")
Back to Journals » Journal of Inflammation Research » Volume 16
Advances in the Study of Immunosuppressive Mechanisms in Sepsis
Received 15 June 2023
Accepted for publication 29 August 2023
Published 8 September 2023 Volume 2023:16 Pages 3967—3981
DOI https://doi.org/10.2147/JIR.S426007
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Xuzhe Fu,1 Zhi Liu,2 Yu Wang1
1Department of Emergency Medicine, Shengjing Hospital of China Medical University, Shenyang, People’s Republic of China; 2Department of Ophthalmology, Shengjing Hospital of China Medical University, Shenyang, People’s Republic of China
Correspondence: Yu Wang, Email [email protected]
Abstract: Sepsis is a life-threatening disease caused by a systemic infection that triggers a dysregulated immune response. Sepsis is an important cause of death in intensive care units (ICUs), poses a major threat to human health, and is a common cause of death in ICUs worldwide. The pathogenesis of sepsis is intricate and involves a complex interplay of pro- and anti-inflammatory mechanisms that can lead to excessive inflammation, immunosuppression, and potentially long-term immune disorders. Recent evidence highlights the importance of immunosuppression in sepsis. Immunosuppression is recognized as a predisposing factor for increased susceptibility to secondary infections and mortality in patients. Immunosuppression due to sepsis increases a patient’s chance of re-infection and increases organ load. In addition, antibiotics, fluid resuscitation, and organ support therapy have limited impact on the prognosis of septic patients. Therapeutic approaches by suppressing excessive inflammation have not achieved the desired results in clinical trials. Research into immunosuppression has brought new hope for the treatment of sepsis, and a number of therapeutic approaches have demonstrated the potential of immunostimulatory therapies. In this article, we will focus on the mechanisms of immunosuppression and markers of immune monitoring in sepsis and describe various targets for immunostimulatory therapy in sepsis.
Keywords: sepsis, hyperinflammation, immunosuppression, immune cell
Introduction
Sepsis and infectious shock are leading causes of death in intensive care units (ICUs) worldwide, and their treatment incurs a substantial financial burden.1,2 The most recent definition of sepsis describes it as a life-threatening condition wherein organ dysfunction arises from a dysregulated host response to infection.3 The pathogenesis of severe sepsis is characterized by an uncontrolled immune response, resulting in the excessive release of inflammatory mediators and subsequent immune dysfunction, which can persist even after the infection has been treated.4 While there have been significant advancements in understanding the pathogenesis and treatment of sepsis, the mortality rate remains high.5 This may be due to over-targeting the inflammatory factor storm caused by excessive inflammation in the early stages of sepsis, just as the previous definition of sepsis was limited to an excessive focus on inflammation.6 In recent years, more and more researchers have shifted their focus towards sepsis-induced immune disorders and immunosuppression due to their roles in the development and prognosis of sepsis.7,8 Consequently, unraveling the mechanisms of immunosuppression and understanding the immune status of the body could provide valuable insights that support the early diagnosis and effective prevention of sepsis.9
Initial Immune Response in Sepsis
Immune dysfunction plays a central role in the development of sepsis, and immunosuppression is closely associated with poor sepsis outcomes. Pattern recognition receptors (PRRs) are a class of receptors that directly recognize specific molecular structures on the surface of pathogens, apoptotic host cells, and damaged senescent cells. PRRs bridge non-specific and specific immunity.10 Through ligand recognition and binding, PRRs can produce non-specific anti-infective and other immune protective effects. Immune cells recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through PRRs. PAMPs are specific and highly conserved molecular structures shared by the same pathogenic microorganisms.11 PAMPs are essential for the survival of pathogens and often have unique molecular or subcellular characteristics not found in host cells. Thus, innate immune cells can recognize PAMPs through PRR, distinguish between “self” and “non-self”, and respond to pathogens and their products and respond to pathogens and their products.12 The PRR can also recognize DAMP, activate natural immunity and cause inflammation. The binding of PRRs to PAMPs or DAMPs directly leads to the phagocytosis of pathogenic microorganisms by intrinsic immune cells, activating the inflammatory response in the body and enhancing the ability of the body to clear invading pathogens. Immune cells such as natural killer cells, macrophages, and dendritic cells, as well as parenchymal cells like epithelial and endothelial cells, are involved in the early local immune response to pathogens. These cells are activated through the interaction of PAMPs and PRRs, triggering intracellular signaling pathways that activate key transcription factors coordinating the inflammatory response, such as NF-κB and activator protein-1 (AP-1).13
Hyperinflammation
In most cases, the intrinsic immune system effectively eliminates invading pathogens through a combination of pro-inflammatory responses and repair mechanisms. The pro-inflammatory response eradicates pathogens by releasing cytokines and chemokines, recruiting phagocytes, and locally activating the complement and coagulation systems.14 Concurrently, the anti-inflammatory mechanism restores homeostasis through local repair processes. However, in severe sepsis, the intrinsic immune system fails to clear pathogens due to disrupted dynamic balance and impaired regulation of physiological processes, resulting in excessive inflammation and immunosuppression.15 The severity of these immune dysfunctions varies among individuals. Severe sepsis manifests as a complex state of immune dysfunction, characterized by continuous release of inflammatory mediators. It begins with an inflammatory response accompanied by immunosuppression, or an intensified anti-inflammatory response leading to immunosuppression.16 The hallmark inflammatory response to sepsis includes complement activation, activation of the coagulation system, activation of the vascular endothelium, neutrophil extracellular traps, endothelial dysfunction, platelet and B-cell actions, whose functions are closely interrelated and cross-regulated.
Persistent immune stimulation in severe sepsis is attributed not only to invading pathogens but also to the release of DAMPs, which activate PRRs. These PRRs often also detect PAMPs, thereby triggering a detrimental cycle of sustained immune system activation and dysfunction.17,18 Systemic activation of the innate immune system by PAMPs and DAMPs results in a severe and persistent inflammatory response, characterized by an excessive release of inflammatory cytokines such as IL-1, TNF, and IL-17, collectively known as a “cytokine storm”. Although the release of high levels of inflammatory factors is transient, robust complement activation and innate immune stimulation enhance the response of the body to infection. However, the excessive inflammatory response leads to tissue and cellular damage, molecular dysregulation, and ultimately organ dysfunction, including multi-organ failure. Patients with sepsis who survive the initial hyperinflammatory phase eventually progress to the subsequent immunosuppressive phase.19 Early mortality in sepsis (often due to cardiovascular collapse and multi-organ dysfunction) may be driven primarily by an excessive inflammatory response.
Regulation of Immune Function
The early phase of sepsis is characterized by an excessive inflammatory response accompanied by a simultaneous decrease in inflammatory factors, notably the cytokine IL-10. IL-10 inhibits the production of IL-6 and IFN-γ while stimulating the production of TNF receptors and IL-1 receptor antagonists. Moreover, during the hyperinflammatory phase of sepsis, autophagy plays a role in packaging pathogen components, damaged organelles, and cellular proteins into lysosomal vesicles for degradation. This process reduces inflammation and cellular activation, preventing the binding of DAMP and PAMP molecules to PRRs and thus suppressing immune activation.20
The period of inflammatory regression in sepsis is a complex process involving the removal of damaged tissues and cells at the site of infection, along with the aforementioned involvement of cellular autophagy. Apoptosis occurs in tissue cells and leukocytes that have lost their function at the site of infection. These apoptotic cells are engulfed by macrophages and cleared from the inflammatory area, triggering the production of IL-10 and transforming growth factor β. Additionally, certain bioactive lipids (eg, lipoxygenin) can promote apoptosis.21 Furthermore, specific groups of cells, such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), contribute to the suppression of cytotoxic effects and help dampen the inflammatory response.22
Immunosuppression of Sepsis
The development of sepsis is not solely associated with excessive inflammation but also with immunosuppression. The relationship between excessive inflammation and immunosuppression is intricate, and contrary to earlier notions, they do not always occur sequentially. Immunosuppression can coexist with excessive inflammation, particularly in cases of viral infections leading to sepsis.23 During this stage, immunosuppression is characterized by lymphocyte depletion and reprogramming of antigen-presenting cells. Advancements in research have provided a deeper understanding of the mechanisms underlying immunosuppression in sepsis, the interplay between hyperinflammation and immunosuppression, and the development of modulatory strategies. As shown in Figure 1.
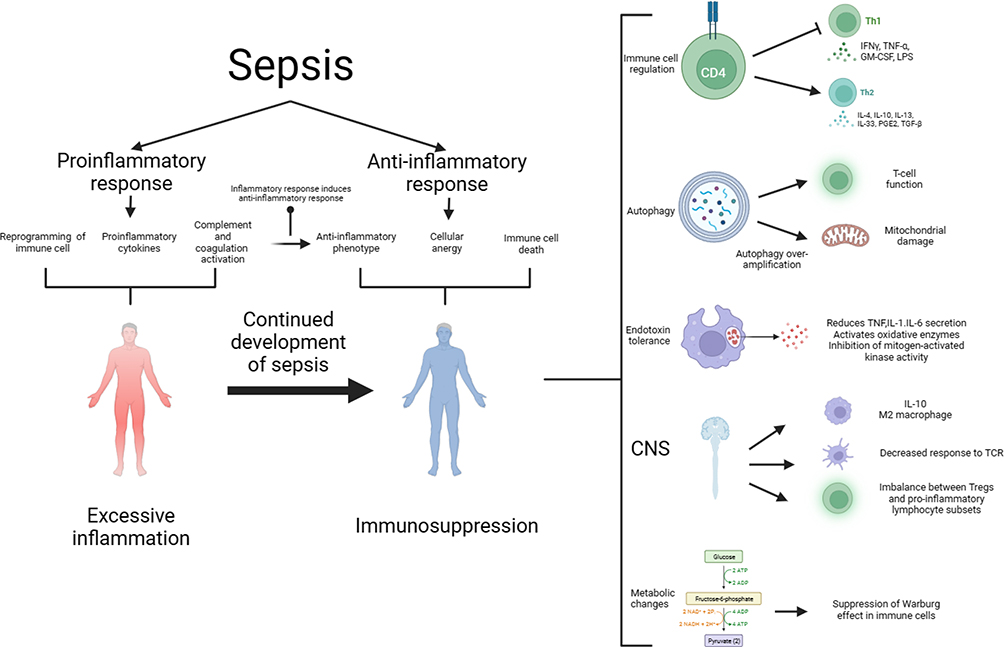 |
Figure 1 The pathogenesis of sepsis is characterized by both proinflammatory and anti-inflammatory mechanisms, and it is now believed that proinflammatory and anti-inflammatory can coexist, and that normally, proinflammatory and anti-inflammatory are in a dynamic equilibrium, which is disrupted by sepsis, which early on leads to the massive secretion of proinflammatory factors, reprogramming of the immune cells, and activation of the complement and coagulation systems, which further leads to a cytokine storm. Anti-inflammatory cytokines may be released subsequently or concurrently, and as the disease progresses, over-activation of the immune system leads to depletion of immune cells and sepsis progresses to an immunosuppressed state. The mechanisms of immunosuppression in sepsis and its complexity involve immune cell regulation, apoptosis autophagy, endotoxin tolerance, central nervous system, and metabolic changes. Therapies targeting immunosuppression in sepsis have the potential to be realized through these mechanisms. Abbreviations: CNS, central nervous system; GM-CSF, granulocyte-macrophage; IFN-γ, interferon gamma; IL, interleukin. |
Immune Cell Regulation and Cell Anergy
CD4+ T cells play an important role in immunosuppression in sepsis, whereas the TH1, TH2, Treg, and Th17 subsets of T cells play an important role in the regulation of inflammation.10 During sepsis, the acquired immune response shifts from a TH1 cell-mediated immune response (a pro-inflammatory response characterized by the production of IFN-γ and IL-12) to a TH2 cell-mediated immune response (an anti-inflammatory response with the production of IL-4, IL-5, IL-10, and IL-13).24 Th17 cells play an important role in the chemotaxis and activation of neutrophils to clear extracellular fungal pathogens, and the Th17 transcription factor retinoic acid receptor-associated orphan receptor-γt was found to remain at its highest level for the first 7 days after infection, after which it began to decline. This suggests that patients with sepsis-induced immunosuppression have an increased susceptibility to secondary fungal infections.22 Moreover, there is a decrease in T cell CD28 expression and T cell receptor (TCR) diversity and the development of an exhausted phenotype characterized by increased expression of programmed cell death 1 and reduced cytokine secretion. Moreover, CD4+ T cells and CD8+ T cells during sepsis exhibit features of incompetence or exhaustion (reduced T cell proliferation and functional defects), resulting in immunosuppression.25 T cells during sepsis also experience reduced adenosine triphosphate (ATP) concentration, along with suppressed oxidative phosphorylation (OXPHOS) and glycolytic pathways, resulting in altered basal metabolic content within these cells. Consequently, their capacity to expand and execute effector functions becomes impaired. Nevertheless, these T-cell dysfunctions are reversible, opening up new possibilities for the treatment of sepsis-induced immunosuppression.26
Immune cell incompetence is mainly seen in T cells, B cells, and NK cells, where there is also an induced relationship, eg, NK cells can induce T cell incompetence (depending on the lymphocyte incompetence gene - GRAIL).27 Immune cell incompetence is the intrinsic functional inactivation of immune cells after encountering antigens, which may be due to a lack of co-stimulation, defective T-cell receptors, etc., leading to reduced T-cell proliferation and cytokine production. The most studied form of immune cell incompetence is the T cell, which is categorized into clonal incompetence and in vivo incompetence.28,29 The first is incomplete T-cell activation, which leads to T-cell growth arrest but does not usually result in functional suppression. The second is activation in the absence of co-stimulation in the naïve T-cell state, leading to varying degrees of T-cell proliferation and differentiation, which are further down-regulated in the immune-tolerant state.30 During the immunosuppressive phase of sepsis, the TNFR1-mediated signaling pathway plays a key role in T-cell dysfunction during S. aureus sepsis by regulating immunomodulatory mediators in MDSC.31 Sema3A plays an important role in sepsis-induced immunosuppression. Sema3A exacerbates sepsis-induced T-cell immunosuppression, which is significantly inhibited and Improvement.32 Lymphoid and myeloid lineages secreted high concentrations of Sema3A in LPS-induced sepsis, especially in the lymphoid lineage, and inhibition of Sema3A alleviated T-cell incompetence, in which the NF-κB signaling pathway is involved in the Sema3A-mediated autocrine loop and is a key signaling pathway, while Tim-3 regulates sepsis-induced by inhibiting the NF-κB signaling pathway in CD4 + T cells.33
B cell incompetence, Anergy refers to a state in which self-reactive B cells exist in the periphery but are quiescent and unresponsive to antigen stimulation. Incompetent B cells are unable to respond to antigen by secreting immunoglobulins.34 Many incompetent B cells also exhibit attenuated B cell receptor signaling in vitro in response to antigen or Toll-like receptor stimulation.35
The definition of NK cell incompetence is still ambiguous, and some studies have proposed a sequential signaling model of NK cell activation similar to that of naïve T cell activation, in which CD137 signaling is absent upon NK-aAPC contact and NK cells shift to an “incompetent state”.36 Other studies have suggested that NK cells are “division-incompetent”. In addition to affecting T-cell incompetence, the effect of NK cell incompetence itself on sepsis remains to be investigated, especially since it has been suggested that NK cell incompetence may be beneficial to the host.37
Depletion of Immune Cells and Apoptosis
Immunosuppression observed in sepsis is strongly associated with a significant depletion of key immune cell populations, including CD4+ and CD8+ T cells, dendritic cells, cross-presenting dendritic cells, and B cells. The loss of these lymphocytes severely compromises the immune system’s ability to effectively combat and eliminate pathogens.23 Sepsis triggers delayed apoptosis of neutrophils (which correlates with the severity of the condition) and a rapid increase in neutrophil levels. While neutrophil apoptosis is delayed, accelerated apoptosis of other immune cells may undermine the host immune system by inducing dephosphorylation of epithelial caspase-8. As the disease progresses, persistent neutrophil dysfunction, coupled with the release of immature neutrophils, eventually leads to neutrophil deficiency.26,38,39
The detrimental effect of apoptosis extends beyond the significant loss of immune cells. It also involves the clearance of apoptotic immune cells by surviving immune cells. The clearance process, mediated by monocytes, macrophages, and dendritic cells, is further compounded by immunosuppression, which is characterized by increased production of the anti-inflammatory cytokine IL-10 or immune cell unresponsiveness. Consequently, surviving phagocytes are rendered incapable of effectively combating pathogens. Remarkably, the clearance of apoptotic cells by mesenchymal stem cells (MSCs) promotes immunosuppression through enhanced prostaglandin E2 production. Additionally, MSC-mediated clearance of apoptotic immune cells induces the generation of immunosuppressive phenotypes in monocytes and macrophages, resulting in reduced inflammation and facilitating the repair and regeneration of damaged tissues.20,40
Apoptosis-induced reduction in the number and function of dendritic cells (DCs), which are highly efficient antigen-presenting cells (APCs), can result in compromised innate and adaptive immune responses. This includes diminished expression of HLA-DR, induction of endotoxin tolerance, and decreased cytokine production, collectively impairing the ability of APCs to stimulate lymphocytes and drive immune function. Various factors such as steroids and cytokines (eg, TNF-α, high mobility group box-1 protein, and heat shock proteins) can directly regulate apoptosis by influencing Caspase-8 activity.41 Apoptosis of immune cells in lymphoid and gut-associated lymphoid tissues can contribute to secondary infections, further exacerbating immunosuppression. Therefore, sepsis-induced apoptosis operates through multiple pathways to compromise host immune defenses. It is important to note that apoptosis of immune cells does not solely have negative effects. For instance, increased PD-L1 expression of neutrophils, which delays apoptosis through PI3K and AKT phosphorylation, can lead to augmented lung injury and mortality. The recent discovery that waveform proteins (VIM) regulate lymphocyte apoptosis suggests that VIM may serve as a novel target for diagnosing and predicting prognosis in patients with sepsis or septic shock.42 Thus, apoptosis exacerbates sepsis-induced immunosuppression in both the innate and adaptive immune systems. Consequently, exploring potential therapeutic targets associated with apoptosis to inhibit immune cell apoptosis holds significant promise for reversing sepsis-induced immunosuppression.
Immune Cytokines
Cytokines are categorized into anti-inflammatory and pro-inflammatory cytokines. The immunosuppressive state of sepsis is associated with elevated secretion of anti-inflammatory cytokines, and the most prominent anti-inflammatory cytokines at this stage are IL-4, IL-10, IL-33, IL-35, IL-37 and IL-38. As shown in Figure 2. IL-4 induces the differentiation of CD4+ T cells into TH2, promotes the secretion of other anti-inflammatory cytokines, and inhibits the release of pro-inflammatory cytokines (including IL-1 and IFN-γ).43 IL- 10 inhibits T cell proliferation and immune function, suppresses the release of TNF-α, and promotes the proliferation of immunosuppressive cells, Treg, and MDSCs. IL-10 circulates in the bloodstream of patients with sepsis, and elevated concentrations of IL-10 have been associated with poor clinical outcomes.44 IL-33 is released during septic tissue injury, activates type 2 innate lymphocytes, and promotes polarization of M2 macrophages, which in turn, via IL-10 enhances the expansion of Treg cell populations.45 IL-38 enhances the immunosuppressive activity of Tregs.46 IL-37 correlates with the severity of sepsis-induced immunosuppression, inhibits the secretion of the proinflammatory cytokines TNF-α, IFN-γ, and IL-2, down-regulates the expression of HLA-DR and CD86 in septic mice, and inhibits antigen presentation.47,48
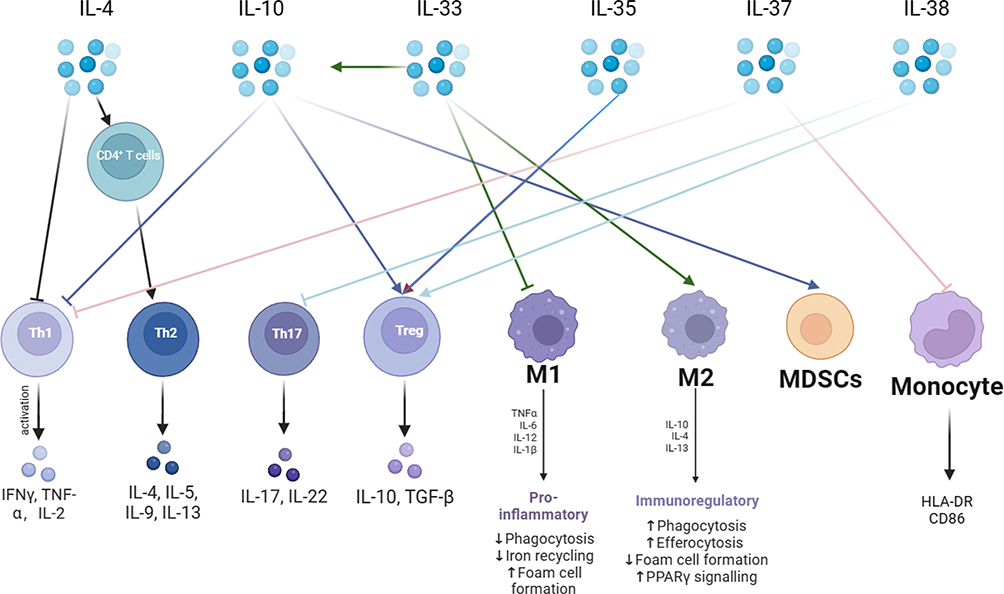 |
Figure 2 Anti-inflammatory cytokines are a very critical part of the immunosuppressive mechanism in sepsis. The mechanism of action of anti-inflammatory cytokines is very complex. The main anti-inflammatory cytokines are IL-4, IL-10, IL-33, IL-35, IL-37, and IL-38. IL-4 promotes the differentiation of CD4+ T lymphocytes into TH2 cells and releases anti-inflammatory cytokines. IL-10 can inhibit pro-inflammatory cytokine secretion and promote immunosuppressive cells Tregs with MDSCs. IL-33 promotes the immunosuppressive effects of IL-10 and promotes macrophage like M2 transformation. IL-35 promotes Tregs. IL-37 inhibits T lymphocyte and monocyte immune function. IL-38 inhibits TH17 and promotes Tregs. Abbreviations: IFN-γ, interferon gamma; IL, interleukin; TGF-β, Transforming growth factor-β; HLA-DR, Human leukocyte antigen; CD86, Cluster of Differentiation 86; PPAR γ, Peroxisome proliferator-activated receptor γ. |
Autophagy of Immune Cells
Autophagy is observed in almost all cell types involved in adaptive immunity, such as lymphocytes, APCs, dying cells, and myeloid cells, all of which promote the formation of an inflammatory microenvironment. In sepsis, lipopolysaccharide (LPS) activates selective autophagy through TLR4-MyD88-dependent or MyD88-independent pathways and also through NF-kB.49 Autophagy is required for the survival and proliferation of CD4+ T, CD8+ T, and NK cells, the memory response of CD8+ T cells, and the maintenance of NK cell activation.50 Autophagy is an important mechanism for the clearance of intracellular bacteria that acts on T and B cells.51 In a recent study, researchers discovered that T cell-specific autophagy promotes survival and enhances T cell function, including apoptosis and cell death. Moreover, it attenuates macrophage phagocytosis in septic mice with an elevated splenic bacterial load in a mouse model of cecum ligation and perforation (CLP). Incomplete autophagy is observed in septic hearts and may contribute to autophagy-induced tissue damage.52 These findings indicate that autophagy plays a protective role against sepsis-induced T-lymphocyte apoptosis and immunosuppression. Furthermore, activation of lymphocyte autophagy appears to alleviate the severe inflammatory response associated with sepsis.53 In addition to lymphocytes, autophagy negatively regulates the abnormal activation of macrophages and reduces the inflammatory response.54
Mitofusin 2 (Mfn2), a negative regulator of autophagy, is an integral mitochondrial outer membrane protein. Increased expression of Mfn2 in sepsis inhibits autophagy and increases the apoptosis of CD4+ T cells, leading to immunosuppression. Autophagy plays a crucial role in controlling B-cell terminal activation. ATG5 is involved in the transport and recruitment of B-cell receptors to lysosomes and MHC class II-enriched compartments (MIIC) in polarized B-cells, facilitating the acquisition and presentation of specific antigens.55 Moreover, autophagy can also regulate the adaptive and innate immune systems through interferon (IFN) and TNF-α. Recent findings demonstrate that artesunate interacts with vitamin D receptors, leading to enhanced autophagy, promotion of proinflammatory cytokine release and bacterial clearance, and reversal of sepsis-induced immunosuppression in vivo and in vitro.56 Other studies have proposed a different perspective and found that autophagy is involved in mitochondrial damage in sepsis and that autophagosomes released into the circulation due to cytosolic emesis cause organ damage.57 The effect of immune cell autophagy on an organism is a complex process, and if immune cells can initiate self-sacrificial programmed death before failure, it reduces inflammation in the organism. However, if autophagy is excessively amplified, its harmful effects may outweigh its protective effects.
Lipopolysaccharide (LPS) Immune Tolerance
LPS immune tolerance is a state of tolerance induced when cells are pre-exposed to endotoxins after initial exposure and then encounter an LPS attack again.58 Endotoxin tolerance is one of the mechanisms underlying immunosuppression. Endotoxin tolerance occurs relatively early in sepsis and peaks at 24–48 h. Recovery began 24–48 h after sepsis, with complete recovery by day 7. In the endotoxin-tolerant state, the inflammatory response-induced cytokine release and cellular and tissue damage were significantly reduced. The main features of endotoxin tolerance are inhibition of LPS-stimulated TNF production, reduction of IL-1 and IL-6 release, activation of cyclooxygenase, inhibition of mitogen-activated protein kinase activation, and impairment of NF-κβ translocation. Therefore, LPS immune tolerance reduces the response of an organism to excessive inflammation.59 Monocytes rendered tolerant to LPS exhibit reduced antigen-presenting capacity and chemotaxis, while their phagocytosis is significantly enhanced. LPS tolerance also brings about changes in metabolic transcription, with diminished expression of genes related to pro-inflammatory cytokines and relatively enhanced expression of some genes, such as the intolerant genes CXCL-5, FCN1, and PID, which protect the organism from systemic infections in an immunosuppressed state.60,61 Acute activation of bone marrow cells triggers clathrate synthesis, which subsequently mediates innate immune tolerance in human monocytes. Studies targeting endotoxin tolerance have shown that β-glucan reverses LPS-induced tolerance. β-Glucan-induced trained immunity counteracts the induced tolerance in sepsis models by inhibiting the expression of immune response gene 1, an enzyme that controls clathrate synthesis, isolated β-glucan-treated monocytes from patients with endotoxemia and restored cytokine production. -Glucan also increases the expression of succinate dehydrogenase, which contributes to the integrity of the tricarboxylic acid cycle and leads to an enhanced innate immune response following secondary stimulation. Inhibition of Drp1/Fis1-dependent mitochondrial fragmentation improves macrophage function and immune responses in both in vitro and in vivo models of endotoxin tolerance, and the reversal of endotoxin tolerance facilitates the restoration of immune function.62
Central Nervous System Regulation
Central nervous system (CNS)-mediated immune regulation occurs through systemic, regional, and local pathways; locally through the sympathetic and parasympathetic nervous systems, and systemically through the hypothalamic-pituitary-adrenal axis to release glucocorticoids.63 In the relationship between the nervous system and immunity, sensory neurons, such as macrophages, are at the vanguard of the defense system of the body and directly interact with pathogens and inflammatory products released by the host to initiate action potentials. The rapid initiation of action potentials allows the nervous system to communicate with the reticuloendothelial system, facilitating reflexive regulation of innate and acquired immune responses.64 In sepsis, N-oleoyldopamine induces the anti-inflammatory cytokine IL-10 via the CNS TRPV10.65 Moreover, sepsis-associated encephalopathy and immunosuppression can form a vicious circle, where the impaired activity of central neuromodulatory mechanisms leads to a decrease in the number and abnormal response of peripheral immune cells, and immune heterosuppression leads to uncontrolled neurological inflammation, which in turn exacerbates brain damage.66 Due to immunosuppression and dysregulation of the gut microbiota, susceptibility to sepsis is significantly increased in patients with cerebral hemorrhage.67 Septic brain dysfunction exerts immunosuppressive effects on peripheral immune cells, such as monocytes/macrophages (increased IL-10 production and M2-type polarization), dendritic cells (reduced responsiveness to TLR stimulation), and T lymphocytes (an imbalance between Tregs and proinflammatory lymphocyte subsets). The CNS plays a key role in altering immune status in sepsis; however, there are no treatments available to improve immune function in sepsis via the central or peripheral nervous systems. The endogenous cannabinoid system implicated is in numerous physiological processes and is believed to play a significant role in the immune response and CNS function. However, the precise immunomodulatory role of the endogenous cannabinoid system in sepsis remains a topic of debate. The vicious circle between sepsis-related encephalopathy and immunosuppression suggests the need for increased vigilance against CNS infections in the context of immunosuppression, to prevent serious side effects.
Epigenetic Regulation
Epigenetics refer to the regulation of gene expression and the persistence of the extracellular environment that specifically affect genomic regulatory sequences. Long-term cellular repression is caused by epigenetic alterations, resulting in the repression of pro-inflammatory gene transcription.68 Regarding the previously mentioned endotoxin tolerance, in vitro evidence suggests that epigenetic modifications play a crucial role in its regulation. Macrophages stimulated by LPS can be classified into two categories based on gene activation through the TLR pathway: tolerant and intolerant. The expression of tolerant genes was repressed, and the expression of non-tolerant genes was increased. Transcriptional activation of non-tolerant genes is associated with the acetylation of their promoters.69,70 The diminished phagocytic capacity of macrophages is not caused by direct exposure to the pathogen but by secondary immunosuppressive signals established locally after the primary infection subsides. This is where signal-regulated protein α (SIRPα) plays a key role in the establishment of a microenvironment that induces tolerance training, and macrophages undergo epigenetic alterations following inflammation, leading to long-term immune paralysis.71
Three main mechanisms underlie epigenetic inheritance: DNA methylation, post-translational histone modifications (methylation, acetylation, ubiquitination, and phosphorylation), and translational regulation. The first two genes regulate gene expression through their action on the chromatin. Post-translational histone modifications and DNA methylation can alter the phenotype of immune cells.72 In animal models of sepsis, histone modifications in the promoter regions of IL-1β and TNF-α by macrophages inhibit their expression. In contrast, DNA methylation in the TNF-α promoter region silences gene expression.73,74 The use of bacterial stimulation by TLR to increase methylation can increase IL-10 levels, and the DNA methylation pattern of monocytes in patients with sepsis may be associated with immunosuppression.75 In addition, a study comparing bone marrow-derived macrophages and wound macrophages in septic mice found that wound repair was impaired and secretion of repair-critical cytokines (IL-12 and IL-23) was reduced after recovery in septic mice; further studies found that reduced expression of the epigenetic enzyme Mll1 in macrophages as well as histone 3 on the inflammatory gene promoter lysine 4 trimethylation was impaired.76
Translation regulation involves ncRNAs; miRNAs are small ncRNAs that can regulate the expression of multiple mRNAs by targeting their messages and play an important role in the epigenetic regulation of gene expression in immune cells.77 Recent studies have identified miRNAs as potential biomarkers of sepsis severity and possible mediators of LPS tolerance. miR-221, miR-579, and miR125b contain binding sites in the 3’ untranslated region of TNF and can block TNF translation, while miR146a can bind to TNF mRNA by inducing a silencing complex and inhibit TNF expression.78–80 Circulating histones play a key role in immunosuppression in sepsis, and changes in histone activity induce chromatin mutations that mediate immunosuppression in the chronic phase in sepsis survivors, a process that involves substantial alterations in histone lysine acetylation, methylation, and DNA methylation.81–83 In addition, immunosuppression persists for some time after sepsis is cured; however, epigenetic modulation persists, which may lead to a poor long-term prognosis for patients with sepsis. Clinical investigations have found that some patients with sepsis are re-hospitalized within 90 days of discharge due to accelerated progression of preexisting chronic disease, residual organ damage, and impaired immune function. Modulation of epigenetic alterations is critical for immunotherapy in sepsis, and the reversal of endotoxin tolerance by β-glucan, as mentioned earlier, is achieved by reversing distal element histone modifications and transcriptional reactivation at the genetic level.
Treatments targeting histone modifications have primarily focused on histone deacetylases (HDACs). Inhibitors of HDACs such as SAHA and TSA, as well as specific inhibitors like TubA (HDAC6 inhibitor), EX527 (selective SIRT1 inhibitor), and AGK2 (selective SIRT2 inhibitor), have demonstrated protective effects against sepsis.84–88 An increasing number of studies have investigated interventions targeting methylation. The catalytic component of histone methyltransferase, zeste homolog (EZH2), has shown promise in regulating immune dysfunction in sepsis. EZH2 levels in lymphocytes can serve as a marker for assessing the risk of secondary infection in patients. Both EZH2 inhibitors, GSK343, and 3-deazolidine strobilurin A, have exhibited protective effects and the ability to inhibit organ damage.89–91 However, inhibiting the histone lysine demethylase JMJD3 has been shown to reduce early mortality in septic mice with excessive inflammation.92 However, it should be noted that treatment approaches involving histone modification using these enzyme inhibitors are still in the early stages of development. The treatment of sepsis with histone modification requires careful adjustment of the immune status of individual patients, with the potential for personalized and tailored treatment strategies.
Altered Metabolism
Several studies have identified alterations in immune cell metabolism as important drivers of sepsis-induced immunosuppression.93 Under normal aerobic conditions, immune cells rely mainly on oxidative phosphorylation for energy supply, a process that yields 36 ATP molecules per molecule of glucose with a high ATP output. However, under the stimulation of pathogens, immune cell metabolism is altered to rely on aerobic glycolysis for energy supply under normal oxygen supply, which yields two ATP molecules per molecule of glucose, has a low ATP output, is highly efficient, and can produce large amounts of energy in a short period to meet immune cell activation, signaling, and proliferation.94 This metabolic switch from oxidative phosphorylation to anaerobic glycolysis, known as the Warburg effect, was first observed in tumor cells.95
Metabolic alterations are also linked to epigenetics. Trained immunity is coordinated by epigenetic markers and continuous changes in metabolic pathways, in which the metabolic sensor sirtuin 1 (SIRT1) plays a key role.96 SIRT1 is an NAD+-dependent histone deacetylase that links transcriptional regulation to intracellular energy and is involved in the coordination of cellular functions. It is capable of histone deacetylation to regulate chromatin function and promote alterations in histone and DNA methylation, leading to transcriptional repression through its anti-inflammatory and antioxidant properties and regulation of the sepsis-induced immune response.97 Further, metabolic shifts are controlled by AMPK disruption of mTOR-dependent protein synthesis and combined sirtuin 1, 3, and 6 reactions that support catabolic energizers.98 SIRT1 promotes heterochromatin formation and NF-κB inactivation of key inflammatory genes such as TNF and IL-1β. Moreover, SIRT1 inhibition can reverse dendritic cells and CD4+ T cells from a tolerant phenotype to an active phenotype.99
Monocytes in an immune-tolerant state exhibit reduced lactate production upon re-stimulation, indicating a diminished Warburg effect. Additionally, their oxidative phosphorylation capacity is impaired, highlighting the occurrence of both immune paralysis and metabolic paralysis in sepsis. The mTOR pathway plays a pivotal role in mediating these alterations, which are observed in the T-cells of patients with sepsis. The dysregulated mTOR pathway leads to a state of metabolic paralysis characterized by inhibited glycolysis, oxidative phosphorylation, glucose transporter protein (Glut) expression, and glucose uptake, ultimately compromising cell proliferation. Restoring immune function in sepsis may be achieved by reversing metabolic paralysis through mTOR pathway modulation. However, the Warburg effect exhibits a dual role, as certain studies indicate that inhibiting the Warburg effect, such as through modulation of the PKM2 pathway or aerobic exercise, can ameliorate sepsis inflammation. Conversely, the epidermal growth factor receptor-induced Warburg effect via TBK1/Glut1 promotes CD4+ T lymphocyte apoptosis and HMGB1 release.100 Research on targeting the Warburg effect for sepsis treatment has primarily focused on the hyperinflammatory phase and improving inflammatory manifestations. Immune restoration therapy during the immunosuppressed phase of sepsis should aim to restore the Warburg effect. These findings emphasize the criticality of timing and intensity in potential therapeutic interventions.
Itaconic acid, which serves as the central regulatory node between immune tolerance and trained immunity (the reprogramming of innate immune cells in response to stimulation, resulting in an enhanced response to subsequent challenges), also impacts metabolism.101 The synthesis of itaconic acid in immune cells is triggered by the inducible enzyme immune response gene 1 (IRG1), which activates the inflammatory suppressor NRF2, thereby promoting immune tolerance in monocytes. However, β-glucan inhibits the expression of IRG1, preventing immune tolerance and inducing trained immunity. This leads to a reduction in itaconic acid production and an increase in the expression of succinate dehydrogenase, which maintains the integrity of the tricarboxylic acid cycle. Consequently, there is an augmented innate immune response following secondary stimulation.102 In conclusion, patients with sepsis-induced immunosuppression exhibit widespread metabolic impairments in immune cells, necessitating further investigation to identify potential therapeutic targets.
Biomarkers to Monitor Immune Cell Status
Innate Immunity Monitoring
Neutrophil function monitoring: Reduced neutrophil bactericidal activity correlates with the severity of sepsis-induced immunosuppression, especially in patients with a poor prognosis. Common markers of neutrophil function are CD64, myelocyte-1 (TREM-1), and CD88.103,104 CD64 and TREM-1 are neutrophil proteins whose activation responds to neutrophil function. Decreased CD88 expression on neutrophils is strongly associated with increased subsequent secondary infections and is a strong predictor of immunosuppression.105
Monocytes/macrophages: HLA-DR expression is a reliable marker of monocyte antigen-presenting capacity.106 HLA-DR not only represents a co-stimulatory molecule, but also a surrogate marker of monocyte incompetence.107 There are also alternative methods for HLA-DR expression, and quantitative reverse transcription PCR (qRT-PCR) assessment of MHC II-related gene expression represents a promising approach.108 Monocyte distribution width (MDW) is an important indicator of the response of monocytes to pathogenic invasion of the blood stream, and elevated MDW has been documented for early recognition of sepsis.109 Monitoring CD1 and IL-3 on day 10 after hospital admission predicts the development of hospital-acquired infections. Septic patients with less IL-10 release from peripheral blood mononuclear cells have a better prognosis.110 Production of various pro-inflammatory cytokines was significantly reduced in monocytes isolated from immunosuppressed sepsis patients, including TNF-α, IL-1β, IL-6 and IL-12. Regardless of the source, they are important components of the cytokine storm and produce different immune functions depending on the concentration.111
Monitoring the function and proportion of myeloid-derived suppressor cells: sepsis itself can lead to a massive increase in peripheral blood MDSC, which is one of the hallmarks of an immunosuppressive response.112
Monitoring of NK cell function: sepsis patients have a significant decrease in NK cell counts, with both CD56+ and CD56− NK cell subsets persistently affected, which is associated with an increased risk of death, with a better prognosis in patients with high NK cell/lymphocyte ratios.113 NK cells exert their cytotoxic effects through the production of a variety of cytokines, the most representative of which is IFN-γ. Serum concentrations of IFN-γ reflects the function of NK cells.114
Dendritic cell assay: significant reductions in splenic DCs can be seen in patients dying of sepsis, and depletion of circulating DC counts is frequently reported in cases of sepsis, which is closely related to the development of septic shock and sepsis-induced immunosuppression.115 Importantly, the reduction in DC counts can persist for weeks and is observed more markedly in patients who die of sepsis.116
Adaptive Immune Tests
T cells: T cell counts are also predictive of sepsis-induced immunosuppression.117 CD4+ T cell counts and CD8+ T cell counts can be assayed by flow cytometry, and changes in T-cell subsets in patients with sepsis also have clinical predictive value.118,119 Sepsis infection affects the adaptive immune response, which is characterized by decreased T cell proliferation, increased apoptosis, and abnormal cytokine secretion.120 Monitoring the level of cytokine secretion is one of the most important indicators of T cell function and differentiation, and the induction of a wide range of cytokines is greatly reduced in patients with sepsis.121 During post-injury sepsis, T cell dysfunction reduces the proliferation of CD4+ T cells and induces a shift toward a TH2-type response with loss of TH1-type response.122 The TH17/Treg ratio shows a very strong positive correlation with the SOFA score, suggesting that the higher the TH17/Treg ratio, the worse the prognosis of patients with sepsis.123
B cells: similar to T cells, septic shock is associated with B lymphocyte failure.124 Serum IgG, IgA and IgM concentrations directly reflect B cell status and activity. Peripheral blood immunoglobulin concentrations in septic patients can be used to assess B-cell immune status, and the combined use of serum IgG1, IgM, and IgA has shown good performance in predicting clinical outcomes in septic patients.125 In addition to the loss of numbers, patients with sepsis develop significant B-cell dysfunction. Enhanced expression of CD80 and CD95 on the surface of B lymphocytes has been associated with an increased risk of death in patients with sepsis.126
Conclusion
In 2017, the World Health Organization recognized sepsis as a global health priority due to its increasing morbidity, mortality, and treatment costs. The controversy regarding the dominant pathogenesis of sepsis, whether it is characterized by sustained immune activation with excessive inflammation or immunosuppression, persists. Nevertheless, the significance of immunosuppression is well supported. While significant progress has been made in immunotherapy research, the translation of these findings into clinical practice is yet to be realized.
The notable importance of the Warburg effect in sepsis presents an opportunity to leverage insights from individualized cancer therapy and develop customized treatment approaches targeting specific immune cell impairments, metabolic dysregulation, and gene expression alterations in patients with sepsis. The successful implementation of personalized interventions necessitates the identification of additional biomarkers to accurately evaluate the immune status of patients. Although some biomarkers are still in the experimental stage, they demonstrate promising potential for future clinical applications. Furthermore, continuous advancements in modern biological technology are expected to refine methods for monitoring immune status. By unraveling the pathogenesis of immune dysfunction underlying sepsis and attaining a comprehensive understanding of the immune status of patients, novel avenues can be explored for early diagnosis, rational prevention, and effective treatment of sepsis.
Data Sharing Statement
This is a narrative review based on published data.
Acknowledgments
This study was supported by grants from the Department of Science and Technology of Liaoning Province (2021JH2/10300107) and Department of Education of Liaoning Province (JC2019015).
Author Contributions
XF: design, text, ZL: design, YW: design, text revision, and final approval. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
Funding was supported by grants from the Department of Science and Technology of Liaoning Province (2021JH2/10300107) and Department of Education of Liaoning Province (JC2019015).
Disclosure
The authors declare no competing interests in this work.
References
1. Bauer M, Gerlach H, Vogelmann T, et al. Mortality in sepsis and septic shock in Europe, North America and Australia between 2009 and 2019- results from a systematic review and meta-analysis. Crit Care. 2020;24(1):239. doi:10.1186/s13054-020-02950-2
2. Rhee C, Klompas M. Sepsis trends: increasing incidence and decreasing mortality, or changing denominator? J Thorac Dis. 2020;12(Suppl 1):S89. doi:10.21037/jtd.2019.12.51
3. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
4. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. 2016;353:i1585. doi:10.1136/bmj.i1585
5. Cecconi M, Evans L, Levy M, et al. Sepsis and septic shock. Lancet. 2018;392(10141):75–87. doi:10.1016/S0140-6736(18)30696-2
6. Liu D, Huang S-Y, Sun J-H, et al. Sepsis-induced immunosuppression: mechanisms, diagnosis and current treatment options. Mil Med Res. 2022;9(1):56. doi:10.1186/s40779-022-00422-y
7. Boyd JH. Toll-like receptors and opportunities for new sepsis therapeutics. Curr Infect Dis Rep. 2012;14(5):455–461. doi:10.1007/s11908-012-0273-5
8. Vincent JL. Current sepsis therapeutics. EBioMedicine. 2022;86:104318. doi:10.1016/j.ebiom.2022.104318
9. Esposito S, De Simone G, Boccia G, et al. Sepsis and septic shock: new definitions, new diagnostic and therapeutic approaches. J Glob Antimicrob Resist. 2017;10:204–212. doi:10.1016/j.jgar.2017.06.013
10. Denning NL, Aziz M, Gurien SD, et al. DAMPs and NETs in sepsis. Front Immunol. 2019;10:2536. doi:10.3389/fimmu.2019.02536
11. Joffre J, Hellman J, Ince C, et al. Endothelial responses in sepsis. Am J Respir Crit Care Med. 2020;202(3):361–370. doi:10.1164/rccm.201910-1911TR
12. Kumar V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int Immunopharmacol. 2020;89(Pt B):107087. doi:10.1016/j.intimp.2020.107087
13. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. doi:10.1016/j.cell.2010.01.022
14. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39(5):517–528. doi:10.1007/s00281-017-0639-8
15. Frimpong A, Owusu EDA, Amponsah JA, et al. Cytokines as potential biomarkers for differential diagnosis of sepsis and other non-septic disease conditions. Front Cell Infect Microbiol. 2022;12:901433. doi:10.3389/fcimb.2022.901433
16. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13(3):260–268. doi:10.1016/S1473-3099(13)70001-X
17. Chan JK, Roth J, Oppenheim JJ, et al. Alarmins: awaiting a clinical response. J Clin Invest. 2012;122(8):2711–2719. doi:10.1172/JCI62423
18. Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity. 2014;40(4):463–475. doi:10.1016/j.immuni.2014.04.001
19. Hotchkiss RS, Schmieg RE, Swanson PE, et al. Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit Care Med. 2000;28(9):3207–3217. doi:10.1097/00003246-200009000-00016
20. Zhang Z, Huang S, Wu S, et al. Clearance of apoptotic cells by mesenchymal stem cells contributes to immunosuppression via PGE2. EBioMedicine. 2019;45:341–350. doi:10.1016/j.ebiom.2019.06.016
21. Xu J, Li J, Xiao K, et al. Dynamic changes in human HLA-DRA gene expression and Th cell subsets in sepsis: indications of immunosuppression and associated outcomes. Scand J Immunol. 2020;91(1):e12813. doi:10.1111/sji.12813
22. Cabrera-Perez J, Condotta SA, Badovinac VP, et al. Impact of sepsis on CD4 T cell immunity. J Leukoc Biol. 2014;96(5):767–777. doi:10.1189/jlb.5MR0114-067R
23. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. doi:10.1038/nri3552
24. Boomer JS, To K, Chang KC, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306(23):2594–2605. doi:10.1001/jama.2011.1829
25. Grailer JJ, Kalbitz M, Zetoune FS, et al. Persistent neutrophil dysfunction and suppression of acute lung injury in mice following cecal ligation and puncture sepsis. J Innate Immun. 2014;6(5):695–705. doi:10.1159/000362554
26. Jia SH, Parodo J, Charbonney E, et al. Activated neutrophils induce epithelial cell apoptosis through oxidant-dependent tyrosine dephosphorylation of caspase-8. Am J Pathol. 2014;184(4):1030–1040. doi:10.1016/j.ajpath.2013.12.031
27. Sloan-Lancaster J, Evavold BD, Allen PM. Induction of T-cell anergy by altered T-cell-receptor ligand on live antigen-presenting cells. Nature. 1993;363(6425):156–159. doi:10.1038/363156a0
28. Lechler R, Chai J-G, Marelli-Berg F, et al. T-cell anergy and peripheral T-cell tolerance. Philos Trans R Soc Lond B Biol Sci. 2001;356(1409):625–637. doi:10.1098/rstb.2001.0844
29. Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165(2):302–319. doi:10.1084/jem.165.2.302
30. Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–334. doi:10.1146/annurev.immunol.21.120601.141110
31. Ledo C, Gonzalez CD, Poncini CV, et al. TNFR1 signaling contributes to T cell anergy during staphylococcus aureus sepsis. Front Cell Infect Microbiol. 2018;8:259. doi:10.3389/fcimb.2018.00259
32. Huang S, Liu D, Sun J, et al. Tim-3 regulates sepsis-induced immunosuppression by inhibiting the NF-kappaB signaling pathway in CD4 T cells. Mol Ther. 2022;30(3):1227–1238. doi:10.1016/j.ymthe.2021.12.013
33. Gao Y, Wang C, Wang Z, et al. Semaphorin 3A contributes to sepsis‑induced immunosuppression by impairing CD4 + T cell anergy. Mol Med Rep. 2021;23(4). doi:10.3892/mmr.2021.11941
34. Cambier JC, Gauld SB, Merrell KT, et al. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol. 2007;7(8):633–643. doi:10.1038/nri2133
35. Cambier JC, Getahun A. B cell activation versus anergy; the antigen receptor as a molecular switch. Immunol Lett. 2010;128(1):6–7. doi:10.1016/j.imlet.2009.09.006
36. Vidard L, Dureuil C, Baudhuin J, et al. CD137(4-1BB) engagement fine-tunes synergistic IL-15- and IL-21-Driven NK cell proliferation. J Immunol. 2019;203(3):676–685. doi:10.4049/jimmunol.1801137
37. Tseng HC, Cacalano N, Jewett A. Split anergized Natural Killer cells halt inflammation by inducing stem cell differentiation, resistance to NK cell cytotoxicity and prevention of cytokine and chemokine secretion. Oncotarget. 2015;6(11):8947–8959. doi:10.18632/oncotarget.3250
38. Taneja R, Parodo J, Jia SH, et al. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med. 2004;32(7):1460–1469. doi:10.1097/01.CCM.0000129975.26905.77
39. Fialkow L, Fochesatto Filho L, Bozzetti MC, et al. Neutrophil apoptosis: a marker of disease severity in sepsis and sepsis-induced acute respiratory distress syndrome. Crit Care. 2006;10(6):R155. doi:10.1186/cc5090
40. Harrell CR, Volarevic V. Apoptosis: a friend or foe in mesenchymal stem cell-based immunosuppression. Adv Protein Chem Struct Biol. 2021;126:39–62.
41. Cao C, Yu M, Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. 2019;10(10):782. doi:10.1038/s41419-019-2015-1
42. Su L, Pan P, Yan P, et al. Role of vimentin in modulating immune cell apoptosis and inflammatory responses in sepsis. Sci Rep. 2019;9(1):5747. doi:10.1038/s41598-019-42287-7
43. Gao K, Jin J, Huang C, et al. Exosomes derived from septic mouse serum modulate immune responses via exosome-associated cytokines. Front Immunol. 2019;10:1560. doi:10.3389/fimmu.2019.01560
44. Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: a complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med. 2002;30(1 Supp):S58–S63. doi:10.1097/00003246-200201001-00008
45. Nascimento DC, Melo PH, Piñeros AR, et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun. 2017;8:14919. doi:10.1038/ncomms14919
46. Ge Y, Huang M, Wu Y, et al. Interleukin-38 protects against sepsis by augmenting immunosuppressive activity of CD4 + CD25 + regulatory T cells. J Cell Mol Med. 2020;24(2):2027–2039. doi:10.1111/jcmm.14902
47. Wang YC, Weng G-P, Liu J-P, et al. Elevated serum IL-37 concentrations in patients with sepsis. Medicine. 2019;98(10):e14756. doi:10.1097/MD.0000000000014756
48. Ge Y, Huang M, Yao YM. Recent advances in the biology of IL-1 family cytokines and their potential roles in development of sepsis. Cytokine Growth Factor Rev. 2019;45:24–34. doi:10.1016/j.cytogfr.2018.12.004
49. Su Y, Qu Y, Zhao F, et al. Regulation of autophagy by the nuclear factor kappaB signaling pathway in the hippocampus of rats with sepsis. J Neuroinflammation. 2015;12:116. doi:10.1186/s12974-015-0336-2
50. Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16(11):661–675.
51. Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120(2):159–162. doi:10.1016/j.cell.2005.01.005
52. Hsieh CH, Pai P-Y, Hsueh H-W, et al. Complete induction of autophagy is essential for cardioprotection in sepsis. Ann Surg. 2011;253(6):1190–1200. doi:10.1097/SLA.0b013e318214b67e
53. Lin CW, Lo S, Hsu C, et al. T-cell autophagy deficiency increases mortality and suppresses immune responses after sepsis. PLoS One. 2014;9(7):e102066. doi:10.1371/journal.pone.0102066
54. Wang Z, Li Y, Yang X, et al. Protective effects of rapamycin induced autophagy on CLP septic mice. Comp Immunol Microbiol Infect Dis. 2019;64:47–52. doi:10.1016/j.cimid.2019.01.009
55. Yin X, Xin H, Mao S, et al. The role of autophagy in sepsis: protection and injury to organs. Front Physiol. 2019;10:1071. doi:10.3389/fphys.2019.01071
56. Shang S, Wu J, Li X, et al. Artesunate interacts with the vitamin D receptor to reverse sepsis-induced immunosuppression in a mouse model via enhancing autophagy. Br J Pharmacol. 2020;177(18):4147–4165. doi:10.1111/bph.15158
57. Unuma K, Aki T, Funakoshi T, et al. Extrusion of mitochondrial contents from lipopolysaccharide-stimulated cells: involvement of autophagy. Autophagy. 2015;11(9):1520–1536. doi:10.1080/15548627.2015.1063765
58. Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol. 2018;14(2):121–137. doi:10.1038/nrneph.2017.165
59. Liu D, Cao S, Zhou Y, et al. Recent advances in endotoxin tolerance. J Cell Biochem. 2019;120(1):56–70. doi:10.1002/jcb.27547
60. Shalova IN, Lim J, Chittezhath M, et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1alpha. Immunity. 2015;42(3):484–498. doi:10.1016/j.immuni.2015.02.001
61. Allantaz-Frager F, Turrel-Davin F, Venet F, et al. Identification of biomarkers of response to IFNg during endotoxin tolerance: application to septic shock. PLoS One. 2013;8(7):e68218. doi:10.1371/journal.pone.0068218
62. Mukherjee R, Tompkins CA, Ostberg NP, et al. Drp1/Fis1-dependent pathologic fission and associated damaged extracellular mitochondria contribute to macrophage dysfunction in endotoxin tolerance. Crit Care Med. 2022;50(6):e504–e515. doi:10.1097/CCM.0000000000005437
63. Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6(4):318–328. doi:10.1038/nri1810
64. Chavan SS, Tracey KJ. Essential neuroscience in immunology. J Immunol. 2017;198(9):3389–3397. doi:10.4049/jimmunol.1601613
65. Joffre J, Wong E, Lawton S, et al. N-Oleoyl dopamine induces IL-10 via central nervous system TRPV1 and improves endotoxemia and sepsis outcomes. J Neuroinflammation. 2022;19(1):118. doi:10.1186/s12974-022-02485-z
66. Ren C, Yao R-Q, Zhang H, et al. Sepsis-associated encephalopathy: a vicious cycle of immunosuppression. J Neuroinflammation. 2020;17(1):14. doi:10.1186/s12974-020-1701-3
67. Lin J, Tan B, Li Y, et al. Sepsis-exacerbated brain dysfunction after intracerebral hemorrhage. Front Cell Neurosci. 2021;15:819182. doi:10.3389/fncel.2021.819182
68. Carson WF, Cavassani KA, Dou Y, et al. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics. 2011;6(3):273–283. doi:10.4161/epi.6.3.14017
69. Binnie A, Tsang JLY, Hu P, et al. Epigenetics of Sepsis. Crit Care Med. 2020;48(5):745–756. doi:10.1097/CCM.0000000000004247
70. Takebe M, Oishi H, Taguchi K, et al. Inhibition of histone deacetylases protects septic mice from lung and splenic apoptosis. J Surg Res. 2014;187(2):559–570. doi:10.1016/j.jss.2013.10.050
71. Roquilly A, Jacqueline C, Davieau M, et al. Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat Immunol. 2020;21(6):636–648. doi:10.1038/s41590-020-0673-x
72. Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447(7143):425–432. doi:10.1038/nature05918
73. Chan C, Li L, McCall CE, et al. Endotoxin tolerance disrupts chromatin remodeling and NF-kappaB transactivation at the IL-1beta promoter. J Immunol. 2005;175(1):461–468. doi:10.4049/jimmunol.175.1.461
74. El GM, Yoza BK, Chen X, et al. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol Cell Biol. 2009;29(7):1959–1971. doi:10.1128/MCB.01862-08
75. Lorente-Sorolla C, Garcia-Gomez A, Català-Moll F, et al. Inflammatory cytokines and organ dysfunction associate with the aberrant DNA methylome of monocytes in sepsis. Genome Med. 2019;11(1):66. doi:10.1186/s13073-019-0674-2
76. Davis FM, Schaller MA, Dendekker A, et al. Sepsis induces prolonged epigenetic modifications in bone marrow and peripheral macrophages impairing inflammation and wound healing. Arterioscler Thromb Vasc Biol. 2019;39(11):2353–2366. doi:10.1161/ATVBAHA.119.312754
77. O’Connell RM, Rao DS, Chaudhuri AA, et al. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10(2):111–122. doi:10.1038/nri2708
78. Wang JF, Yu M-L, Yu G, et al. Serum miR-146a and miR-223 as potential new biomarkers for sepsis. Biochem Biophys Res Commun. 2010;394(1):184–188. doi:10.1016/j.bbrc.2010.02.145
79. El GM, Church A, Liu T, et al. MicroRNA-146a regulates both transcription silencing and translation disruption of TNF-alpha during TLR4-induced gene reprogramming. J Leukoc Biol. 2011;90(3):509–519. doi:10.1189/jlb.0211074
80. El GM, McCall CE. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J Biol Chem. 2010;285(27):20940–20951. doi:10.1074/jbc.M110.115063
81. Ibanez-Cabellos JS, Aguado C, Pérez-Cremades D, et al. Extracellular histones activate autophagy and apoptosis via mTOR signaling in human endothelial cells. Biochim Biophys Acta Mol Basis Dis. 2018;1864(10):3234–3246. doi:10.1016/j.bbadis.2018.07.010
82. Carson WT, Kunkel SL. Regulation of cellular immune responses in sepsis by histone modifications. Adv Protein Chem Struct Biol. 2017;106:191–225.
83. Bomsztyk K, Mar D, An D, et al. Experimental acute lung injury induces multi-organ epigenetic modifications in key angiogenic genes implicated in sepsis-associated endothelial dysfunction. Crit Care. 2015;19(1):225. doi:10.1186/s13054-015-0943-4
84. Li Y, Liu Z, Liu B, et al. Citrullinated histone H3: a novel target for the treatment of sepsis. Surgery. 2014;156(2):229–234. doi:10.1016/j.surg.2014.04.009
85. Shen MJ, Sun L-C, Liu X-Y, et al. Trichostatin A improves the inflammatory response and liver injury in septic mice through the FoxO3a/autophagy signaling pathway. World J Emerg Med. 2022;13(3):182–188. doi:10.5847/wjem.j.1920-8642.2022.056
86. Zhao T, Alam HB, Liu B, et al. Selective inhibition of SIRT2 improves outcomes in a lethal septic model. Curr Mol Med. 2015;15(7):634–641. doi:10.2174/156652401507150903185852
87. Zhao T, Li Y, Bronson RT, et al. Selective histone deacetylase-6 inhibition attenuates stress responses and prevents immune organ atrophy in a lethal septic model. Surgery. 2014;156(2):235–242. doi:10.1016/j.surg.2014.03.033
88. Williams AM, Dennahy IS, Bhatti UF, et al. Histone deacetylase inhibitors: a novel strategy in trauma and sepsis. Shock. 2019;52(3):300–306. doi:10.1097/SHK.0000000000001308
89. Yong H, Wu G, Chen J, et al. lncRNA MALAT1 accelerates skeletal muscle cell apoptosis and inflammatory response in sepsis by decreasing BRCA1 expression by recruiting EZH2. Mol Ther Nucleic Acids. 2020;19:97–108. doi:10.1016/j.omtn.2019.10.028
90. Zhao D, Li Z, Liu X, et al. Lymphocyte expression of EZH2 is associated with mortality and secondary infectious complications in sepsis. Int Immunopharmacol. 2020;89(Pt B):107042. doi:10.1016/j.intimp.2020.107042
91. Yue D, Wang Z, Yang Y, et al. EZH2 inhibitor GSK343 inhibits sepsis-induced intestinal disorders. Exp Ther Med. 2021;21(5):437. doi:10.3892/etm.2021.9854
92. Pan Y, Wang J, Xue Y, et al. GSKJ4 protects mice against early sepsis via reducing proinflammatory factors and up-regulating MiR-146a. Front Immunol. 2018;9:2272. doi:10.3389/fimmu.2018.02272
93. Cheng SC, Scicluna BP, Arts RJW, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. 2016;17(4):406–413. doi:10.1038/ni.3398
94. O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–565. doi:10.1038/nri.2016.70
95. Stienstra R, Netea-Maier RT, Riksen NP, et al. Specific and complex reprogramming of cellular metabolism in myeloid cells during innate immune responses. Cell Metab. 2017;26(1):142–156. doi:10.1016/j.cmet.2017.06.001
96. Zhang B, Moorlag SJCFM, Dominguez-Andres J, et al. Single-cell RNA sequencing reveals induction of distinct trained-immunity programs in human monocytes. J Clin Invest. 2022;132(7). doi:10.1172/JCI147719
97. Gandhirajan A, Roychowdhury S, Vachharajani V. Sirtuins and sepsis: cross talk between redox and epigenetic pathways. Antioxidants. 2021;11(1):3. doi:10.3390/antiox11010003
98. Liu TF, Vachharajani V, Millet P, et al. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem. 2015;290(1):396–408. doi:10.1074/jbc.M114.566349
99. Martin AN, Alexander-Miller M, Yoza BK, et al. Sirtuin1 targeting reverses innate and adaptive immune tolerance in septic mice. J Immunol Res. 2018;2018:2402593. doi:10.1155/2018/2402593
100. Wang X, Wang Z, Tang D. Aerobic exercise improves LPS-induced sepsis via regulating the Warburg effect in mice. Sci Rep. 2021;11(1):1–12. doi:10.1038/s41598-020-79139-8
101. Dominguez-Andres J, Novakovic B, Li Y, et al. The itaconate pathway is a central regulatory node linking innate immune tolerance and trained immunity. Cell Metab. 2019;29(1):211–220.e5. doi:10.1016/j.cmet.2018.09.003
102. Novakovic B, Habibi E, Wang S-Y, et al. β-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. 2016;167(5):1354–1368.e14. doi:10.1016/j.cell.2016.09.034
103. Kaufmann I, Hoelzl A, Schliephake F, et al. Polymorphonuclear leukocyte dysfunction syndrome in patients with increasing sepsis severity. Shock. 2006;26(3):254–261. doi:10.1097/01.shk.0000223131.64512.7a
104. Dimoula A, Pradier O, Kassengera Z, et al. Serial determinations of neutrophil CD64 expression for the diagnosis and monitoring of sepsis in critically ill patients. Clin Infect Dis. 2014;58(6):820–829. doi:10.1093/cid/cit936
105. Conway MA, Datta D, Shankar-Hari M, et al. Cell-surface signatures of immune dysfunction risk-stratify critically ill patients: INFECT study. Intensive Care Med. 2018;44(5):627–635. doi:10.1007/s00134-018-5247-0
106. Winkler MS, Rissiek A, Priefler M, et al. Human leucocyte antigen (HLA-DR) gene expression is reduced in sepsis and correlates with impaired TNFalpha response: a diagnostic tool for immunosuppression? PLoS One. 2017;12(8):e0182427. doi:10.1371/journal.pone.0182427
107. Yin J, Chen Y, Huang J-L, et al. Prognosis-related classification and dynamic monitoring of immune status in patients with sepsis: a prospective observational study. World J Emerg Med. 2021;12(3):185–191. doi:10.5847/wjem.j.1920-8642.2021.03.004
108. Peronnet E, Venet F, Maucort-Boulch D, et al. Association between mRNA expression of CD74 and IL10 and risk of ICU-acquired infections: a multicenter cohort study. Intensive Care Med. 2017;43(7):1013–1020. doi:10.1007/s00134-017-4805-1
109. Polilli E, Frattari A, Esposito JE, et al. Monocyte distribution width (MDW) as a new tool for the prediction of sepsis in critically ill patients: a preliminary investigation in an intensive care unit. BMC Emerg Med. 2021;21(1):147. doi:10.1186/s12873-021-00521-4
110. Hall MW, Knatz NL, Vetterly C, et al. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med. 2011;37(3):525–532. doi:10.1007/s00134-010-2088-x
111. Pachot A, Monneret G, Voirin N, et al. Longitudinal study of cytokine and immune transcription factor mRNA expression in septic shock. Clin Immunol. 2005;114(1):61–69. doi:10.1016/j.clim.2004.08.015
112. Coudereau R, Waeckel L, Cour M, et al. Emergence of immunosuppressive LOX-1+ PMN-MDSC in septic shock and severe COVID-19 patients with acute respiratory distress syndrome. J Leukoc Biol. 2022;111(2):489–496. doi:10.1002/JLB.4COVBCR0321-129R
113. Feng T, Liao X, Yang X, et al. A shift toward inhibitory receptors and impaired effector functions on NK cells contribute to immunosuppression during sepsis. J Leukoc Biol. 2020;107(1):57–67. doi:10.1002/JLB.4A0818-313RR
114. Guo Y, Patil NK, Luan L, et al. The biology of natural killer cells during sepsis. Immunology. 2018;153(2):190–202. doi:10.1111/imm.12854
115. Poehlmann H, Schefold JC, Zuckermann-Becker H, et al. Phenotype changes and impaired function of dendritic cell subsets in patients with sepsis: a prospective observational analysis. Crit Care. 2009;13(4):R119. doi:10.1186/cc7969
116. Guisset O, Dilhuydy M-S, Thiébaut R, et al. Decrease in circulating dendritic cells predicts fatal outcome in septic shock. Intensive Care Med. 2007;33(1):148–152. doi:10.1007/s00134-006-0436-7
117. Jimenez-Aguilar R, Sánchez-Zauco N, Tiburcio-Felix R, et al. Effects of cardiopulmonary bypass on the development of lymphopenia and sepsis after cardiac surgery in children with congenital cardiopathy. Exp Ther Med. 2020;19(1):435–442. doi:10.3892/etm.2019.8241
118. Wilson JK, Zhao Y, Singer M, et al. Lymphocyte subset expression and serum concentrations of PD-1/PD-L1 in sepsis - pilot study. Crit Care. 2018;22(1):95. doi:10.1186/s13054-018-2020-2
119. Markwart R, Condotta SA, Requardt RP, et al. Immunosuppression after sepsis: systemic inflammation and sepsis induce a loss of naive T-cells but no enduring cell-autonomous defects in T-cell function. PLoS One. 2014;9(12):e115094. doi:10.1371/journal.pone.0115094
120. Huang LF, Yao Y-M, Dong N, et al. Association between regulatory T cell activity and sepsis and outcome of severely burned patients: a prospective, observational study. Crit Care. 2010;14(1):R3. doi:10.1186/cc8232
121. Skirecki T, Swacha P, Hoser G, et al. Bone marrow is the preferred site of memory CD4+ T cell proliferation during recovery from sepsis. JCI Insight. 2020;5(10). doi:10.1172/jci.insight.134475
122. Gupta DL, Bhoi S, Mohan T, et al. Coexistence of Th1/Th2 and Th17/Treg imbalances in patients with post traumatic sepsis. Cytokine. 2016;88:214–221. doi:10.1016/j.cyto.2016.09.010
123. Hou YC, Pai M-H, Liu -J-J, et al. Alanyl-glutamine resolves lipopolysaccharide-induced lung injury in mice by modulating the polarization of regulatory T cells and T helper 17 cells. J Nutr Biochem. 2013;24(9):1555–1563. doi:10.1016/j.jnutbio.2013.01.004
124. Rauch PJ, Chudnovskiy A, Robbins CS, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335(6068):597–601. doi:10.1126/science.1215173
125. Bermejo-Martin JF, Rodriguez-Fernandez A, Herrán-Monge R, et al. Immunoglobulins IgG1, IgM and IgA: a synergistic team influencing survival in sepsis. J Intern Med. 2014;276(4):404–412. doi:10.1111/joim.12265
126. Monserrat J, de Pablo R, Diaz-Martín D, et al. Early alterations of B cells in patients with septic shock. Crit Care. 2013;17(3):R105. doi:10.1186/cc12750
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.