")
Back to Journals » OncoTargets and Therapy » Volume 10
AKT inhibition is an effective treatment strategy in ARID1A-deficient gastric cancer cells
Authors Lee D, Yu EJ, Ham I, Hur H, Kim Y
Received 14 April 2017
Accepted for publication 8 July 2017
Published 22 August 2017 Volume 2017:10 Pages 4153—4159
DOI https://doi.org/10.2147/OTT.S139664
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Dakeun Lee,1 Eun Ji Yu,1 In-Hye Ham,2,3 Hoon Hur,2,3 You-Sun Kim4,5
1Department of Pathology, Ajou University School of Medicine, Suwon, Republic of Korea; 2Department of Surgery, Ajou University School of Medicine, Suwon, Republic of Korea; 3Brain Korea 21 Plus Research Center for Biomedical Sciences, Ajou University, Suwon, Republic of Korea; 4Department of Biochemistry, Ajou University School of Medicine, Suwon, Republic of Korea; 5Department of Biomedical Sciences, Graduate School, Ajou University, Suwon, Republic of Korea
Background: The At-rich interactive domain 1A (ARID1A) is frequently mutated in gastric cancers (GCs) with a poor prognosis. Growing evidence indicates that loss of ARID1A expression leads to activation of the phosphatidylinositol 3-kinase (PI3K)/AKT pathway by AKT phosphorylation. We aim to investigate the different sensitivity for the AKT inhibitor in ARID1A-deficient GC cells.
Methods: After transfection using siRNA or shRNA, the effect of ARID1A knockdown on the PI3K/AKT signaling pathway was evaluated by Western blot analysis. ARID1A-knockdown cells were treated with AKT inhibitor (GSK690693), 5-fluorouracil, or cisplatin, alone or in combination. Viability and apoptosis were analyzed using EZ-CYTOX cell viability assay and flow cytometry, respectively.
Results: ARID1A depletion accelerated the phosphorylation of AKT and S6 in a dose-dependent manner and led to an increased proliferation of MKN-1, MKN-28, and KATO-III GC cells (P<0.001). ARID1A-deficient cells were more vulnerable to GSK690693 in comparison to the controls (P<0.001), even at very low doses. Flow cytometry confirmed the increased apoptosis in ARID1A-deficient cells treated with GSK690693 (0.01 µmol/L; P<0.001). In contrast to our expectations, ARID1A depletion did not cause resistance to 5-fluorouracil or cisplatin. Addition of GSK690693 to the conventional chemotherapy induced more decreased cell viability in ARID1A-knockdown cells (P<0.01).
Conclusion: Loss of ARID1A expression is a surrogate marker for the activation of the AKT signaling pathway and is also a reliable biomarker to predict the response for the AKT inhibitor. We anticipate that appropriate patient selection based on ARID1A expression in the tumor tissue will increase the drug sensitivity for the AKT inhibition and improve the clinical outcome.
Keywords: gastric cancer, ARID1A, AKT inhibitor, chemotherapy
Introduction
Despite recent improvements in surgical and chemotherapeutic approaches, about half of the patients diagnosed with advanced gastric cancer (GC) still die from recurrent disease after curative surgery or distant metastasis.1,2 Fluoropyrimidine- or platinum-based conventional cytotoxic chemotherapy is still widely used as the first-line treatment of GC.3 However, nearly all patients develop resistance to these drugs with adverse effects, limiting the efficacy of this approach.4,5 The only targeted therapeutic agent, trastuzumab (Roche, Basel, Switzerland), increases survival by only a few months, even in a selected biomarker-positive population.1 Thus, identification of efficacious therapeutic interventions that sensitize GC cells is required to improve the management of GC patients.
The AT-rich interactive domain 1A (ARID1A) is an ARID family member that encodes a subunit of the Switch/Sucrose nonfermentable chromatin remodeling family.6 ARID1A is involved in regulating diverse cellular processes, including development, differentiation, proliferation, and DNA repair.7 Recent genome-wide sequencing studies demonstrated that ARID1A is frequently mutated in ovarian and endometrial cancers, and also in GCs, ranging from 8% to 33%.8–12 Genetic alterations in ARID1A in GCs are mostly insertion/deletion mutations, which lead to truncation of ARID1A protein.10 Loss of ARID1A protein is considered a poor prognostic factor in a variety of cancers, including GC.13
Growing evidence indicates that loss of ARID1A expression leads to the activation of the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, mainly due to AKT phosphorylation.14–16 This appears to be true in GCs, as we previously demonstrated in the human GC tissues by immunohistochemistry.17 If ARID1A-deficient tumor cells depend on constitutive activation of the AKT pathway, it is assumed that tumor cells lacking ARID1A expression may be more vulnerable to the selective AKT pathway inhibitors.18 In support of this notion, MCF7 breast cancer cells and primary MRC5 cells exhibit significantly increased sensitivity to the AKT inhibitors, MK-2206 and perifosine, as well as the PI3K inhibitor, buparlisib, after ARID1A knockdown.19
In this study, we demonstrate that loss of ARID1A promotes proliferation of cancer cells by activating the AKT signaling pathway in GC cell lines. The promising preclinical activity of selective AKT pathway inhibitors was demonstrated in ARID1A-deficient GC cells.
Materials and methods
Cell lines and cell culture
MKN-1, MKN-28, MKN-45, KATO-III, AGS, MKN-74, and NCI-N87 human GC cell lines were purchased from the Korean Cell Line Bank and were maintained in RPMI 1640 (Hyclone, South Lagan, UT, USA) supplemented with 10% fetal bovine serum, 1% penicillin, and streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Reagents and antibodies
The following reagents and antibodies were used: GSK690693 (Cell Signalling Technology, Danvers, MA, USA), 5-fluorouracil (5-FU) and cisplatin (both from Sigma-Aldrich, St Louis, MO, USA), ARID1A, p-AKT, AKT, p-S6, poly-ADP ribose polymerase (1:1,000; Cell Signalling Technology), and β-actin (1:5,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Western blot analysis
After treatment with the specified drugs, cells were washed with PBS and lysed in the lysis buffer. Lysates were centrifuged at 13,000 rpm for 20 min at 4°C and then incubated on ice for 20 min. Protein concentration was determined by the Bradford assay (Bio-Rad, Hercules, CA, USA). Equal amounts of protein from each sample were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA, USA). The immunoblots were blocked by incubation in 5% skim milk, 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 0.1% Tween 20 for 1 hour at room temperature. The membrane was then incubated with primary antibodies on a shaker overnight at 4°C. Protein detection was performed using the ECL kit (Abclon, Seoul, Korea).
RNA interference
ARID1A depletion was done by transfection of MKN-1, MKN-28, and KATO-III cells with SMARTpool: ON-TARGETplus ARID1A siRNA (L-017263-00-0005; GE Dharmacon, Lafayette, CO, USA). siRNAs were transfected using DharmaFECT1 (GE Dharmacon). In addition, MKN-1, MKN-28, and KATO-III cells were infected with a lentivirus encoding a nonspecific (NS) scramble shRNA (#1864; Addgene, Cambridge, MA, USA), Mission ARID1A shRNA#2 (NM00615.3-TRCN0000059089; Sigma-Aldrich), or Mission ARID1A shRNA#3 (NM00615.3-TRCN0000059090; Sigma-Aldrich) using polybrene (EMD Millipore). After infection, cells were selected with puromycin (Sigma-Aldrich) and ARID1A expression levels were checked by Western blot.
Cell viability assay
After 48-hour exposure of specified drugs, cells were analyzed using EZ-CYTOX cell viability assay kit (Daeillab Services, Seoul, Korea). Cells were plated in 96-well plates. EZ-CYTOX solution was added to each well of the plates and incubated at 37°C for 1.5–2 hours. The absorbance was measured at 450 nm.
Cell apoptosis
To detect synergistic effects of the chemotherapeutic drugs on cell apoptosis, treated cells were analyzed using the fluorescein isothiocyanate-Annexin V apoptosis kit (BD Biosciences, San Jose, CA, USA). The results were analyzed by flow cytometry using an FACSAria III device (BD Biosciences). All the experiments were repeated independently in triplicate.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 5.0 (GraphPad Software Inc., La Jolla, CA, USA). The half inhibitory concentration (IC50) values were calculated with an F-test. For two-group comparisons, a two-tailed unpaired t-test was used. For multiple group comparisons, one-way analysis of variance was used. Two-sided P<0.05 was considered statistically significant.
Results
ARID1A knockdown activates AKT pathway and promotes growth of GC cells
ARID1A protein levels in different GC cell lines were evaluated using Western blotting. Most of the GC cells showed intact ARID1A expression, while MKN-45 was ARID1A deficient (Figure 1A). Then, we selected MKN-1, MKN-28, and KATO-III and silenced endogenous ARID1A using siRNAs in these cells. The siRNA remained as effective till 48 hours after transfection. ARID1A depletion increased cell proliferation compared to the controls (P<0.001), and it accelerated the phosphorylation of AKT and its downstream S6 proteins in a dose-dependent manner (Figure 1B–D).
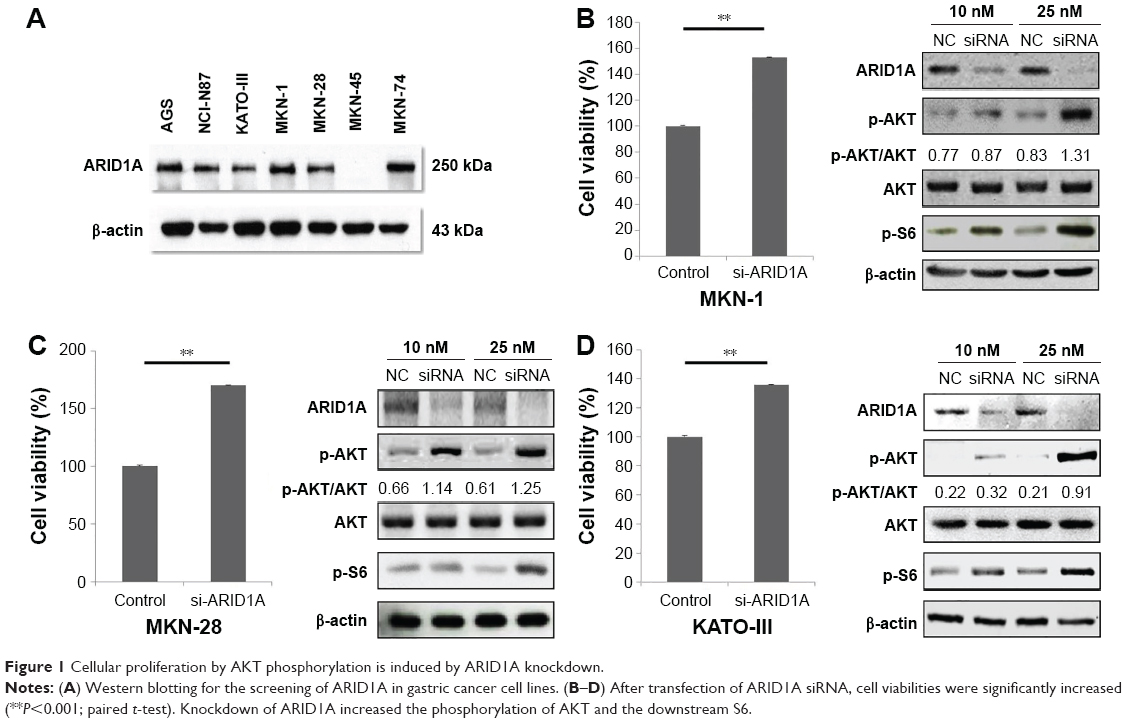 | Figure 1 Cellular proliferation by AKT phosphorylation is induced by ARID1A knockdown. |
ARID1A-deficient GC cells are vulnerable to select AKT inhibitors
After transfection with control-siRNA and siARID1A, GC cells were incubated with increasing concentrations (0.01–10 μmol/L) of the AKT inhibitor, GSK690693, for 48 hours, followed by cell viability detection (Figure 2). ARID1A-deficient cells were more vulnerable to the AKT inhibitor compared to the controls (P<0.001), even at very low doses. Consistent with this, ARID1A-deficient MKN-45 cells were most sensitive to GSK690693. The IC50 value of the wild-type (ARID1A-loss) MKN-45 was 0.043, while the IC50 values of the wild-type (ARID1A-intact) MKN-1, MKN-28, and KATO-III cells were 0.132, 0.084, and 4.521, respectively (P<0.001; Figure 3).
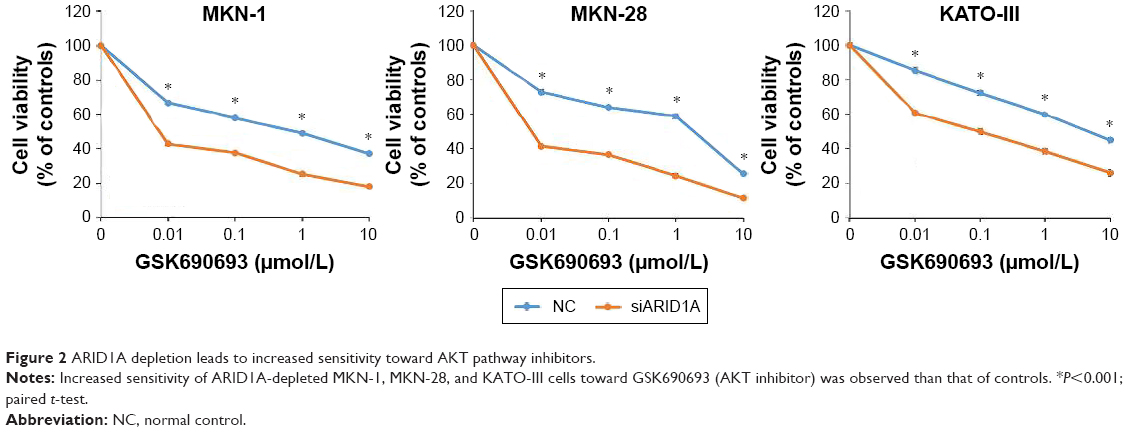 | Figure 2 ARID1A depletion leads to increased sensitivity toward AKT pathway inhibitors. |
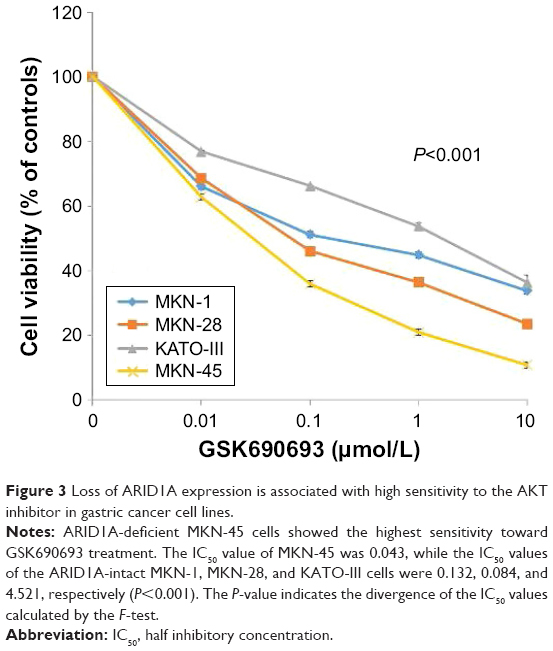 | Figure 3 Loss of ARID1A expression is associated with high sensitivity to the AKT inhibitor in gastric cancer cell lines. |
AKT inhibition leads to increased apoptosis in ARID1A-deficient cells
Treatment with the GSK690693 (10 μmol/L) completely abrogated p-Akt induced by ARID1A knockdown in ARID1A-deficient MKN-28 cells and led to reduced p-S6, in contrast to the controls (Figure 4A). Poly-ADP ribose polymerase cleavage was increased in ARID1A-knockdown cells treated with GSK690693. Flow cytometry confirmed the increased apoptosis in ARID1A-deficient cells treated with even low dose (0.01 μmol/L) of GSK690693, compared to the controls (P<0.001; Figure 4B).
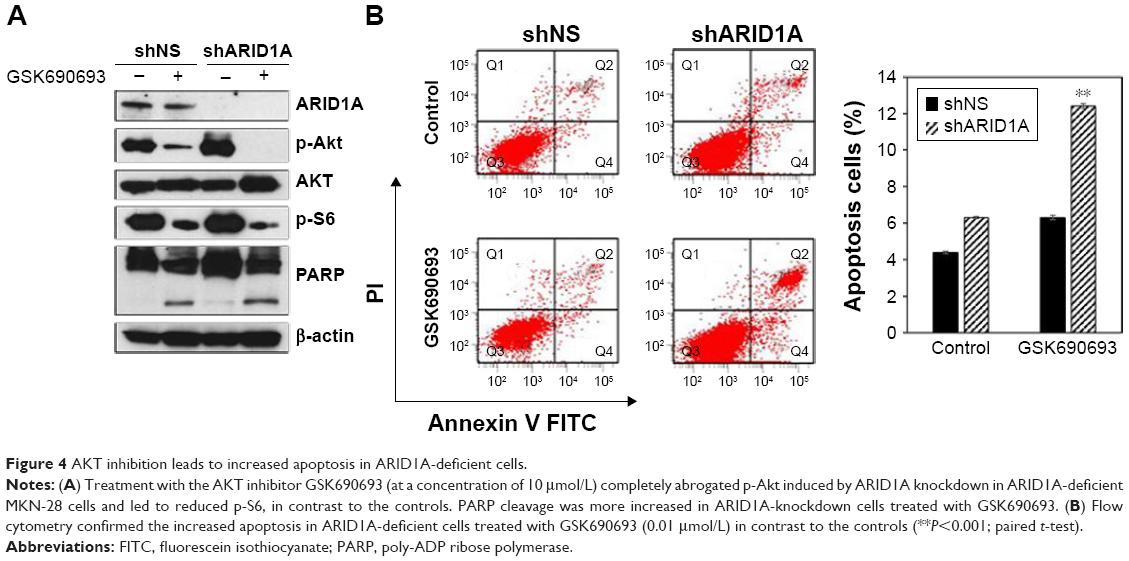 | Figure 4 AKT inhibition leads to increased apoptosis in ARID1A-deficient cells. |
Addition of AKT inhibitors to conventional chemotherapy exerts increased antitumor activity in ARID1A-deficient cancer cells
Since ARID1A knockdown increased the proliferation of cancer cells by activating AKT signaling, we hypothesized that ARID1A-depleted cells might be resistant to conventional chemotherapy. The antiproliferative effect of conventional chemotherapy was examined in ARID1A-depleted GC cells. 5-FU or cisplatin was applied at different concentrations (10–60 μmol/L) for 48 hours to MKN-1, MKN-28, and KATO-III cells transfected with control-shRNA and shARID1A. In contrast to our expectations, ARID1A depletion did not cause resistance to these drugs (Figure 5). Next, to further explore the synergistic effect of GSK690693 with conventional chemotherapy, the cells were treated with 5-FU (10 μmol/L) or cisplatin (10 μmol/L) in the presence of a minimal drug concentration of GSK690693 (0.01 μmol/L). Compared with single agent alone, addition of GSK690693 to the conventional chemotherapy induced more decreased cell viability in ARID1A-knockdown MKN-28 and KATO-III cells, compared to wild-type cells (P<0.01; Figure 6A). Consistent with this observation, GSK690693 in combination with 5-FU or cisplatin induced a significant increase in apoptosis compared with 5-FU or cisplatin alone in ARID1A-knockdown cells (P<0.01; Figure 6B)
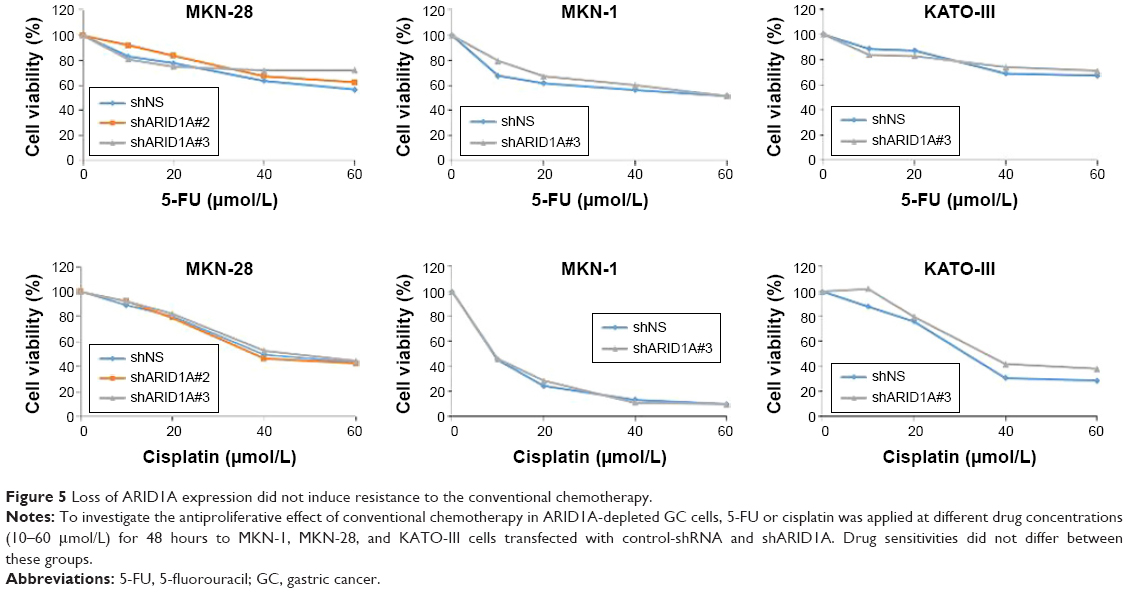 | Figure 5 Loss of ARID1A expression did not induce resistance to the conventional chemotherapy. |
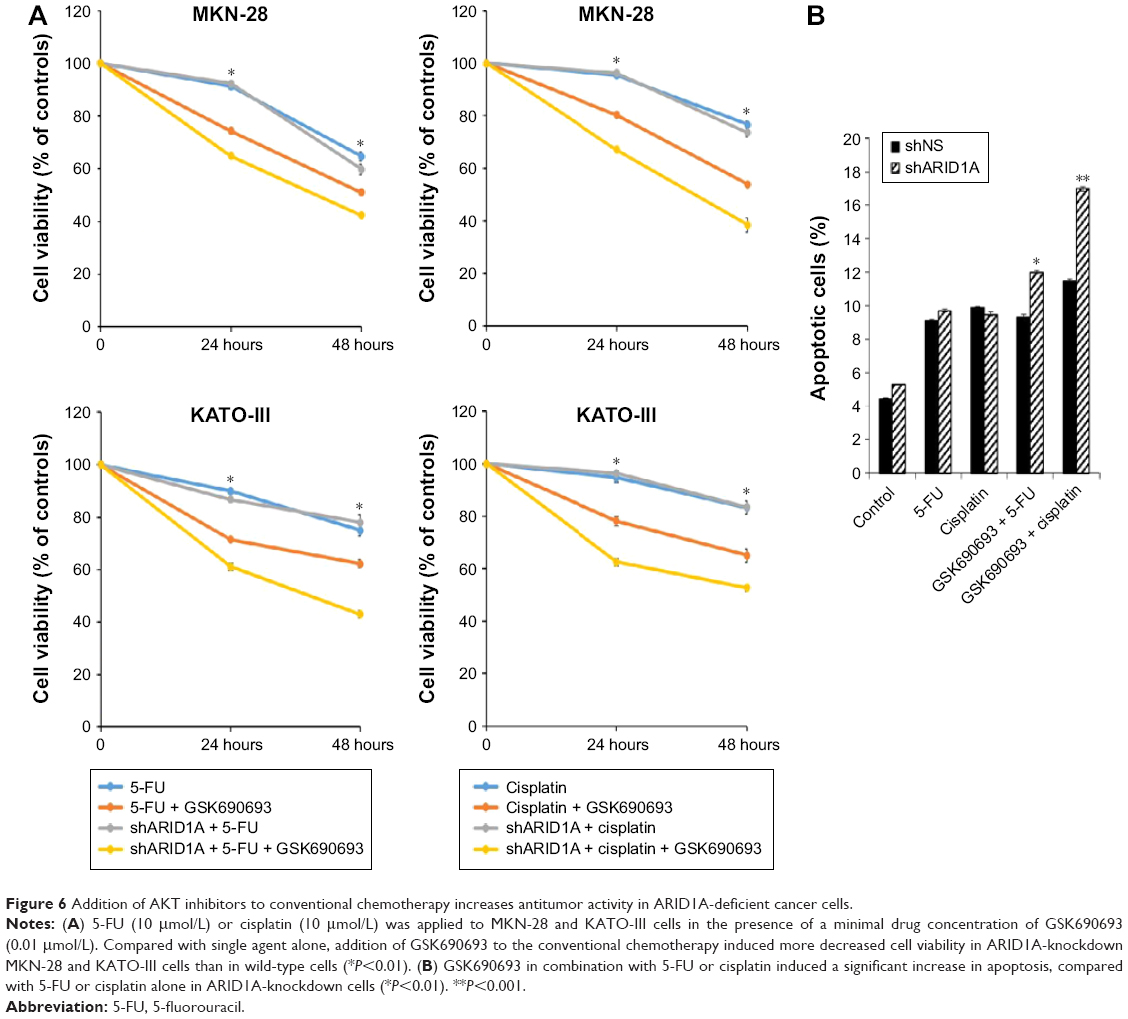 | Figure 6 Addition of AKT inhibitors to conventional chemotherapy increases antitumor activity in ARID1A-deficient cancer cells. |
Discussion
We demonstrate that loss of ARID1A induces cellular proliferation by activating the PI3K/AKT signaling pathway. ARID1A-deficient cancer cells were much more vulnerable to the AKT inhibitor, possibly due to the more significant abrogation of p-AKT and its downstream node p-S6 in ARID1A-depleted cancer cells treated with GSK690693. ARID1A knockdown sensitizes MCF7 breast cancer cells and HGC-27/SGC-7901 GC cells to PI3K and AKT inhibition.19,20 We confirmed this using different GC cell lines, suggesting that this phenomenon is ubiquitous regardless of cancer types. In addition, combination treatment of AKT inhibitor with conventional chemotherapy had an additive effect in ARID1A-knockdown GC cells, which might have clinical utility in the future.
The PI3K/AKT pathway is frequently altered in cancers, promoting growth, proliferation, and survival.21,22 Targeting its major nodes, such as PI3K or AKT, may represent a key therapeutic opportunity.21 Since the PI3K/AKT pathway has also been implicated in carcinogenesis and progression of GC, clinical investigations have targeted the PI3K/AKT pathway in patients with GC.23 However, the clinical efficacy of these inhibitors as monotherapy in particular has, so far, been limited, showing very low response rate.24 These failures possibly stem from the lack of appropriate patient selection based on a reliable biomarker.
Here, we present ARID1A as a surrogate marker for the activation of AKT pathway and may also anticipate more improved clinical response when patients with ARID1A-negative GC are treated with AKT inhibitors. Moreover, unlike other protein biomarkers, the expression status of ARID1A in tumor tissue can be clearly distinguished and leaves no concern for interobserver variability in most cases.17
PI3K/AKT has been implicated for chemotherapy resistance, including resistance for trastuzumab treatment.25 A clinical investigation revealed that the loss of ARID1A expression was related to shorter progression-free survival and chemoresistance in ovarian clear cell carcinoma.26 ARID1A gene silencing reduced the sensitivity of ovarian clear cell carcinoma to cisplatin.27 However, in contrast to our expectation, ARID1A-deficient GC cells did not show resistance when treated with conventional chemotherapy, such as 5-FU and cisplatin. Viability of ARID1A-knockdown MKN-28 cells treated with paclitaxel was also similar to that of wild-type cells (data not shown). The exact relationship between ARID1A loss and chemoresistance needs to be investigated further.
The limitations of our study are that only one AKT inhibitor was used and the lack of in vivo experiments. Further assessment of the mechanism of action could shed light on the importance and clinical utility of using AKT inhibitors for treating patients having ARID1A-deficient GC, either singly or in combination.
Conclusion
Loss of ARID1A expression is a surrogate marker for the activation of AKT signaling pathway and also a reliable biomarker to predict the response for the AKT inhibitor. We anticipate that appropriate patient selection based on ARID1A expression in the tumor tissue will increase the drug sensitivity for AKT inhibition and improve the clinical outcome. Future mechanistic and in vivo studies should be performed prior to exploring this pathway in clinical trials.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2017R1C1B2003970). This research was also supported by the Bio & Medical Technology Development Program of the NRF funded by the Korean government, MSIP (2015M3A9B6073827).
Disclosure
The authors report no conflicts of interest in this work.
References
Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697. | ||
Lee J, Lim DH, Kim S, et al. Phase III trial comparing capecitabine plus cisplatin versus capecitabine plus cisplatin with concurrent capecitabine radiotherapy in completely resected gastric cancer with D2 lymph node dissection: the ARTIST trial. J Clin Oncol. 2012;30(3):268–273. | ||
Wadhwa R, Song S, Lee JS, Yao Y, Wei Q, Ajani JA. Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol. 2013;10(11):643–655. | ||
Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358(1):36–46. | ||
Cervantes A, Roda D, Tarazona N, Rosello S, Perez-Fidalgo JA. Current questions for the treatment of advanced gastric cancer. Cancer Treat Rev. 2013;39(1):60–67. | ||
Dallas PB, Pacchione S, Wilsker D, Bowrin V, Kobayashi R, Moran E. The human SWI-SNF complex protein p270 is an ARID family member with non-sequence-specific DNA binding activity. Mol Cell Biol. 2000;20(9):3137–3146. | ||
Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28(14):1653–1668. | ||
Cancer Genome Atlas Research, Network, Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. | ||
Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363(16):1532–1543. | ||
Wang K, Kan J, Yuen ST, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43(12):1219–1223. | ||
Wang K, Yuen ST, Xu J, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46(6):573–582. | ||
Zang ZJ, Cutcutache I, Poon SL, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44(5):570–574. | ||
Yang L, Wei S, Zhao R, Wu Y, Qiu H, Xiong H. Loss of ARID1A expression predicts poor survival prognosis in gastric cancer: a systematic meta-analysis from 14 studies. Sci Rep. 2016;6:28919. | ||
Liang H, Cheung LW, Li J, et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012;22(11):2120–2129. | ||
Bosse T, ter Haar NT, Seeber LM, et al. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol. 2013;26(11):1525–1535. | ||
Wiegand KC, Hennessy BT, Leung S, et al. A functional proteogenomic analysis of endometrioid and clear cell carcinomas using reverse phase protein array and mutation analysis: protein expression is histotype-specific and loss of ARID1A/BAF250a is associated with AKT phosphorylation. BMC Cancer. 2014;14:120. | ||
Kim YB, Ham IH, Hur H, Lee D. Various ARID1A expression patterns and their clinical significance in gastric cancers. Hum Pathol. 2016;49:61–70. | ||
Samartzis EP, Noske A, Dedes KJ, Fink D, Imesch P. ARID1A mutations and PI3K/AKT pathway alterations in endometriosis and endometriosis-associated ovarian carcinomas. Int J Mol Sci. 2013;14(9):18824–18849. | ||
Samartzis EP, Gutsche K, Dedes KJ, Fink D, Stucki M, Imesch P. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget. 2014;5(14):5295–5303. | ||
Zhang Q, Yan HB, Wang J, et al. Chromatin remodeling gene AT-rich interactive domain-containing protein 1A suppresses gastric cancer cell proliferation by targeting PIK3CA and PDK1. Oncotarget. 2016;7(29):46127–46141. | ||
Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13(5):1021–1031. | ||
Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075–1083. | ||
Tran P, Nguyen C, Klempner SJ. Targeting the phosphatidylinositol-3-kinase pathway in gastric cancer: can omics improve outcomes? Int Neurourol J. 2016;20(Suppl 2):S131–S140. | ||
Ramanathan RK, McDonough SL, Kennecke HF, et al. Phase 2 study of MK-2206, an allosteric inhibitor of AKT, as second-line therapy for advanced gastric and gastroesophageal junction cancer: A SWOG cooperative group trial (S1005). Cancer. 2015;121(13):2193–2197. | ||
Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11(2):263–275. | ||
Katagiri A, Nakayama K, Rahman MT, et al. Loss of ARID1A expression is related to shorter progression-free survival and chemoresistance in ovarian clear cell carcinoma. Mod Pathol. 2012;25(2):282–288. | ||
Lyu C, Zhang Y, Zhou X, Lang J. ARID1A gene silencing reduces the sensitivity of ovarian clear cell carcinoma to cisplatin. Exp Ther Med. 2016;12(6):4067–4071. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.