi X, Huang T, et al. Dynamic landscape and evolution of m6A methylation in human. Nucleic Acids Res. 2020;48:6251–6264. doi:10.1093/nar/gkaa347
10. Zhou H, Yin K, Zhang Y, Tian J, Wang S. The RNA m6A writer METTL14 in cancers: roles, structures, and applications. Biochim Biophys Acta Rev Cancer. 2021;1876:188609. doi:10.1016/j.bbcan.2021.188609
11. Huang Q, Mo J, Liao Z, Chen X, Zhang B. The RNA m(6)A writer WTAP in diseases: structure, roles, and mechanisms. Cell Death Dis. 2022;13:852. doi:10.1038/s41419-022-05268-9
12. Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Molecular Cell. 2019;74:640–650. doi:10.1016/j.molcel.2019.04.025
13. He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18:176. doi:10.1186/s12943-019-1109-9
14. Fang Z, Mei W, Qu C, et al. Role of m6A writers, erasers and readers in cancer. Exp Hematol Oncol. 2022;11:45. doi:10.1186/s40164-022-00298-7
15. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m⁶A RNA methylation. Nat Rev Genet. 2014;15:293–306. doi:10.1038/nrg3724
16. An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21:14. doi:10.1186/s12943-022-01500-4
17. Chen XY, Zhang J, Zhu JS. The role of m(6)A RNA methylation in human cancer. Mol Cancer. 2019;18:103. doi:10.1186/s12943-019-1033-z
18. Xu Y, Song M, Hong Z, et al. The N6-methyladenosine METTL3 regulates tumorigenesis and glycolysis by mediating m6A methylation of the tumor suppressor LATS1 in breast cancer. J Exp Clin Cancer Res. 2023;42:10. doi:10.1186/s13046-022-02581-1
19. Shi J, Zhang Q, Yin X, et al. Stabilization of IGF2BP1 by USP10 promotes breast cancer metastasis via CPT1A in an m6A-dependent manner. Int J Bio Sci. 2023;19:449–464. doi:10.7150/ijbs.76798
20. Jiang X, Liu B, Nie Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74. doi:10.1038/s41392-020-00450-x
21. Loos RJ, Yeo GS. The bigger picture of FTO: the first GWAS-identified obesity gene. Nat Rev Endocrinol. 2014;10:51–61. doi:10.1038/nrendo.2013.227
22. Qu J, Yan H, Hou Y, et al. RNA demethylase ALKBH5 in cancer: from mechanisms to therapeutic potential. J Hematol Oncol. 2022;15:8. doi:10.1186/s13045-022-01224-4
23. Niu Y, Lin Z, Wan A, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18:46. doi:10.1186/s12943-019-1004-4
24. Ou B, Liu Y, Gao Z, et al. Senescent neutrophils-derived exosomal piRNA-17560 promotes chemoresistance and EMT of breast cancer via FTO-mediated m6A demethylation. Cell Death Dis. 2022;13:905. doi:10.1038/s41419-022-05317-3
25. Xu Y, Ye S, Zhang N, et al. The FTO/miR-181b-3p/ARL5B signaling pathway regulates cell migration and invasion in breast cancer. Cancer Commun. 2020;40:484–500. doi:10.1002/cac2.12075
26. Lin W, Mo CQ, Kong LJ, Chen L, Wu KL, Wu X. FTO-mediated epigenetic upregulation of LINC01559 confers cell resistance to docetaxel in breast carcinoma by suppressing miR-1343-3p. Kaohsiung J Med Sci. 2023;39:873–882. doi:10.1002/kjm2.12728
27. Zhao L, Kang M, Liu X, et al. UBR7 inhibits HCC tumorigenesis by targeting Keap1/Nrf2/Bach1/HK2 and glycolysis. J Exp Clin Cancer Res. 2022;41:330. doi:10.1186/s13046-022-02528-6
28. Liu X, Li P, Huang Y, et al. M(6)A demethylase ALKBH5 regulates FOXO1 mRNA stability and chemoresistance in triple-negative breast cancer. Redox Biol. 2024;69:102993. doi:10.1016/j.redox.2023.102993
29. Wang Y, Zhang L, Sun XL, et al. NRP1 contributes to stemness and potentiates radioresistance via WTAP-mediated m6A methylation of Bcl-2 mRNA in breast cancer. Apoptosis. 2023;28:233–246. doi:10.1007/s10495-022-01784-3
30. Xie J, Ba J, Zhang M, Wan Y, Jin Z, Yao Y. The m6A methyltransferase METTL3 promotes the stemness and malignant progression of breast cancer by mediating m6A modification on SOX2. J BUON. 2021;26:444–449.
31. Sun Y, Dong D, Xia Y, Hao L, Wang W, Zhao C. YTHDF1 promotes breast cancer cell growth, DNA damage repair and chemoresistance. Cell Death Dis. 2022;13:230. doi:10.1038/s41419-022-04672-5
32. Jézéquel P, Frénel JS, Campion L, et al. Bc-GenExMiner 3.0: new mining module computes breast cancer gene expression correlation analyses. Database. 2013;2013:bas060. doi:10.1093/database/bas060
33. Gao W, Wen H, Liang L, et al. IL20RA signaling enhances stemness and promotes the formation of an immunosuppressive microenvironment in breast cancer. Theranostics. 2021;11:2564–2580. doi:10.7150/thno.45280
34. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Method. 2009;347:70–78. doi:10.1016/j.jim.2009.06.008
35. Liu X, Su K, Sun X, et al. Sec62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/β-catenin pathway. J Exp Clin Cancer Res. 2021;40:132. doi:10.1186/s13046-021-01934-6
36. Wei CY, Zhu MX, Yang YW, et al. Downregulation of RNF128 activates Wnt/β-catenin signaling to induce cellular EMT and stemness via CD44 and CTTN ubiquitination in melanoma. J Hematol Oncol. 2019;12:21. doi:10.1186/s13045-019-0711-z
37. Cho YH, Ro EJ, Yoon JS, et al. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat Commun. 2020;11:5321. doi:10.1038/s41467-020-19173-2
38. Tang Q, Chen J, Di Z, et al. TM4SF1 promotes EMT and cancer stemness via the Wnt/β-catenin/SOX2 pathway in colorectal cancer. J Exp Clin Cancer Res. 2020;39:232. doi:10.1186/s13046-020-01690-z
39. Roy S, Kar M, Roy S, et al. Inhibition of CD44 sensitizes cisplatin-resistance and affects Wnt/β-catenin signaling in HNSCC cells. Int J Biol Macromol. 2020;149:501–512. doi:10.1016/j.ijbiomac.2020.01.131
40. Zhang C, Liu L, Li W, et al. Upregulation of FAM83F by c-Myc promotes cervical cancer growth and aerobic glycolysis via Wnt/β-catenin signaling activation. Cell Death Dis. 2023;14:837. doi:10.1038/s41419-023-06377-9
41. Chen YY, Chen Y, Wang WC, et al. Cyclin D1 regulates osteoarthritis chondrocyte apoptosis via WNT3/β-catenin signalling. Artif Cells Nanomed Biotechnol. 2019;47:1971–1977. doi:10.1080/21691401.2019.1593853
42. Sun WJ, Li JH, Liu S, et al. RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016;44:D259–65. doi:10.1093/nar/gkv1036
43. Xuan J, Chen L, Chen Z, et al. RMBase v3.0: decode the landscape, mechanisms and functions of RNA modifications. Nucleic Acids Res. 2024;52:D273–d84. doi:10.1093/nar/gkad1070
44. Xuan JJ, Sun WJ, Lin PH, et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46:D327–d34. doi:10.1093/nar/gkx934
45. Bao X, Zhang Y, Li H, et al. RM2Target: a comprehensive database for targets of writers, erasers and readers of RNA modifications. Nucleic Acids Res. 2023;51:D269–d79. doi:10.1093/nar/gkac945
46. Deng S, Zhang H, Zhu K, et al. M6A2Target: a comprehensive database for targets of m6A writers, erasers and readers. Briefings Bioinf. 2021;22. doi:10.1093/bib/bbaa055
47. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–w102. doi:10.1093/nar/gkx247
48. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. 2012;2:401–404. doi:10.1158/2159-8290.Cd-12-0095
49. Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19:326. doi:10.1186/s12885-019-5538-z
50. Zhang C, Samanta D, Lu H, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA. 2016;113:E2047–56. doi:10.1073/pnas.1602883113
51. Zhang C, Zhi WI, Lu H, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7:64527–64542. doi:10.18632/oncotarget.11743
52. Wang Z, Da Silva TG, Jin K, et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer Res. 2014;74:6364–6374. doi:10.1158/0008-5472.Can-14-2051
53. Xiong YX, Zhang XC, Zhu JH, et al. Collagen I-DDR1 signaling promotes hepatocellular carcinoma cell stemness via hippo signaling repression. Cell Death Differ. 2023;30:1648–1665. doi:10.1038/s41418-023-01166-5
54. Zhu R, Gires O, Zhu L, et al. TSPAN8 promotes cancer cell stemness via activation of sonic hedgehog signaling. Nat Commun. 2019;10:2863. doi:10.1038/s41467-019-10739-3
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.