")
Back to Journals » OncoTargets and Therapy » Volume 11
ALDH1A2 suppresses epithelial ovarian cancer cell proliferation and migration by downregulating STAT3
Authors Wang Y, Shao F, Chen L
Received 8 July 2017
Accepted for publication 17 August 2017
Published 31 January 2018 Volume 2018:11 Pages 599—608
DOI https://doi.org/10.2147/OTT.S145864
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Samir Farghaly
Yichen Wang, Feng Shao, Lu Chen
Department of Gynecologic Oncology, Zhejiang Cancer Hospital, Hangzhou, China
Abstract: Epithelial ovarian cancer is the deadliest gynecological malignancy worldwide. A better understanding of epithelial ovarian cancer pathogenesis and the molecular mechanism underlying its metastasis may increase overall survival rates. Previous studies have indicated that aldehyde dehydrogenase 1 family member A2 (ALDH1A2) is a candidate tumor suppressor in epithelial ovarian cancer. However, the potential role of ALDH1A2 in the molecular mechanisms of epithelial ovarian cancer remains largely unclear. In the present study, we found lower expression of ALDH1A2 in high-grade epithelial ovarian cancer tissues than in low-grade epithelial ovarian cancer tissues. Overexpression of ALDH1A2 decreased the proliferation and migration of epithelial ovarian cancer cell lines, whereas ALDH1A2 knockdown significantly increased cell growth and migration. Moreover, upregulation of ALDH1A2 also reduced the activation of signal transducer and activator of transcription 3 (STAT3). In conclusion, these findings suggest that ALDH1A2 suppresses epithelial ovarian cancer cell proliferation and migration by downregulating STAT3.
Keywords: ALDH1A2, epithelial ovarian cancer cell, STAT3, migration, cell growth
Introduction
Human ovarian cancers are the most lethal malignancies in women, accounting for ~4% of all cancers worldwide.1 Approximately, 22,440 new cases of ovarian cancer are diagnosed, and 14,080 patients die from ovarian cancer each year.2 The most common form of ovarian cancer is epithelial ovarian cancer.3 More than 70% of epithelial ovarian cancer patients are diagnosed at a late stage, resulting in a dismal 30% survival rate.4 Notably, the overall survival rate could be significantly increased if metastasis is prevented.5 More attention should be given to prevention, early detection, and treatment of ovarian cancer to decrease its mortality rate. At present, up to 17% of ovarian cancers are potentially preventable through population-based genetic testing of known ovarian cancer susceptibility genes.5 Thus, understanding ovarian cancer pathogenesis and the molecular mechanism of its metastasis is crucial for the management of this lethal, highly metastatic disease.
Aldehyde dehydrogenase 1 family member A2 (ALDH1A2) is a rate-limiting enzyme involved in the cellular synthesis of retinoic acid, which has prodifferentiation properties.6 Previous studies have demonstrated ALDH1A2 to be a candidate tumor suppressor.7 Low ALDH1A2 expression was associated with an unfavorable prognosis in head and neck squamous cell carcinoma.8 Kim et al7 detected ALDH1A2 expression in normal prostate epithelia but not in prostate cancer tissue. Interestingly, ALDH1A2 was strongly downregulated by 36-fold in 779 ovarian cancer cases compared with 18 normal controls and suspected to contribute to invasiveness and metastasis.9 Ma and Zhao10 also revealed that high mRNA expression of ALDH1A2 is significantly associated with worse overall survival in TP53 wild-type ovarian cancer patients. Therefore, ALDH1A2 may play a pivotal role in ovarian cancer behavior.
Janus-activated kinase (JAK) is a tyrosine kinase, and activation of JAK via autophosphorylation leads to its phosphorylation of specific tyrosine residues on relevant receptors. Signal transducer and activator of transcription (STAT) proteins are recruited to receptors by binding to phosphotyrosine residues in the Src homology 2 domain of the STAT protein.11 STAT is subsequently phosphorylated by JAK2 and translocated into the nucleus. The STAT family is a group of latent cytoplasmic proteins that regulate various metabolic processes.12,13 The STAT family includes seven structurally and functionally related proteins: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6.12 STAT3 is constitutively aberrantly activated in ~70% of human solid tumors, and it modulates the expression of oncogenes controlling the proliferation and metastasis of tumor cells.14,15 STAT3 is commonly present downstream of many oncogenic mutations.16 Substantive evidence indicates that downregulating STAT3 mitigates the malignant behavior of cancer cells.17,18 Upregulation of STAT3 is more commonly associated with clinically aggressive high-grade ovarian cancer than with more indolent low-grade cancers.11 Moreover, a previous study demonstrated that ALDH1A2 regulated the expression of STAT3 in ovarian cancer cells.9 Park et al19 found that ALDH1A2 inhibited the phosphorylation of STAT3.
In this study, we found that ALDH1A2 was significantly upregulated in low-grade epithelial ovarian cancer tissue. Overexpression of ALDH1A2 decreased the proliferation and migration of epithelial ovarian cancer cells. ALDH1A2 knockdown significantly increased cell growth and migration. Moreover, upregulating ALDH1A2 also reduced the activation of STAT3. These novel data indicate that ALDH1A2 inhibits epithelial ovarian cancer cell proliferation and migration by downregulating STAT3.
Materials and methods
Clinical tissues
This study was approved by the Medical Ethics Committee of the Zhejiang Cancer Hospital. All subjects provided written informed consent, and none of them received chemoradiotherapy prior to surgery. The histological grade was classified as low-grade (including well to moderately differentiated endometrioid and mucinous carcinomas) and high-grade (combining high-grade serous, clear cell, and poorly differentiated endometrioid cancer).20 High-grade and low-grade epithelial ovarian cancer tissues were obtained during oophorosalpingectomy or surgical debulking.
Immunohistochemistry
ALDH1A2 expression was detected by immunohistochemistry (IHC) in paraffin-embedded specimens. The slides were incubated overnight with rabbit anti-ALDH1A2 monoclonal antibody (1:100; Abcam, Cambridge, UK) by employing an avidin-biotin complex method. Then, a secondary antibody was applied for 30 min at room temperature. These expressions were confirmed by semiquantitative analyses using ImageJ software (Rasband, WS, ImageJ, US National Institutes of Health, Bethesda, MD, USA, https://imagej.nih.gov/ij/, 1997–2016).
Cell culture
The normal ovarian cell line (HOSEpiC) and the ovarian cancer cell lines (A2780 and SKOV3) were purchased commercially from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma-Aldrich Co., St Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS; HyClone, Jerusalem, Israel).
Lentiviral and retroviral infection
A lentiviral short hairpin RNA (shRNA) construct targeting ALDH1A2 (#SHCLNV-NM_170697) was obtained from Sigma-Aldrich Co. Four shRNA sequences targeting ALDH1A2 were designed (Table 1). The oligonucleotides were phosphorylated, annealed, and cloned into the pLKO.1 vector (Sigma-Aldrich Co.). The overexpression particles were provided by GenePharma (Shanghai, China). For retroviral overexpression of ALDH1A2, full-length ALDH1A2 cDNA was obtained by polymerase chain reaction (PCR). The full-length ALDH1A2 cDNA was then subcloned into the BamHI and XhoI sites of the LV3 retroviral vector. Lentiviral infection was performed according to the manufacturer’s protocols.
 | Table 1 Sequences interfering with ALDH1A2 |
Cell proliferation
To evaluate the effects of ALDH1A2 on ovarian cancer cells, the cells were seeded into 96-well plates on days 3, 6, 9, 12, and 15 postinfection (1,000 cells/well). After 24 h, the medium was removed, and the cells were treated with 10% Cell Counting Kit-8 (CCK8) (Dojindo, Tokyo, Japan) in 100 μL RPMI 1640 medium without FBS for 2 h at 37°C. The absorbance at 450 nm, which is directly proportional to the rate of cell proliferation, was measured using a microplate reader.
Wound migration assay
The cells were seeded into six-well plates and cultured to 100% confluence. A pipette tip was used to scratch a straight line in the cell layer to create a wound. Then, the cells were washed with PBS and treated with RPMI 1640 medium without FBS. Wound images were observed under a light microscope. The wound gap widths were measured using ImageJ software.
Migration assay
Cell culture inserts (24-well, pore size 8 μm; Sigma-Aldrich Co.) were seeded with 2×105 cells in 200 μL of medium supplemented without FBS. Medium with 10% FBS (500 μL) was added to the lower chamber and served as a chemotactic agent. After incubation for 8 h, nonmigrating cells were removed from the upper side of the membrane, and the cells on the lower side of the membrane were fixed with 4% paraformaldehyde. The cells were stained with crystal violet staining, and cell numbers were counted. Each individual experiment was performed with triplicate inserts, and five microscopic fields were counted per insert.
RNA isolation and reverse-transcription quantitative PCR
Total cellular RNA was isolated using RNAiso reagent (Takara, Tokyo, Japan). Total RNA (≤500 ng) was reverse-transcribed into cDNA in a reaction volume of 10 μL using the Double-Strand cDNA Synthesis Kit (Takara). One microliter of cDNA was used as the template for the reverse-transcription quantitative PCR (RT-qPCR) reaction. All gene transcripts were quantified by RT-qPCR using the Power SYBR Green PCR Master Mix on the ABI StepOnePlus System. The ALDH1A2 primer sequences were (forward primer) 5′-GATGCTGACTTGGACTATGCTGT-3′ and (reverse primer) 5′-CTGTTTCTTATCAATCTGGGGAC-3′, and the sequences for β-actin, the internal control, were (reverse primer) 5′-GAAGATGGTGATGGGATTT-3′ and (forward primer) 5′-GAAGGTGAAGGTCGGAGT-3′. The 2−ΔΔCt method was used to calculate relative gene expression.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay lysis buffer supplemented with a protease inhibitor (Beyotime, Haimen, China). Total proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to a polyvinylidene fluoride membrane. After blocking in 5% bovine serum albumin for 1 h, the membranes were incubated overnight at 4°C with antibodies specific to β-actin (1:2,000; CST, Beverly, MA, USA), p-STAT3 (1:1,000; CST), t-STAT3 (1:1,000; CST), p-JAK2 (1:1,000; CST), and t-JAK2 (1:1,000; CST). Horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1,000) was applied as a secondary antibody for 2 h at room temperature. For all Western blots, β-actin served as the internal control. The expression of all proteins was quantified using Bio-Rad Quantity One software (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Statistical analysis
Statistical analysis was performed using SPSS 22.0 software. All experiments were performed at least in triplicate, and the data are presented as mean ± standard deviation. Statistical significance was determined using a two-tailed Student’s t-test when comparing two groups and one-way ANOVA followed by Dunnett’s post hoc test when comparing more than two groups. A P-value ≤0.05 was considered to represent a statistically significant difference.
Results
ALDH1A2 expression is higher in low-grade epithelial ovarian cancer tissues than in high-grade tissues
To compare the expression of ALDH1A2 between high- and low-grade epithelial ovarian cancer tissues, IHC was performed. Significantly higher ALDH1A2 levels were identified in low-grade epithelial ovarian cancer tissues than in high-grade tissues (Figure 1; P<0.05).
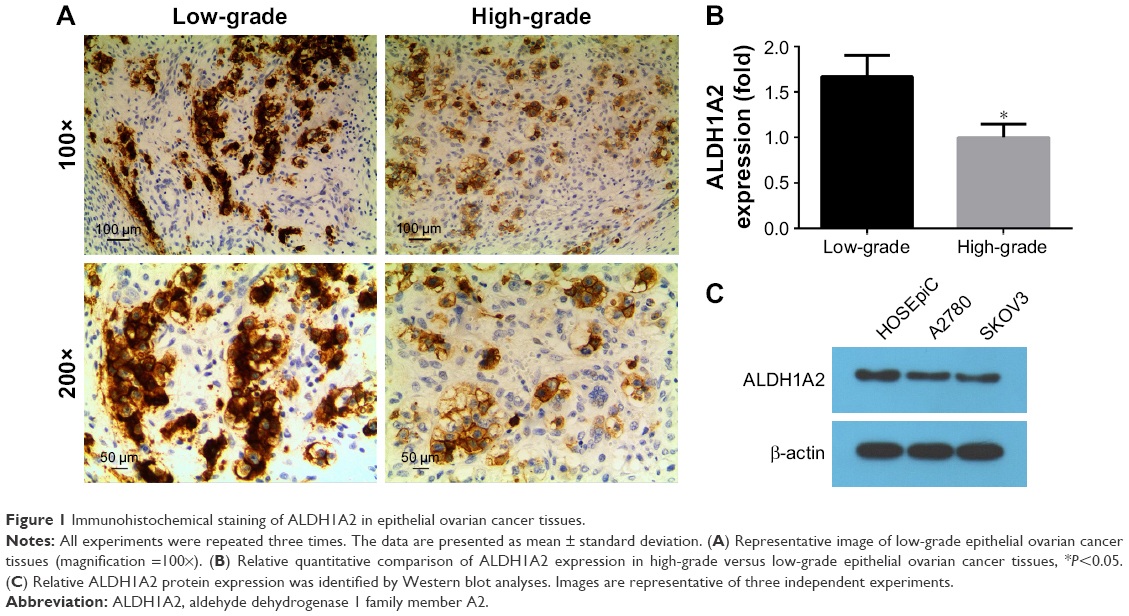 | Figure 1 Immunohistochemical staining of ALDH1A2 in epithelial ovarian cancer tissues. |
Up- and downregulation of ALDH1A2 levels in epithelial ovarian cancer cell lines using lentiviral particles
To investigate the effects of ALDH1A2 on epithelial ovarian cancer cell lines, lentiviral vectors were used to upregulate and downregulate ALDH1A2 levels in epithelial ovarian cancer cell lines. As shown in Figure 2, ALDH1A2 levels were knocked down using four shRNAs. shRNA3 and shRNA4 were chosen for subsequent experiments because of their efficient knockdown of ALDH1A2. Western blotting analysis showed successful upregulation and downregulation of ALDH1A2 levels in epithelial ovarian cancer cell lines.
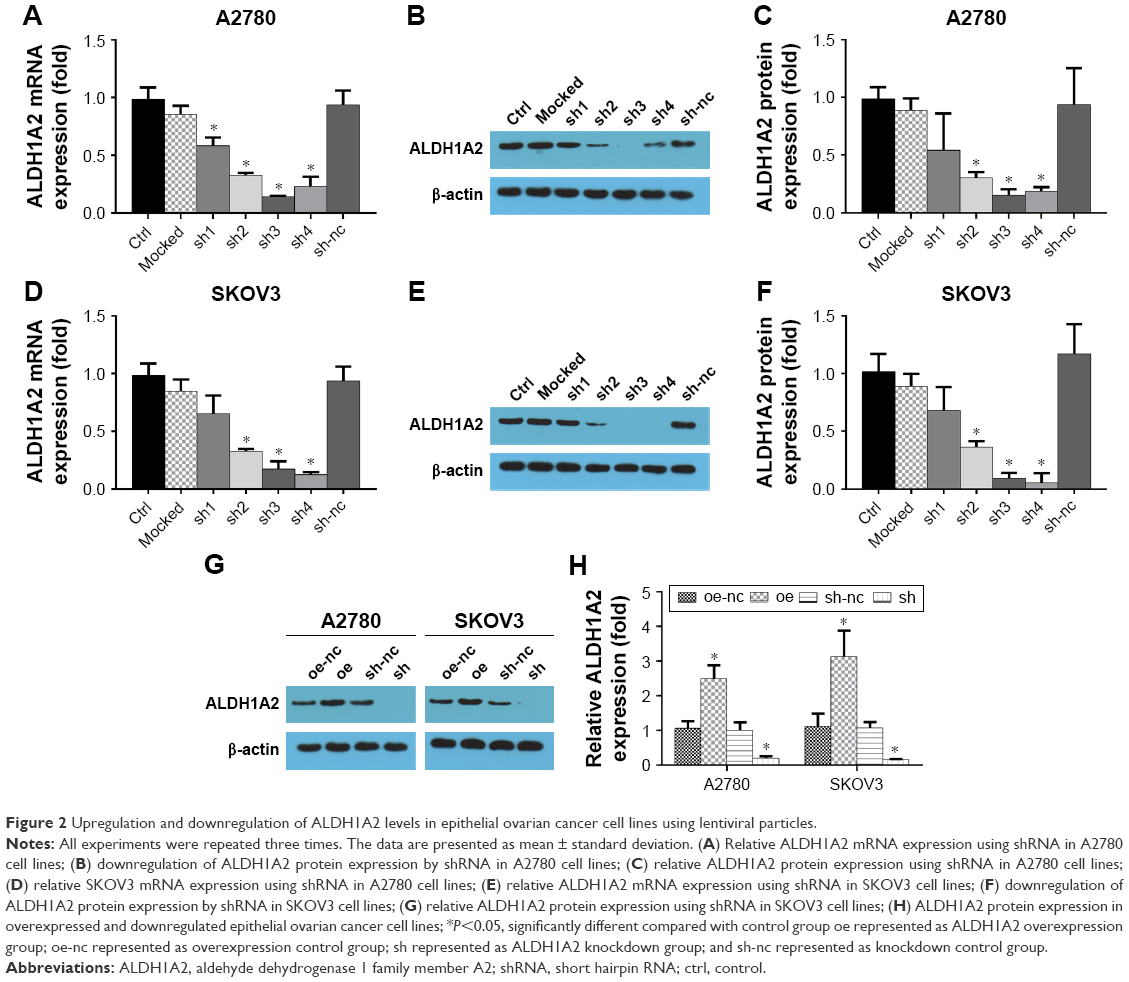 | Figure 2 Upregulation and downregulation of ALDH1A2 levels in epithelial ovarian cancer cell lines using lentiviral particles. |
Overexpression of ALDH1A2 inhibits proliferation, and ALDH1A2 knockdown promotes proliferation in epithelial ovarian cancer cells
The effects of ALDH1A2 on the proliferation rate of epithelial ovarian cancer cells were identified by CCK8 assay, which revealed significantly higher proliferation in the ALDH1A2 knockdown group compared with the other groups. When ALDH1A2 was overexpressed, the proliferation rate decreased significantly (Figure 3).
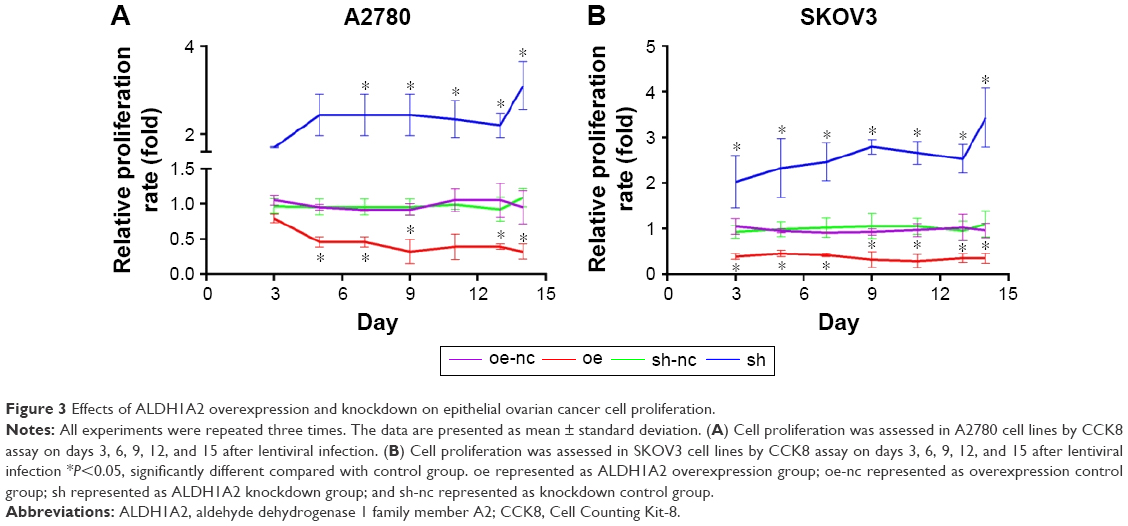 | Figure 3 Effects of ALDH1A2 overexpression and knockdown on epithelial ovarian cancer cell proliferation. |
ALDH1A2 depletion enhances migration, and ALDH1A2 upregulation decreases migration of epithelial ovarian cancer cells
To assess the impact of ALDH1A2 on cell migration, a wound migration assay was performed. Relevant photographs were taken immediately after wound induction and on the following day. The wound-healing abilities of ALDH1A2-overexpressing epithelial ovarian cancer cells were significantly lower than those of the control group. In contrast, ALDH1A2 depletion markedly increased the wound-closure capacity (Figure 4A).
 | Figure 4 The effects of ALDH1A2 knockdown and overexpression on the migration of epithelial ovarian cancer cells. |
A transwell migration assay was also performed to determine the effects of ALDH1A2 on the migration ability of epithelial ovarian cancer cells. As shown in Figure 4B, migration was markedly decreased in cells overexpressing ALDH1A2. In contrast, ALDH1A2 downregulation increased the migration of epithelial ovarian cancer cells.
The JAK2/STAT3 signaling pathway is upregulated by ALDH1A2 knockdown, and ALDH1A2 overexpression inhibits activation of the JAK2/STAT3 signaling pathway
To explore the molecular mechanism underlying the effects of ALDH1A2 on epithelial ovarian cancer cells, members of the JAK2/STAT3 signaling pathway, a key regulator of epithelial ovarian cancer cell migration and proliferation, were evaluated. ALDH1A2 knockdown significantly upregulated the levels of p-JAK2 and p-STAT3. In addition, p-JAK2 and p-STAT3 levels were decreased in ALDH1A2-overexpressing cells (Figure 5).
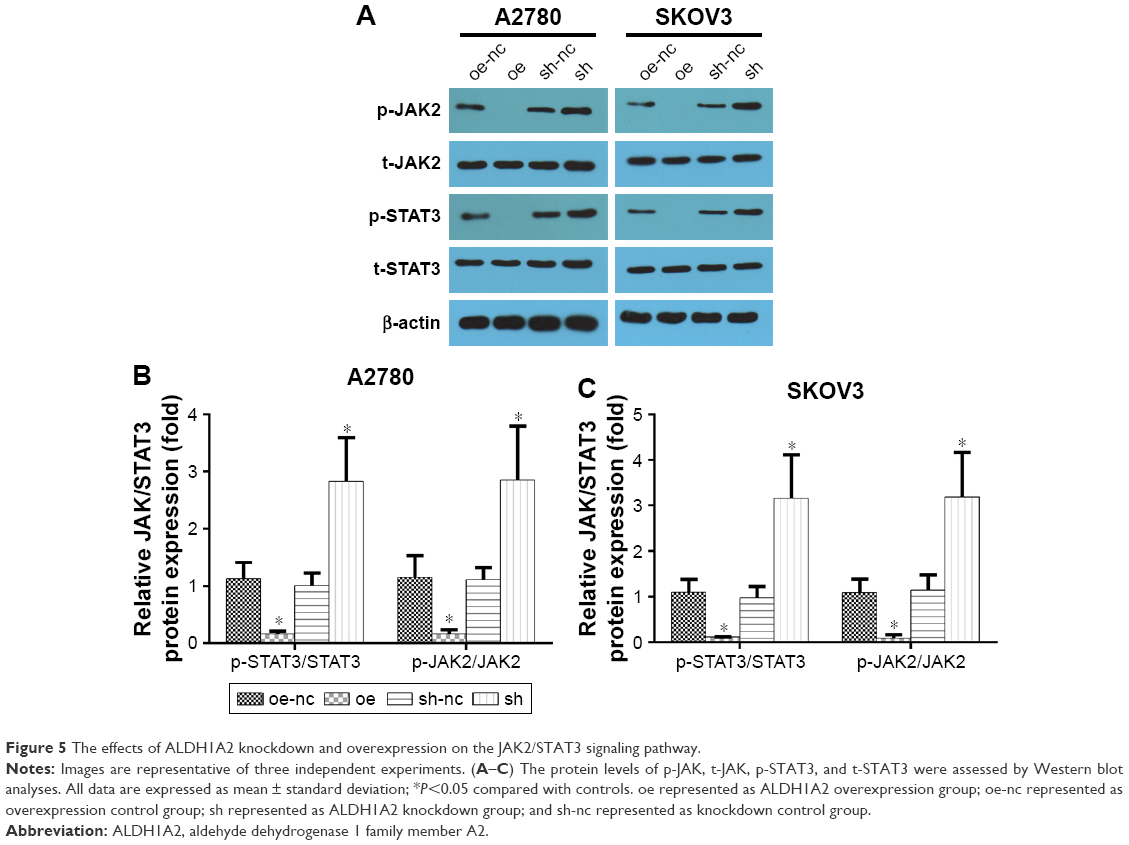 | Figure 5 The effects of ALDH1A2 knockdown and overexpression on the JAK2/STAT3 signaling pathway. |
Blocking the STAT3 signaling pathway rescues the increased cell migration and proliferation induced by ALDH1A2 knockdown
To verify the role of the STAT3 signaling pathway, ADZ1480, a specific STAT3 inhibitor, was used to inhibit STAT3 expression. The concentration of ADZ1480 used was 5 μg/mL, based on a previous study.21 ADZ1480 significantly inhibited the activation of STAT3 (Figure 6). The proliferation rate and migration ability of epithelial ovarian cancer cells exposed to ADZ1480 were markedly decreased. The increased proliferation and migration induced by ALDH1A2 knockdown were also reduced by ADZ1480 exposure.
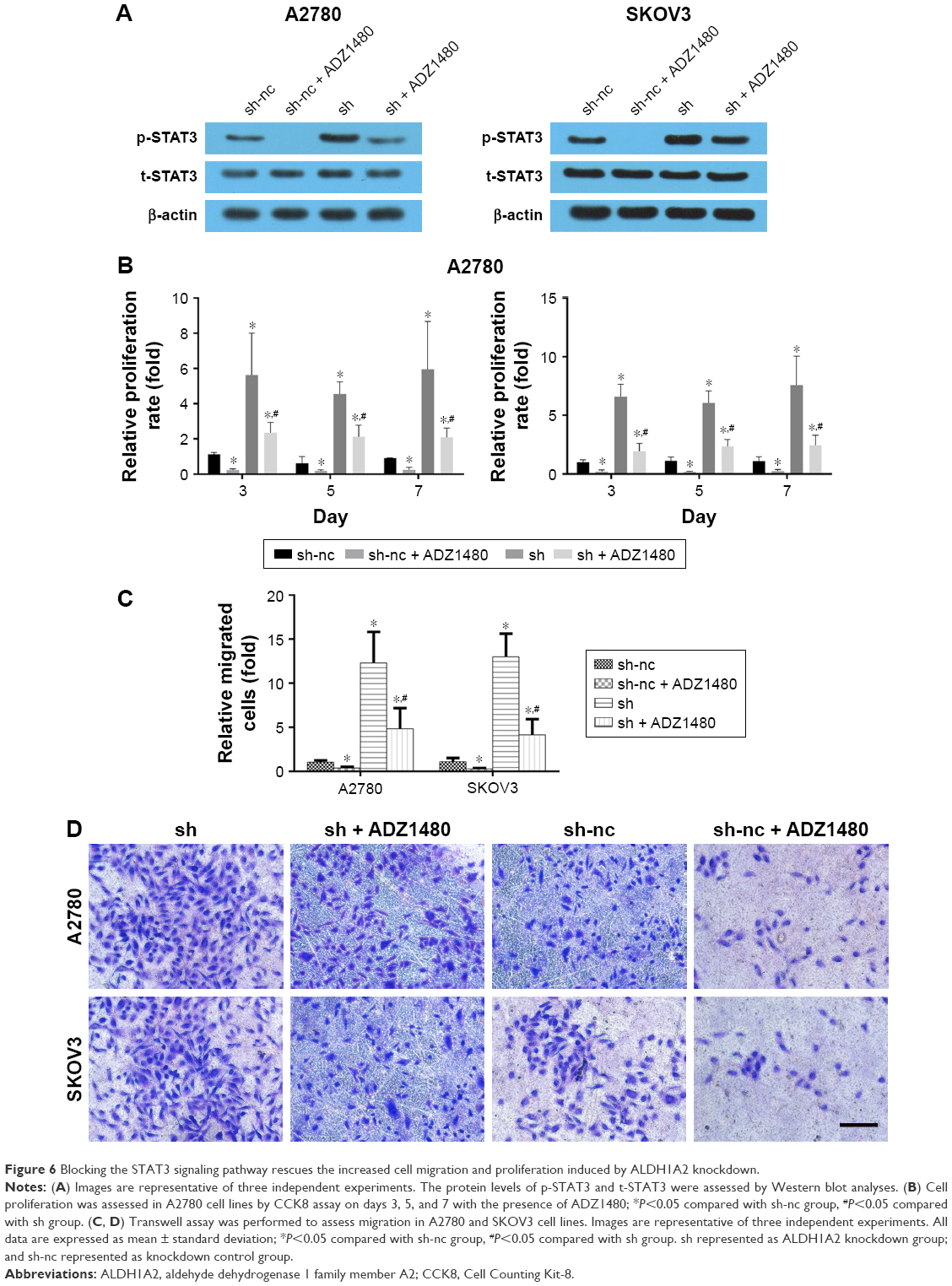 | Figure 6 Blocking the STAT3 signaling pathway rescues the increased cell migration and proliferation induced by ALDH1A2 knockdown. |
Discussion
In this study, we found that ALDH1A2 was significantly upregulated in well-differentiated epithelial ovarian cancer tissues. Overexpression of ALDH1A2 decreased the proliferation and migration of epithelial ovarian cancer cells, while ALDH1A2 knockdown significantly increased cell growth and migration. Moreover, downregulation of ALDH1A2 activated STAT3 expression. Interestingly, the increased migration and proliferation observed after ALDH1A2 knockdown were rescued by inhibition of STAT3. These novel findings indicate that ALDH1A2 suppresses epithelial ovarian cancer cell proliferation and migration by upregulating STAT3 expression.
Previous studies have strongly indicated that ALDH1A2 is a candidate tumor suppressor in epithelial ovarian cancer.7,8 ALDH1A2 was strongly downregulated by 36-fold in 779 epithelial ovarian cancer cases compared with 18 normal controls, and it is thought to contribute to invasiveness and metastasis.9 Ma and Zhao found that higher ALDH1A2 expression in epithelial ovarian cancer cells was significantly associated with lower overall survival rate.10 Similarly, in our study, IHC revealed lower ALDH1A2 expression in high-grade than in low-grade epithelial ovarian cancer tissues, suggesting that high ALDH1A2 expression may be associated with a favorable prognosis. Overexpression of ALDH1A2 decreased the proliferation and migration of two different epithelial ovarian cancer cell lines, indicating that high ALDH1A2 levels in epithelial ovarian cancers might be associated with favorable prognosis.
Inappropriate activation of STAT3 plays a crucial role in the proliferation and metastasis of tumor cells. JAK2 is expressed in ovarian carcinomas but not in normal ovaries, suggesting an important role for JAK2 activation in ovarian tumorigenesis.22 Moreover, previous studies have suggested that inhibition of the JAK2/STAT3 pathway disrupts functions essential for epithelial ovarian cancer growth and progression.11,21 Gritsina et al21 found that the tumor suppressor gene ARHI (DIRAS3) inhibited epithelial ovarian cancer cell migration by downregulating STAT. Brandt et al20 revealed that epidermal growth factor induced mesenchymal transition by activating the JAK2/STAT3 signaling pathway in epithelial ovarian cancer cells, and this pathway may play a role in enhancing the migration of epithelial ovarian cancer cells. Inhibiting STAT3 phosphorylation using a JAK2 inhibitor preferentially decreased the number and migration of epithelial ovarian cancer cells.23 Interestingly, a previous study indicated that ALDH1A2 suppresses STAT3 activation.19 Considering the critical role of STAT3 in cell proliferation and migration, we assessed the levels of JAK2/STAT3 signaling pathway members in epithelial ovarian cancer cells. Our results were consistent with previous studies. According to the Western blot analyses, STAT3 expression was upregulated by ALDH1A2 knockdown and downregulated by ALDH1A2 overexpression. Furthermore, the increased migration and proliferation induced by ALDH1A2 knockdown was rescued by a STAT3 inhibitor. Thus, we inferred that downregulating ALDH1A2 causes aberrant activation of STAT3 in epithelial ovarian cancer cells.
The present study had several limitations. First, although the results were obtained in vitro, they need to be further verified in epithelial ovarian cancer cell lines and in animal models. Second, the data obtained revealed that ALDH1A2 suppresses epithelial ovarian cancer cell proliferation and migration by downregulating STAT3. However, other signaling pathways remain unclear, and further studies are therefore required. Thirdly, the dose-dependent effect of ALDH1A2 should be checked, as it may be associated with the prognosis.
Conclusion
Taken together, our data suggest that ALDH1A2 suppresses epithelial ovarian cancer cell proliferation and migration via downregulation of STAT3.
Disclosure
The authors report no conflicts of interest in this work.
References
Nezhat FR, Apostol R, Nezhat C, et al. New insights in the pathophysiology of ovarian cancer and implications for screening and prevention. Am J Obstet Gynecol. 2015;213(3):262–267. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Fleury H, Communal L, Carmona E, et al. Novel high-grade serous epithelial ovarian cancer cell lines that reflect the molecular diversity of both the sporadic and hereditary disease. Genes Cancer. 2015;6(9–10):378–398. | ||
Yeung TL, Leung CS, Yip KP, et al. Cellular and molecular processes in ovarian cancer metastasis. A review in the theme: cell and molecular processes in cancer metastasis. Am J Physiol Cell Physiol. 2015;309(7):C444–C456. | ||
Sopik V, Rosen B, Giannakeas V, et al. Why have ovarian cancer mortality rates declined? Part III. Prospects for the future. Gynecol Oncol. 2015;138(3):757–761. | ||
Theodosiou M, Laudet V, Schubert M. From carrot to clinic: an overview of the retinoic acid signaling pathway. Cell Mol Life Sci. 2010;67(9):1423–1445. | ||
Kim H, Lapointe J, Kaygusuz G, et al. The retinoic acid synthesis gene ALDH1a2 is a candidate tumor suppressor in prostate cancer. Cancer Res. 2005;65(18):8118–8124. | ||
Seidensaal K, Nollert A, Feige AH, et al. Impaired aldehyde dehydrogenase 1 subfamily member 2A-dependent retinoic acid signaling is related with a mesenchymal-like phenotype and an unfavorable prognosis of head and neck squamous cell carcinoma. Mol Cancer. 2015;14:204. | ||
Huang R, Li X, Holm R, et al. The expression of aldehyde dehydrogenase 1 (ALDH1) in ovarian carcinomas and its clinicopathological associations: a retrospective study. BMC Cancer. 2015;15:502. | ||
Ma YM, Zhao S. Prognostic values of aldehyde dehydrogenase 1 isoenzymes in ovarian cancer. Onco Targets Ther. 2016;9:1981–1988. | ||
Silver DL, Naora H, Liu J, et al. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004;64(10):3550–3558. | ||
Darnell JE Jr. STATs and gene regulation. Science. 1997;277(5332):1630–1635. | ||
Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–662. | ||
Yao X, Liu H, Zhang X, et al. Cell surface GRP78 accelerated breast cancer cell proliferation and migration by activating STAT3. PLoS One. 2015;10(5):e0125634. | ||
Yeh JE, Frank DA. STAT3-interacting proteins as modulators of transcription factor function: implications to targeted cancer therapy. ChemMedChem. 2016;11(8):795–801. | ||
Chai EZ, Shanmugam MK, Arfuso F, et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol Ther. 2016;162:86–97. | ||
Furtek SL, Backos DS, Matheson CJ, et al. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol. 2016;11(2):308–318. | ||
Nelson EA, Walker SR, Kepich A, et al. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood. 2008;112(13):5095–5102. | ||
Park JH, Choi AJ, Kim SJ, et al. 3,3′-Diindolylmethane inhibits Flt3L/GM-CSF-induced-bone marrow-derived CD103(+) dendritic cell differentiation regulating phosphorylation of STAT3 and STAT5. Immune Netw. 2015;15(6):278–290. | ||
Brandt S, Samartzis EP, Zimmermann AK, et al. Lack of MRE11-RAD50-NBS1 (MRN) complex detection occurs frequently in low-grade epithelial ovarian cancer. BMC Cancer. 2017;17(1):44. | ||
Gritsina G, Xiao F, O’Brien SW, et al. Targeted blockade of JAK/STAT3 signaling inhibits ovarian carcinoma growth. Mol Cancer Ther. 2015;14(4):1035–1047. | ||
Colomiere M, Findlay J, Ackland L, et al. Epidermal growth factor-induced ovarian carcinoma cell migration is associated with JAK2/STAT3 signals and changes in the abundance and localization of alpha6beta1 integrin. Int J Biochem Cell Biol. 2009;41(5):1034–1045. | ||
Burgos-Ojeda D, Wu R, McLean K, et al. CD24+ ovarian cancer cells are enriched for cancer-initiating cells and dependent on JAK2 signaling for growth and metastasis. Mol Cancer Ther. 2015;14(7):1717–1727. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.