Back to Journals » The Application of Clinical Genetics » Volume 9
Alagille syndrome: clinical perspectives
Authors Saleh M, Kamath B, Chitayat D
Received 2 December 2015
Accepted for publication 13 February 2016
Published 30 June 2016 Volume 2016:9 Pages 75—82
DOI https://doi.org/10.2147/TACG.S86420
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Maha Saleh,1 Binita M Kamath,2 David Chitayat1,3
1Division of Clinical and Metabolic Genetics, 2Division of Gastroenterology, Hepatology and Nutrition, Department of Pediatrics, The Hospital for Sick Children, 3Department of Obstetrics and Gynecology, Prenatal Diagnosis and Medical Genetics Program, Mount Sinai Hospital, University of Toronto, Toronto, ON, Canada
Abstract: Alagille syndrome is an autosomal dominant, complex multisystem disorder characterized by the presence of three out of five major clinical criteria: cholestasis with bile duct paucity on liver biopsy, congenital cardiac defects (with particular involvement of the pulmonary arteries), posterior embryotoxon in the eye, characteristic facial features, and butterfly vertebrae. Renal and vascular abnormalities can also occur. Inter- and intrafamilial variabilities in the clinical manifestations are common. We reviewed the clinical features and management as well as the molecular basis of Alagille syndrome.
Keywords: Alagille syndrome, ALGS, genetics, liver
Introduction
Alagille syndrome (ALGS; MIM118450) is a highly variable, autosomal dominant multisystem condition with an estimated frequency of one in 30,000.1 ALGS is caused by mutations in one of two genes: JAG1 and NOTCH2. It was initially described as a hepatic disease, but molecular testing has shown that individuals with ALGS and JAG1 or NOTCH2 mutations may present without overt liver disease.2,3
ALGS has been defined by a paucity of intrahepatic bile ducts, in association with at least three of five main clinical abnormalities: cholestasis, cardiac disease, skeletal abnormalities, ocular abnormalities, and characteristic facial features.2 The cholestasis is a consequence of the paucity of bile ducts.
The prevalence of ALGS was originally estimated at 1:70,000 live births; however, this is most likely an underestimation, as cases were ascertained exclusively on the basis of presence of neonatal liver disease2 and childhood and adult cases without overt liver disease were not included in this frequency.
Clinical diagnosis and diagnostic criteria
Traditionally, the clinical diagnostic criteria for ALGS included liver histology showing bile duct paucity (an increased portal tract-to-bile duct ratio) and three of five major clinical features: cholestasis; ophthalmologic abnormalities (commonly posterior embryotoxon); characteristic facial features (prominent forehead, deep-set eyes with moderate hypertelorism, pointed chin, and straight nose with a bulbous tip); cardiac defect (commonly stenosis of the peripheral/pulmonary arteries); and skeletal abnormalities (commonly butterfly vertebrae).
In addition, abnormalities of the kidneys and vasculature (often in the head and neck) are important manifestations of ALGS.3–5 These recent observations have led to an expansion of the phenotypic criteria of ALGS such that three of seven characteristic clinical criteria are sufficient for a clinical diagnosis. Two classic criteria with confirmed ALGS in a first-degree relative may also be sufficient for a diagnosis of ALGS. Finally, a liver biopsy is no longer considered mandatory to make a diagnosis of ALGS, and the presence of cholestasis is acceptable to fulfill this criterion.
Systemic manifestations and clinical description
Disease history and presentation
ALGS is a multisystem disorder with a wide spectrum of clinical variability ranging from life-threatening liver or cardiac disease to only subclinical manifestations, such as mildly abnormal liver enzymes, a heart murmur, butterfly vertebrae, posterior embryotoxon, or characteristic facial features. The diagnosis may be difficult because of the variable expressivity of the clinical manifestations.6 This variability is present even among individuals from the same family sharing the same mutation.7 In a study of 53 mutation-positive relatives of affected individuals, 25 (47%) did not meet classic clinical diagnostic criteria. Thus, without a molecular diagnosis, those individuals would have likely been missed altogether.7
Disease prognosis and risk of mortality depend on the severity of organ involvement. Early mortality is typically caused by cardiac disease or severe liver disease and later mortality is often caused by vascular accidents.8,9
A few case reports have documented the prenatal findings in ALGS using a detailed ultrasound scan, including fetal hemi vertebrae in the lower thoracic region, cardiac changes, and absent gallbladder, in a fetus with JAG1 mutation10 (Figure 1A–C).
 | Figure 1 Prenatal diagnosis of Alagille syndrome. |
Two studies, one by Emerick et al8 and another by Subramaniam et al,11 discuss the frequency of clinical manifestations in individuals with ALGS (Table 1).
 | Table 1 A summary of the clinical features and the frequency reported among individuals with ALGS |
Hepatic manifestations
In the majority of cases, individuals with ALGS present in infancy with cholestasis (conjugated hyperbilirubinemia with high GGT, increased serum bile acids, and elevated cholesterol and triglycerides), which manifest as jaundice, intense pruritus, xanthomas (fatty deposits on the extensor surfaces), and failure to thrive due to fat malabsorption.7,12,13
A liver biopsy typically shows paucity of the intrahepatic bile ducts. In newborns with ALGS, bile duct paucity is not always present and the liver biopsy may demonstrate ductal proliferation, resulting in the possible misdiagnosis of ALGS as biliary atresia.
Unremitting cholestasis and progressive liver disease necessitating liver transplantation occur in ~15% of individuals with ALGS.8 To date, it has been difficult to predict which infants will progress to end-stage liver disease. There are no genotypic, histologic, or radiologic predictors of liver disease severity. However, a recent study by Mouzaki et al demonstrated that a high serum total bilirubin between the ages of 12–24 months combined with fibrosis on liver biopsy and the presence of xanthomata on physical examination can predict poor long-term hepatic outcomes in patients with ALGS12 (Figure 2).
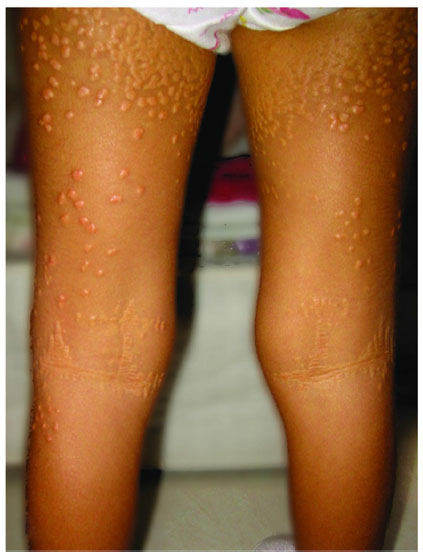 | Figure 2 Xanthomas present on the extensor surface of the buttocks and thighs. |
Cardiac manifestations
Cardiac findings ranging from benign heart murmurs to significant structural defects occur in 90%–97% of individuals with ALGS.8,14 The pulmonary vasculature is most commonly involved. Pulmonic stenosis (peripheral and branch) is the most common cardiac finding (67%).8 The most common complex cardiac defect is tetralogy of Fallot, which is seen in 7%–16% of individuals.8 Other cardiac malformations include ventricular septal defect, atrial septal defect, aortic stenosis, and coarctation of the aorta (in order of decreasing frequency).
Ocular manifestations
The most common ocular finding in individuals with ALGS is posterior embryotoxon, diagnosed by a slit-lamp examination. Posterior embryotoxon (a prominent Schwalbe’s ring) is a defect in the anterior chamber of the eye and has been reported in 78%–89% of individuals with ALGS.8,15 Posterior embryotoxon does not affect visual acuity and has an incidence of 8%–15% in the general population.
Other ocular defects seen in ALGS include Axenfeld-Rieger anomaly, which is characterized by an abnormal pupil that is off-center (corectopia) or by extra holes in the iris that look like multiple pupils (polycoria).
Ocular ultrasonographic examination in 20 children with ALGS found optic disk drusen in 90%. Retinal pigmentary changes are also common (32% in one study).15 Although these changes were initially thought to be the result of dietary deficiency, they have been seen in individuals with normal serum concentrations of vitamins A and E.
Skeletal manifestations
The most common radiographic finding is butterfly shaped thoracic vertebrae, secondary to clefting abnormality of the vertebral bodies. The reported frequency of butterfly vertebrae ranges from 33% to 93%.9,16,17 Additional skeletal features include a square shape of the proximal part of the fingers with tapering distal phalanges and extra digital flexion creases.18 There also appears to be an increase in pathological long bone fractures in ALGS, which may be due to cholestasis and/or an intrinsic defect of the bones.19
Facial features
The pattern of facial features observed in children with ALGS includes a high forehead with frontal bossing or flattening, deep-set eyes with moderate hypertelorism, pointed chin, and saddle or straight nose with a bulbous tip (Figure 3). These features give the face the appearance of an inverted triangle. The typical facial features are almost universally present in ALGS due to JAG1 mutations.1,20 The typical facial features do not seem to be as prevalent in individuals with ALGS carrying a NOTCH2 mutation.
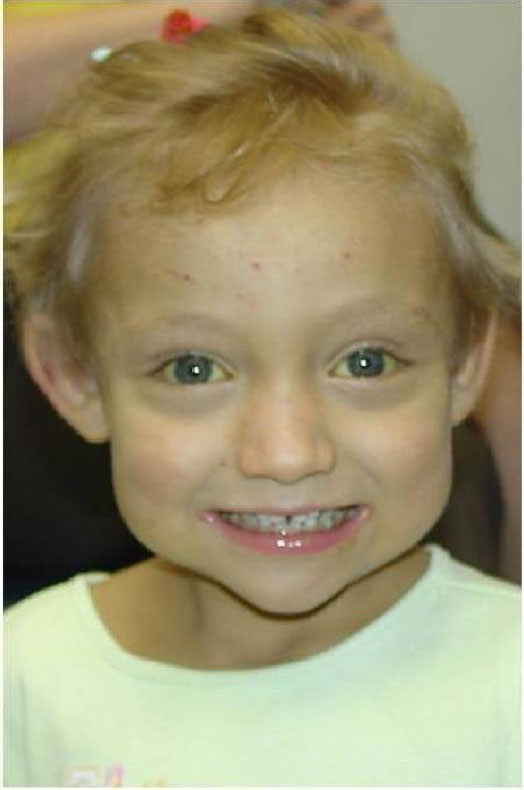 | Figure 3 Characteristic facial features seen in ALGS. |
Renal abnormalities
Structural renal anomalies, such as small hyperechoic kidney and renal cysts, as well as functional abnormalities, such as renal tubular acidosis, have been reported in up to 39% of individuals with ALGS.4 Hypertension and renal artery stenosis have also been noted in adults with ALGS.21
Vascular abnormalities
Neurovascular accidents (intracranial bleeding) have been reported in 15% of ALGS8 and accounted for 34% of mortality in one large study.7 These are potentially devastating complications, and there are reports of adults with ALGS with sudden subarachnoid hemorrhage following the rupture of intracranial aneurysms.22 Renovascular anomalies, middle aortic syndrome, and other intra-abdominal vascular anomalies have also been reported.21
Additional features
The following additional features have been observed: short stature: this may be due to poor growth associated with cholestasis, a severe cardiac defect, and/or intrinsic bone defects (50%–90%);9,23 and immunodeficiency and recurrent infections.24
Pregnancy and ALGS
A few cases of successful pregnancy in AGS have been described by different groups (Table 2), and stemming from these cases, several issues should be considered before attempting embarking on a pregnancy:
- the severity of liver disease and the degree of portal hypertension, which could further worsen during pregnancy;
- the degree of cardiac dysfunction and, in particular, the severity of pulmonary hypertension; and
- the 50% chance for the fetus to inherit the maternal or paternal ALGS mutation, although the severity of the clinical manifestations cannot be predicted in view of the observed intrafamilial variability.
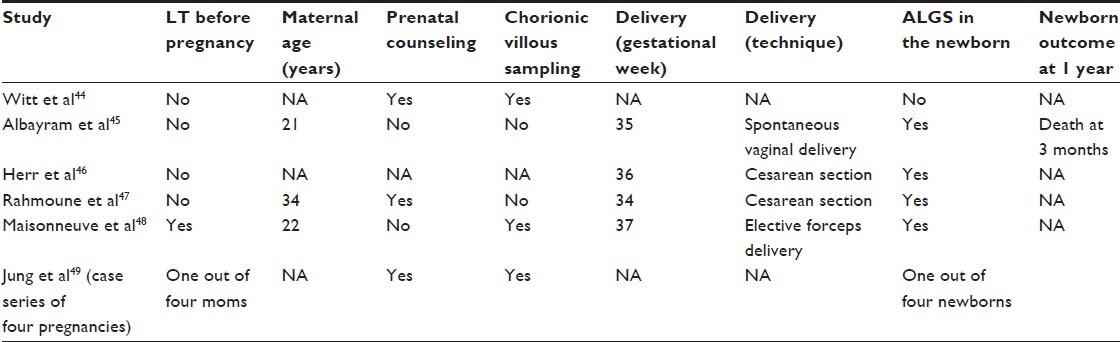 | Table 2 Case reports of successful pregnancies in mothers with ALGS |
Thus, genetic counseling is crucial before conception.25 This includes the discussion of preimplantation and prenatal diagnosis as well as the use of surrogate mother if the pregnancy can be associated with maternal risk for deterioration.
Genetics of ALGS
ALGS is caused by mutations in one of two genes: JAG1 and NOTCH2. Up to 98% are caused by mutations in JAG1 and 2% are caused by mutations in NOTCH2 (Table 3).
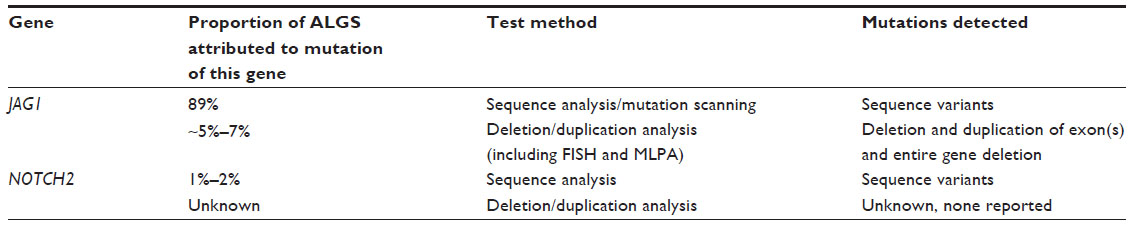 | Table 3 Summary of molecular genetic testing used in ALGS (GeneReviews) |
JAG1 (chromosome 20p12.2)
JAG1 is a cell surface protein that functions as a ligand for one of four Notch transmembrane receptors, which are key signaling molecules in the Notch signaling pathway, an evolutionarily conserved pathway that is crucial in development.
More than 500 pathogenic mutations have been identified so far in individuals with ALGS. In all, 69% are protein-truncating variants (frameshift and nonsense).26,27
NOTCH2 (chromosome 1p12-p11)
NOTCH2 encodes a member of the Notch family of transmembrane receptors. The Notch receptors (NOTCH1, NOTCH2, NOTCH3, and NOTCH4 in humans) share structural characteristics, including an extracellular domain consisting of multiple epidermal growth factor-like repeats and an intracellular domain consisting of multiple, different domain types. The Notch family members play a role in a variety of developmental processes by controlling cell fate decisions.
Twelve different pathogenic variants have been identified in eleven unrelated families with clinical features of ALGS, including one splice site alteration, one frameshift variant, one nonsense variant, and seven missense variants.28,29
20p12.2 microdeletion
The presence of developmental delay and/or hearing loss in addition to the features commonly seen in ALGS may increase the suspicion of chromosome 20p12.2 interstitial microdeletion, which encompasses the JAG1 gene (the ALGS critical region). Kamath et al studied 21 patients with deletions of the short arm of chromosome 20. Eleven patients who had normal development with no anomalies outside of those associated with ALGS had deletions between 95 kb and 4 Mb. The proximal and distal boundaries of these eleven deletions constitute a 5.4 Mb region that defines the JAG1-associated critical region. The other ten patients had bigger deletions between 3.28 Mb and 14.62 Mb, which extended outside the critical region, and, notably, all of these patients had developmental delay and other associated features.30
Genotype–phenotype correlations
No genotype–phenotype correlations exist between the clinical manifestations of ALGS and the specific JAG1 pathogenic variant types or the location of the mutation within the gene.31,32 However, two families with JAG1 pathogenic missense variants had cardiac disease without liver involvement.33,34
Patients with disease-causing variants in NOTCH2 may have an increased incidence of kidney disease.29 However, the number of individuals identified with ALGS caused by mutations of NOTCH2 is still too small to draw conclusions.
In general, understanding the genetic basis of ALGS has assisted the molecular diagnosis and broadened our understanding of the phenotype associated with JAG1 and NOTCH2 mutations. However, a lack of genotype–phenotype correlations has limited the use of these genetic data to impact clinical management.
Treatment of manifestations
Given the multisystem involvement of ALGS, patients with ALGS require evaluation by a multidisciplinary team, including specialists in hepatology, medical genetics, cardiology, nephrology, nutrition, and ophthalmology; liver transplantation team; neurologists; neurosurgeons; and specialists in child development depending on the findings.35 Here, we will concentrate on the hepatic and neurovascular complications of ALGS.
Hepatic manifestations
The liver disease of ALGS typically manifests with severe debilitating pruritus and disfiguring xanthomas. The management is largely supportive with choleretic agents (ursodeoxycholic acid) and other medications (cholestyramine, rifampin, naltrexone) for the itch. Surgical biliary diversion procedures (partial internal biliary diversion and ileal exclusion) have also been used in ALGS to ameliorate itch and xanthomas.36,37 A Kasai procedure (hepatic portoenterostomy), as used in biliary atresia, seems to worsen the outcome in ALGS and is therefore not recommended.38
Liver transplantation for ALGS has an 80% 5-year survival rate, and results in some catch-up growth in 90% of affected individuals.39 Kamath et al showed that the 1-year survival rate for individuals with ALGS was 87%, compared to a 96% 1-year survival rate for individuals with biliary atresia. This reduction in survival was attributed to the vascular and systemic involvement in ALGS.40
Neurovascular manifestations
Neurovascular manifestations are treated in a standard manner. For ALGS individuals with symptomatic moyamoya disease, revascularization can prevent ischemic events and neurologic disability. A recent study by Baird et al on a group of five children with ALGS and symptomatic moyamoya revealed good postsurgical outcomes. These patients remained clinically and radiologically stroke free during long-term follow-up despite progression of moyamoya arteriopathy on angiographic studies.41
No cerebrovascular screening guidelines exist for ALGS. Given this good outcome, it was suggested by Kamath et al that the same standards of care applied to children with moyamoya in the absence of ALGS should be applied to children with ALGS.
Genetic counseling
ALGS is inherited in an autosomal dominant manner. Therefore, offspring of an individual with ALGS have a 50% chance of inheriting the causative gene mutation in the JAG1 or NOTCH2 gene.
In ~50%–70% of affected individuals, the mutation is de novo,3 while up to 50% of individuals with ALGS have an affected parent.
If the proband has an identifiable JAG1 or NOTCH2 pathogenic variant, molecular genetic testing of the parents is recommended. If the proband shows a microdeletion of 20p12 on fluorescence in situ hybridization (FISH) testing, FISH testing of both parents is indicated.
If one of the parents is affected, their risk for having an affected child in their future pregnancies is 50%. However, when the parents are clinically unaffected, the risk to the siblings of a proband appears to be low, but multiple instances of a child inheriting ALGS from an apparently unaffected, phenotypically normal parent with mosaicism for a 20p microdeletion have been reported.42 If the JAG1 or NOTCH2 pathogenic variant or deletion present in the proband cannot be found in either parent, the risk to the siblings is low, but higher than that of the general population because of the possibility of germline mosaicism.43
Conclusion
ALGS is a multisystem disorder associated with liver, cardiac, eyes, face, renal, central nervous, and skeletal abnormalities. The condition has an autosomal dominant mode of inheritance, and thus, the recurrence risk for an affected person is 50% in each subsequent pregnancy. The mortality is ~10%, with vascular accidents, cardiac disease, and liver disease being the most frequent cause of death. The two causative genes associated with ALGS are JAG1 and NOTCH2. The management of an affected patient should be conducted by a multidisciplinary team, which includes specialists in medical genetics, gastroenterology, hepatology, nutrition, cardiology, ophthalmology, nephrology, liver transplantation, and when necessary, neurosurgery.
Disclosure
The authors report no conflicts of interest in this work.
References
Spinner N, Leonard L, Krantz I. Alagille syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews. Seattle, WA: University of Washington; 2013. | |
Danks DM, Campbell PE, Jack I, Rogers J, Smith AL. Studies of the aetiology of neonatal hepatitis and biliary atresia. Arch Dis Child. 1977;52:360–367. | |
Krantz ID, Colliton RP, Genin A, et al. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet. 1998;62:1361–1369. | |
Kamath BM, Podkameni G, Hutchinson AL, et al. Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A. 2012;158A:85–89. | |
Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2012;3:251–257. | |
Guegan K, Stals K, Day M, Turnpenny P, Ellard S. JAG1 mutations are found in approximately one third of patients presenting with only one or two clinical features of Alagille syndrome. Clin Genet. 2012;82:33–40. | |
Kamath BM, Bason L, Piccoli DA, Krantz ID, Spinner NB. Consequences of JAG1 mutations. J Med Genet. 2003;40:891–895. | |
Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29:822–829. | |
Kamath BM, Spinner NB, Emerick KM, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109:1354–1358. | |
Alessandro G, Incerti M, Andreani M. Alagille syndrome: prenatal sonographic findings. J Clin Ultrasound. 2007;35(3):156–158. | |
Subramaniam P, Knisely A, Portmann B, et al. Diagnosis of Alagille syndrome-25 years of experience at King’s College Hospital. J Pediatr Gastroenterol Nutr. 2011;52:84–89. | |
Mouzaki M, Bass LM, Sokol RJ, Piccoli DA, Quammie C, Kamath BM. Early life predictive markers of liver disease outcome in an International, Multicentre Cohort of children with Alagille syndrome. Liver Int. Epub 2015 Jul 22. | |
McElhinney DB, Krantz ID, Bason L, et al. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106:2567–2574. | |
Krantz ID, Smith R, Colliton RP, et al. Jagged1 mutations in patients ascertained with isolated congenital heart defects. Am J Med Genet. 1999;84:56–60. | |
Hingorani M, Nischal KK, Davies A, et al. Ocular abnormalities in Alagille syndrome. Ophthalmology. 1999;106:330–337. | |
Sanderson E, Newman V, Haigh SF, Baker A, Sidhu PS. Vertebral anomalies in children with Alagille syndrome: an analysis of 50 consecutive patients. Pediatr Radiol. 2002;32:114–119. | |
Zanotti S, Canalis E. Notch and the skeleton. Mol Cell Biol. 2012; 30:886–896. | |
Kamath BM, Loomes KM, Oakey RJ, Krantz ID. Supernumerary digital flexion creases: an additional clinical manifestation of Alagille syndrome. Am J Med Genet. 2002;112:171–175. | |
Bales CB, Kamath BM, Munoz PS, et al. Pathologic lower extremity fractures in children with Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;51:66–70. | |
Lin HC, Hoang PL, Hutchinson A, et al. Alagille syndrome in a Vietnamese cohort: mutation analysis and assessment of facial features. Am J Med Genet A. 2012;158A:1005–1013. | |
Salem JE, Bruguiere E, Iserin L, Guiochon-Mantel A, Plouin PF. Hypertension and aortorenal disease in Alagille syndrome. J Hypertens. 2012;30:1300–1306. | |
Doberentz E, Kuchelmeister K, Madea B. Subarachnoid hemorrhage due to aneurysm rupture in a young woman with Alagille syndrome – a rare cause of sudden death. Leg Med (Tokyo). 2015;17(5):309–312. | |
Arvay JL, Zemel BS, Gallagher PR, et al. Body composition of children aged 1 to 12 years with biliary atresia or Alagille syndrome. J Pediatr Gastroenterol Nutr. 2005;40:146–150. | |
Tilib Shamoun S, Le Friec G, Spinner N, Kemper C, Baker AJ. Immune dysregulation in Alagille syndrome: a new feature of the evolving phenotype. Clin Res Hepatol Gastroenterol. 2015;39(5):566–569. | |
Ferrarese A, Senzolo M, Burra P. Successful pregnancy in Alagille Syndrome. Dig Liver Dis. 2015;47(1):86–87. | |
Colliton RP, Bason L, Lu FM, Piccoli DA, Krantz ID, Spinner NB. Mutation analysis of Jagged1 (JAG1) in Alagille syndrome patients. Hum Mutat. 2001;17:151–152. | |
Ropke A, Kujat A, Graber M, Giannakudis J, Hansmann I. Identification of 36 novel Jagged1 (JAG1) mutations in patients with Alagille syndrome. Hum Mutat. 2003;21:100. | |
Kamath BM, Baur RC, Loomes KM, et al. NOTCH2 mutations in Alagille syndrome. J Med Genet. 2012;49(2):138–144. | |
McDaniell R, Warthen DM, Sanchez-Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. | |
Kamath BM, Thiel BD, Gai X, et al. SNP array mapping of chromosome 20p deletions: genotypes, phenotypes, and copy number variation. Hum Mutat. 2009;30(3):371–378. | |
Spinner NB, Colliton RP, Crosnier C, et al. Jagged1 mutations in Alagille syndrome. Hum Mutat. 2001;17:18–33. | |
Crosnier C, Driancourt C, Raynaud N, et al. Mutations in JAGGED1 gene are predominantly sporadic in Alagille syndrome. Gastroenterology. 1999;116:1141–1148. | |
Eldadah ZA, Hamosh A, Biery NJ, et al. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Hum Mol Genet. 2001;10:163–169. | |
Le Caignec C, Lefevre M, Schott JJ, et al. Familial deafness, congenital heart defects, and posterior embryotoxon caused by cysteine substitution in the first epidermal-growth-factor-like domain of jagged 1. Am J Hum Genet. 2002;71:180–186. | |
Kamath BM, Loomes KM, Piccoli DA. Medical management of Alagille syndrome. J Pediatr Gastroenterol Nutr. 2010;50:580–586. | |
Emerick KM, Whitington PF. Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. Hepatology. 2002;35:1501–1506. | |
Mattei P, von Allmen D, Piccoli D, Rand E. Relief of intractable pruritus in Alagille syndrome by partial external biliary diversion. J Pediatr Surg. 2006;41:104–107. | |
Sheflin-Findling S, Arnon R, Lee S, et al. Partial internal biliary diversion for Alagille syndrome: case report and review of the literature. J Pediatr Surg. 2012;47:1453–1456. | |
Pawlowska J, Socha P, Jankowska I. Factors affecting catch-up growth after liver transplantation in children with cholestatic liver diseases. Ann Transplant. 2010;15:72–76. | |
Kamath BM, Yin W, Miller H, et al. Studies of pediatric liver transplantation. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl. 2012;18:940–948. | |
Baird LC, Smith ER, Ichord R, et al. Moyamoya syndrome associated with Alagille syndrome: outcome after surgical revascularization. J Pediatr. 2015;166(2):470–473. | |
Laufer-Cahana A, Krantz ID, Bason LD, Lu FM, Piccoli DA, Spinner NB. Alagille syndrome inherited from a phenotypically normal mother with a mosaic 20p microdeletion. Am J Med Genet. 2002;112:190–193. | |
Giannakudis J, Ropke A, Kujat A, et al. Parental mosaicism of JAG1 mutations in families with Alagille syndrome. Eur J Hum Genet. 2001;9:209–216. | |
Witt H, Neumann LM, Grollmuß O, Luck W, Becker M. Prenatal diagnosis of Alagille syndrome. J Pediatric Gastroenetrology Nutr. 2004;38(1):105–106. | |
Albayram F, Stone K, Nagey D, Schwarz KB, Blakemore K. Alagille syndrome: prenatal diagnosis and pregnancy outcome. Fetal Diagn Ther. 2002;17(3):182–184. | |
Herr F, Schreiner I, Baal N, Pfarrer C, Zygmunt M. Expression patterns of Notch receptors and their ligands Jagged and Delta in human placenta. Placenta. 2011;32(8):554–563. | |
Rahmoune FC, Bruyère M, Tecsy M, Benhamou D. Alagille syndrome and pregnancy: anesthetic management for cesarean section. Int J Obstet Anesth. 2011;20(4):355–358. | |
Maisonneuve E, Morin F, Crochetière C, et al. Multidisciplinary management of a hepatic and renal transplant patient with Alagille syndrome. Int J Obstet Anesth. 2012;21(4):382–383. | |
Jung C, Driancourt C, Baussan C, et al. Prenatal molecular diagnosis of inherited cholestatic diseases. J Pediatr Gastroenterol Nutr. 2007;44(4):453–458. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.