Back to Journals » Journal of Hepatocellular Carcinoma » Volume 11
Advances in Targeted Drug Resistance Associated with Dysregulation of Lipid Metabolism in Hepatocellular Carcinoma
Authors Huang X, Wang M, Zhang D, Zhang C, Liu P
Received 31 October 2023
Accepted for publication 20 December 2023
Published 16 January 2024 Volume 2024:11 Pages 113—129
DOI https://doi.org/10.2147/JHC.S447578
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Xiaoju Huang,1– 3 Mengmeng Wang,1– 3 Dan Zhang,1– 3 Chen Zhang,4 Pian Liu1– 3
1Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, People’s Republic of China; 2Institute of Radiation Oncology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, People’s Republic of China; 3Hubei Key Laboratory of Precision Radiation Oncology, Wuhan, 430022, People’s Republic of China; 4Liver Transplant Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, 430022, People’s Republic of China
Correspondence: Chen Zhang; Pian Liu, Email [email protected]; [email protected]
Abstract: Hepatocellular carcinoma is the prevailing malignant neoplasm affecting the liver, often diagnosed at an advanced stage and associated with an unfavorable overall prognosis. Sorafenib and Lenvatinib have emerged as first-line therapeutic drugs for advanced hepatocellular carcinoma, improving the prognosis for these patients. Nevertheless, the issue of tyrosine kinase inhibitor (TKI) resistance poses a substantial obstacle in the management of advanced hepatocellular carcinoma. The pathogenesis and advancement of hepatocellular carcinoma exhibit a close association with metabolic reprogramming, yet the attention given to lipid metabolism dysregulation in hepatocellular carcinoma development remains relatively restricted. This review summarizes the potential significance and research progress of lipid metabolism dysfunction in Sorafenib and Lenvatinib resistance in hepatocellular carcinoma. Targeting hepatocellular carcinoma lipid metabolism holds promising potential as an effective strategy to overcome hepatocellular carcinoma drug resistance in the future.
Keywords: hepatocellular carcinoma, lipid metabolism, sorafenib, lenvatinib, targeted drug resistance
Introduction
Liver cancer ranks as the sixth most prevalent cancer globally and stands as the third principal contributor to cancer-related mortality.1 The most common subtype of primary liver cancer is hepatocellular carcinoma, accounting for 75–85% of liver cancer cases.2 Owing to its subtle onset, considerable invasiveness, and absence of early manifestations, a significant proportion of patients receive diagnoses during the intermediate to advanced stages, with approximately 70% experiencing recurrent metastasis following surgical resection.3 Notably, tyrosine kinase inhibitors (TKIs) such as Sorafenib and Lenvatinib have emerged as pivotal agents for first-line molecular targeted therapy in advanced hepatocellular carcinoma.4,5 Additionally, Regorafenib, Cabozantinib, and Ramucirumab have been endorsed as second-line therapeutic options for hepatocellular carcinoma.6–8 Furthermore, the clinical efficacy of immune checkpoint inhibitors (ICI),9 anti-angiogenic agents,10 and other targeted therapies, such as EGFR inhibitors,11 has progressively manifested, thereby broadening the therapeutic possibilities for advanced hepatocellular carcinoma.
The presence of clinical drug resistance continues to pose a significant obstacle in the management of advanced hepatocellular carcinoma. Most hepatocellular carcinoma patients receiving Sorafenib developing resistance within 6 months.12 Despite Lenvatinib exhibiting non-inferiority to Sorafenib in relation to overall survival, the emergence of acquired resistance remains a substantial impediment to the efficacy of Lenvatinib. In hepatocellular carcinoma, our understanding of the resistance targets and specific mechanisms of Sorafenib and Lenvatinib remains limited. Recent studies have identified a potential association between dysregulation of lipid metabolism homeostasis and the pathogenesis of hepatocellular carcinoma. The interconnection between metabolic reprogramming and the development and advancement of hepatocellular carcinoma has been acknowledged.13–15 Dysregulated lipid metabolism plays a significant role in a variety of liver diseases, including hepatocellular carcinoma.16 In contrast to the extensive research conducted on glucose and amino acid metabolism in hepatocellular carcinoma, there has been relatively less emphasis on investigating the influence of dysregulated lipid metabolism on the occurrence and progression of hepatocellular carcinoma.
The objective of this review is to provide an overview of the dysregulated lipid metabolism in hepatocellular carcinoma, emphasizing its potential significance and impact on the resistance development to Sorafenib and Lenvatinib. Furthermore, the potential of targeting lipid metabolism as a novel strategy to overcome resistance to targeted therapy in hepatocellular carcinoma is discussed. In the future, targeting lipid metabolism pathways holds promise as another emerging strategy to improve targeted therapy resistance in hepatocellular carcinoma.
The Correlation Between Dysregulation of Lipid Metabolism and the Onset of Hepatocellular Carcinoma
The lipidomics investigations in hepatocellular carcinoma predominantly concentrate on the alterations observed in biological markers, specifically key enzymes implicated in lipid metabolism. However, there remains a dearth of more direct and comprehensive approaches and indicators to evaluate modifications in lipid metabolism within the context of hepatocellular carcinoma.
De Novo Synthesis and Uptake of Fatty Acids
The augmented synthesis rate of fatty acids (FAs) in hepatocellular carcinoma cells is correlated with an unfavorable prognosis in hepatocellular carcinoma patients.17 This association is closely tied to the heightened expression of various pivotal enzymes involved in the lipid metabolism pathway.18 In this discourse, we primarily focus on several frequently overexpressed enzymes in the process of fatty acid synthesis in hepatocellular carcinoma, such as fatty acid synthase (FASN), ATP citrate lyase (ACLY), stearoyl-CoA desaturase 1 (SCD1) and fatty acid translocase CD36.
Fatty acid synthase is a crucial enzyme involved in the de novo synthesis of fatty acids, exerting a prominent influence among other fatty acid synthases. Its catalytic function involves the conversion of acetyl-CoA and malonyl-CoA into palmitic acid ester and other long-chain fatty acids.19 The upregulation of fatty acid synthase leads to the augmentation of endogenous fatty acid synthesis and the formation of lipid rafts, thereby facilitating tumor cell proliferation.20 In hepatocellular carcinoma tissues, fatty acid synthase exhibits a high expression level, and the inhibition of fatty acid synthase through knockdown techniques has been shown to impede the proliferation, invasion, and migration of hepatocellular carcinoma cells.21 ATP citrate lyase serves as a catalyst for the production of acetyl-CoA and oxaloacetate through the conversion of citrate and CoA. Moreover, ATP citrate lyase has been observed to be upregulated in hepatocellular carcinoma and is closely correlated with unfavorable prognosis in patients.22
Stearoyl-CoA desaturase 1 is a crucial enzyme responsible for the transformation of saturated fatty acids (SFAs) into monounsaturated fatty acids (MUFAs). The elevated expression of stearoyl-CoA desaturase 1 is significantly correlated with advanced stage of hepatocellular carcinoma and reduced overall survival in comparison to hepatocellular carcinoma patients exhibiting normal stearoyl-CoA desaturase 1 expression.23 The upregulation of stearoyl-CoA desaturase 1 expression has the potential to modify the ratio of monounsaturated fatty acids to saturated fatty acids within tumor cells, and this ratio has a direct impact on the fluidity of tumor cell membranes. Increased membrane fluidity is also a characteristic feature of tumor progression.24 Consequently, the heightened expression of stearoyl-CoA desaturase 1 can facilitate the invasion of hepatocellular carcinoma cells by modifying lipid composition and augmenting membrane fluidity.25
CD36, also referred to as fatty acid translocase, exhibits expression and assumes diverse functions across various cellular populations. While CD36 participates in the absorption of fatty acids (FAs) in numerous cell types, its expression is notably scarce in normal hepatic cells. Conversely, CD36 manifests high expression levels in hepatocellular carcinoma tissues, thereby correlating with reduced overall survival rates among hepatocellular carcinoma patients.26 This observation suggests an augmented dependence on exogenous fatty acids uptake within hepatocellular carcinoma tissues. According to recent research, it has been established that CD36-mediated endocytosis plays a crucial role in facilitating the transportation of fatty acids across the plasma membrane in adipocytes. Furthermore, the process of palmitoylation of CD36 has been found to exert dynamic control over the activity of fatty acid uptake.27
Cholesterol Metabolism
Cholesterol serves as a crucial constituent of lipid composition, and its interaction with sphingolipids facilitates the formation of lipid rafts, thereby regulating membrane fluidity, protein transport, and tumor progression.28,29 Furthermore, disrupted cholesterol homeostasis is implicated in the metabolic dysregulation observed in hepatocellular carcinoma. While certain studies have demonstrated that elevated dietary cholesterol is an independent risk factor for hepatocellular carcinoma,30 the influence of serum cholesterol levels on the prognosis of hepatocellular carcinoma patients remains inconclusive. The upregulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) in hepatocellular carcinoma31 and the promotion of hepatocellular carcinoma growth through cholesterol synthesis in the absence of fatty acid synthase mediated de novo lipogenesis suggest a potential synergistic relationship between de novo lipogenesis and endogenous cholesterol synthesis in facilitating hepatocellular carcinoma growth.32
The cellular uptake of exogenous cholesterol is facilitated by the binding of low density lipoprotein receptors to low density lipoprotein and subsequent endocytosis. Several studies have demonstrated a significant association between reduced expression of low density lipoprotein receptors and unfavorable prognosis in hepatocellular carcinoma. The inhibition of low density lipoprotein receptors mediated uptake of exogenous cholesterol has been shown to activate the MEK/ERK pathway, thereby enhancing endogenous cholesterol synthesis and facilitating the proliferation and migration of hepatocellular carcinoma cells.33 Additionally, ABCA8, a transmembrane transport protein, plays a crucial role in regulating cholesterol efflux and maintaining the homeostasis of high density lipoprotein cholesterol. Decreased expression of ABCA8 can induce oxidative stress and elevate the production of reactive oxygen species (ROS), which subsequently activate the ERK signaling pathway, induce epithelial-mesenchymal transition (EMT), and promote the progression of hepatocellular carcinoma.34
In conclusion, a decrease in exogenous cholesterol utilization may lead to an upregulation of endogenous cholesterol synthesis in hepatocellular carcinoma cells, potentially promoting hepatocellular carcinoma progression. Nevertheless, an excessive accumulation of cholesterol can induce lipotoxicity in tumor cells.35 The synthesis of cholesterol esters may act as an adaptive regulatory mechanism to maintain cholesterol levels within a safe range in cells. However, the specific regulatory mechanisms of cholesterol esters in hepatocellular carcinoma progression have yet to be fully elucidated. The intricate regulatory mechanisms underlying the interplay between cholesterol metabolism in hepatocellular carcinoma and the development of drug resistance specific to hepatocellular carcinoma remain incompletely elucidated.
Fatty Acid β-Oxidation
Furthermore, the dysregulation of lipid metabolism in hepatocellular carcinoma extends beyond lipid synthesis and encompasses fatty acid β-oxidation. Fatty acid β-oxidation, the primary pathway for fatty acid oxidation and decomposition, has received limited attention regarding its significance in hepatocellular carcinoma development. A crucial enzyme in this process is carnitine palmitoyltransferase 1 (CPT1), which facilitates the conversion of acyl-coenzyme A to acylcarnitine for mitochondrial entry. Carnitine palmitoyltransferase 1 also plays a pivotal role in regulating fatty acid oxidation (FAO) and preserving energy homeostasis.36
There exists research evidence that substantiates the notable downregulation of fatty acid oxidation in hepatocellular carcinoma cells, and the diminished expression of carnitine palmitoyltransferase 2 (CPT2) in hepatocellular carcinoma enables evasion of lipotoxicity through the inhibition of JNK activation.37 As a result, the reduction in carnitine palmitoyltransferase 2 expression leads to the accumulation of a substantial quantity of acylcarnitine within the mitochondria. The downregulation of carnitine palmitoyltransferase 2 facilitates the adaptation of hepatocellular carcinoma cells to an environment abundant in lipids, and the excessive presence of acylcarnitine can serve as a mediator for STAT3 activation, thereby promoting the progression of liver cancer.37 ACOX1, known as acyl-CoA oxidase 1, is a crucial enzyme involved in regulating the rate of fatty acid β-oxidation. The downregulation of acyl-CoA oxidase 1 expression has been found to contribute to the development of hepatocellular carcinoma.38 Consequently, the suppression of crucial enzymes implicated in fatty acid β-oxidation can potentially elevate the levels of fatty acids through the inhibition of their oxidation and utilization. This, in turn, creates a conducive lipid milieu that promotes the malignant phenotypic characteristics of hepatocellular carcinoma cells, including enhanced proliferation, invasion, and resistance to therapeutic agents.
In summary, dysregulation of lipid metabolism in hepatocellular carcinoma is characterized by increased endogenous fatty acid synthesis and uptake, upregulated cholesterol synthesis, and changes in fatty acid oxidation (Figure 1). These alterations in lipid metabolism are closely linked to the growth, proliferation, metastasis, and development of drug resistance in hepatocellular carcinoma.
Dysregulation of Hepatocellular Carcinoma Lipid Metabolism and Its Association with Targeted Drug Resistance
The metabolism of lipids is a multifaceted process that is regulated by numerous pathways. The intermediates produced during lipid metabolism have the potential to impact the development of hepatocellular carcinoma and contribute to resistance against targeted drugs via diverse signaling pathways. In recent times, mounting evidence indicates a strong correlation between lipid metabolism and targeted drug resistance in hepatocellular carcinoma, thereby highlighting the potential of targeting lipid metabolism pathways as a promising and emerging strategy to overcome treatment resistance in hepatocellular carcinoma. The purpose of this section is to discuss the impact of dysregulated lipid metabolism on the development of resistance to Sorafenib and Lenvatinib in hepatocellular carcinoma.
Role of Lipid Metabolism Pathways and Key Molecules in Sorafenib Resistance
Sorafenib, a multi-targeted tyrosine kinase inhibitor, exerts its anti-tumor effects by inhibiting tumor proliferation through various receptor tyrosine kinases, including c-Kit, FLT-2, and RET, as well as the Raf/MEK/ERK pathway.39 Moreover, Sorafenib also exhibits indirect anti-tumor activity by targeting VEGFRs and interfering with angiogenesis, thereby inhibiting tumor growth.40 Over the past decade, Sorafenib has consistently been recommended as the first-line treatment with the most robust evidence for advanced hepatocellular carcinoma and has been extensively studied to understand the mechanisms of drug resistance among tyrosine kinase inhibitors. The issue of Sorafenib resistance remains a significant obstacle in the management of advanced hepatocellular carcinoma patients. The underlying mechanisms of Sorafenib resistance are intricate and still uncertain, prompting current research to focus on various hepatocellular carcinoma-related signaling pathways (such as PI3K-AKT, JAK-STAT), excessive activation of epidermal growth factor receptors (EGFR), epithelial-mesenchymal transition (EMT), regulation of cell death pathways (including autophagy, ferroptosis), and excessive activation of hypoxia-inducible factors.41–43
In recent studies, it has been observed that there is an abnormal elevation in the activity of lipid metabolism-related enzymes in Sorafenib-resistant hepatocellular carcinoma cells,44 providing direct evidence for the involvement of lipid metabolism in Sorafenib resistance. Specifically, stearoyl-CoA desaturase 1, a crucial enzyme in unsaturated fatty acid synthesis, has been identified as a contributor to Sorafenib resistance in hepatocellular carcinoma due to its high expression levels. Moreover, the knockdown of stearoyl-CoA desaturase 1 has been shown to induce endoplasmic reticulum stress (ERS) and enhance the sensitivity of hepatocellular carcinoma stem cells to Sorafenib.45 Another relevant gene, SLC27A5, which encodes fatty acid transport protein 5 (FATP5), belongs to the SLC27A gene family. The role of fatty acid transport protein 5 in fatty acid transport and bile acid metabolism is of significant importance. In hepatocellular carcinoma, the downregulation of SLC27A5 has been observed, which consequently facilitates the progression of hepatocellular carcinoma. Specifically, the decrease in SLC27A5 expression leads to elevated levels of intracellular polyunsaturated fatty acids and reactive oxygen species (ROS), thereby activating the KEAP1/NRF2 pathway and promoting the growth of hepatocellular carcinoma cells.46 A potential approach to augment the efficacy of Sorafenib treatment in hepatocellular carcinoma cells could involve the combination of Sorafenib with NRF2/TXNRD1 inhibitors.
Despite the longstanding emphasis on the influence of cholesterol on the progression of hepatocellular carcinoma, the precise mechanisms through which cholesterol metabolism impacts resistance to Sorafenib remain elusive. Niemann-Pick C2 (NPC2), a glycoprotein involved in the regulation of intracellular free cholesterol levels, is found to be downregulated in liver cancer tissues. This downregulation leads to the accumulation of free cholesterol, ultimately inducing resistance to Sorafenib in hepatocellular carcinoma cells by activating the MAPK/AKT pathway.47 NPC2 and free cholesterol can be utilized as biological markers for assessing the treatment response and prognosis evaluation of Sorafenib in hepatocellular carcinoma. Furthermore, Sterol Regulatory Element-Binding Protein 2 (SREBP2) directly interacts with the STARD4 promoter, leading to an elevation in mitochondrial cholesterol levels and facilitating the development of Sorafenib resistance in hepatocellular carcinoma. These specific mechanisms may be attributed to the influence of mitochondrial cholesterol on cell membrane permeability and the release of cytochrome C.48
The novel potential of Maprotiline, an antidepressant medication, as a therapeutic approach for hepatocellular carcinoma is noteworthy. Maprotiline acts as a potent norepinephrine reuptake inhibitor, exhibiting robust antidepressant effects. In hepatocellular carcinoma cells, the activation of the ERK pathway by cellular retinoic acid-binding protein 1 (CRABP1) leads to the phosphorylation of Sterol Regulatory Element-Binding Protein 2, which in turn promotes cholesterol synthesis and hepatocellular carcinoma cell growth.49 The direct binding of Maprotiline to cellular retinoic acid-binding protein 1 inhibits its activity, thereby suppressing the activation of the ERK pathway and the phosphorylation of Sterol Regulatory Element-Binding Protein 2, ultimately resulting in the inhibition of cholesterol synthesis and hepatocellular carcinoma growth.49 Furthermore, Maprotiline has been found to be associated with resistance to Sorafenib in hepatocellular carcinoma. In contrast to Sorafenib alone, low dose Maprotiline combined with Sorafenib inhibits hepatocellular carcinoma cell proliferation more strongly. A strong theoretical basis for the development of lipid metabolism inhibitors has been established by examining key molecules in lipid metabolism pathways for their complex regulatory roles in Sorafenib resistance. There is, however, room for further exploration of lipid metabolism resistance targets.
The Role of Lipid Metabolism Pathways and Their Key Molecules in Lenvatinib Resistance
Researchers have diligently focused on overcoming the clinical hurdle of targeted therapy resistance and creating innovative targeted medications. Consequently, in 2018, the Food and Drug Administration (FDA) granted approval to Lenvatinib as a first-line treatment option for advanced hepatocellular carcinoma.5 Lenvatinib, an orally administered tyrosine kinase inhibitor (TKI), exhibits selective inhibition of VEGFR1-3, FGFR1-4, PDGFR, RET, and KIT. Notably, Lenvatinib has been proven to be non-inferior to Sorafenib, making it the first drug to demonstrate such efficacy.5 Consequently, the approval of Lenvatinib for the treatment of advanced hepatocellular carcinoma has significantly altered the therapeutic landscape, which was dominated by Sorafenib for over a decade.
The recently sanctioned pharmaceutical agent, Lenvatinib, encounters a substantial obstacle in the form of resistance. In recent years, there has been a predominant focus among researchers on the investigation of Lenvatinib resistance in relation to various factors, including the HGF/c-MET axis,50 activation of the MAPK/ERK pathway,51 upregulation of IRF2 and β-catenin expression,52 overexpression of FGFR1 and activation of downstream AKT/mTOR pathway,53 and upregulation of VEGFR2 expression and activation of downstream Ras/MEK/ERK pathway.54 Given the clinical challenges associated with Lenvatinib resistance, it is imperative to further explore the underlying mechanisms of resistance.
In the study conducted, genomic libraries were employed by researchers to conduct a screening for genes associated with resistance to Lenvatinib. The findings revealed a correlation between the activation of EGFR, MEK/ERK, and PI3K/AKT signaling pathways and the development of acquired resistance to Lenvatinib.55 Furthermore, lipid rafts, which are lipid microdomains within the cell membrane, were observed to possess high dynamism and enrichment of sphingolipids and cholesterol. These lipid rafts selectively recruit specific signaling proteins to regulate protein-protein interactions and facilitate cell signaling transduction.56 ABCB1, a member of the ATP-binding cassette transporters family, is acknowledged as a multidrug resistance protein 1 (MDR1) and functions as an efflux pump for numerous anticancer drugs, thereby augmenting drug efflux and exerting a pivotal influence on diverse drug resistance phenotypes.57 Investigations have demonstrated that hepatocellular carcinoma cells possess the ability to activate the epidermal growth factor receptor (EGFR), which subsequently triggers the activation of ABCB1 in a lipid raft-dependent manner. This study provides evidence of a strong correlation between the activation of the EGFR/STAT3/ABCB1 axis and the development of resistance to Lenvatinib. This resistance is accompanied by changes in cholesterol metabolism and the activation of lipid rafts. These findings suggest that Lenvatinib promotes the efflux of drugs and the activation of lipid rafts, leading to the induction of resistance.58 However, the combination of Erlotinib and Lenvatinib can effectively inhibit the EGFR/STAT3/ABCB1 axis, thereby reducing Lenvatinib resistance mediated by drug efflux. This therapeutic approach holds promise for improving the prognosis of hepatocellular carcinoma patients with initially high EGFR expression.59
The Sterol Regulatory Element Binding Protein serves as a pivotal transcription factor responsible for the regulation of lipid synthesis. The activation of Sterol Regulatory Element Binding Protein 2 mediated cholesterol biosynthesis significantly augments hepatocellular carcinoma stemness and confers resistance to therapeutic agents.60 Cholesterol deposition has been found to be elevated in hepatocellular carcinoma cells resistant to Sorafenib and Lenvatinib. Nevertheless, the administration of simvastatin has demonstrated the potential to enhance the susceptibility of hepatocellular carcinoma cells to Sorafenib and Lenvatinib.60 Sterol Regulatory Element Binding Protein 2 exhibits potential as a dependable biological indicator for evaluating the effectiveness of Sorafenib and Lenvatinib.
Hepatocellular carcinoma is a solid tumor characterized by a significant vascularization and a propensity for both intrahepatic and distant metastasis. Sphingosine-1-phosphate (S1P) is the ultimate outcome of sphingolipid metabolism, which constitutes an essential component of biological membranes.61 Moreover, Sphingosine-1-phosphate, a crucial regulatory factor, plays a pivotal role in the regulation of vascular growth and maturation. In the circulatory system, Sphingosine-1-phosphate predominantly associates with high density lipoprotein (HDL) containing apolipoprotein M (ApoM), thereby facilitating the interaction between Sphingosine-1-phosphate and its corresponding Sphingosine-1-phosphate receptor (S1PR).61 Sphingosine-1-phosphate receptor 1 exhibits significant expression in hepatocellular carcinoma and contributes to angiogenesis by suppressing the synthesis of ceramide. The expression of Sphingosine-1-phosphate receptor 1 diminishes upon administration of Lenvatinib to patients with advanced hepatocellular carcinoma.62 Consequently, it is postulated that Lenvatinib may impede hepatocellular carcinoma angiogenesis, thereby potentially delaying the progression of hepatocellular carcinoma. Nevertheless, further investigation is required to furnish corroborative evidence elucidating the comprehensive utilization of lipid metabolism resources by hepatocellular carcinoma for metabolic remodeling and the subsequent augmentation of target resistance.
A Comparative Analysis of the Mechanisms Underlying Resistance to Sorafenib and Lenvatinib
Comparative analysis with Sorafenib has demonstrated enhancements in objective response rate (ORR), progression-free survival (PFS), and time to tumor progression (TTP) in hepatocellular carcinoma patients.5 Nevertheless, the attainment of long-term benefits remains elusive. Consequently, it is imperative to undertake further investigation into the resistance mechanisms shared by Sorafenib and Lenvatinib, as well as their divergences, to facilitate the precise treatment of patients with advanced hepatocellular carcinoma.
Sorafenib and Lenvatinib exhibit targeted therapeutic effects in hepatocellular carcinoma through the inhibition of diverse receptor tyrosine kinases (RTKs). However, their differential selectivity for specific targets results in distinct treatment outcomes. Notably, the Raf/MEK/ERK pathway plays a crucial role in regulating cellular proliferation and differentiation. The pERK protein, a vital downstream element of the MEK/ERK pathway, is implicated in this process. In a clinical study, it was determined that patients with hepatocellular carcinoma who exhibited elevated levels of baseline pERK were more responsive to Sorafenib,63 thereby indicating the potential utility of pERK as a biomarker for assessing the effectiveness of Sorafenib. In vitro experiments further revealed a direct association between the sensitivity of hepatocellular carcinoma cell lines to Sorafenib and the expression level of baseline pERK. Notably, Sorafenib exhibited more pronounced inhibition in hepatocellular carcinoma cell lines characterized by higher baseline pERK expression, as compared to those with lower pERK levels.64 Therefore, it is hypothesized that the Raf/MEK/ERK signaling pathway plays a crucial role in the inhibitory effects of sorafenib on the proliferation of hepatocellular carcinoma cells.
In a recent study, the impact of Lenvatinib on Sorafenib-resistant hepatocellular carcinoma cells was assessed by researchers. Initially, a comparison was made between the kinase profiles of Lenvatinib and Sorafenib, revealing that Lenvatinib demonstrated stronger inhibition of FGFR4 and ERK pathways in comparison to Sorafenib.65 Consequently, it is postulated that Lenvatinib may exhibit superior efficacy in hepatocellular carcinoma patients with elevated FGFR4 expression. Additionally, significantly elevated expression levels of EGFR, p-Akt, and p-ERK were observed in Sorafenib-resistant hepatocellular carcinoma cells when compared to wild-type hepatocellular carcinoma cells.65 This phenomenon could potentially be attributed to the upregulation of EGFR, leading to the activation of downstream AKT and ERK signaling pathways. Suppression of the FGFR4/ERK pathway by Lenvatinib may impede the growth of Sorafenib-resistant hepatocellular carcinoma cells. Consequently, the concurrent administration of Lenvatinib and ERK inhibitors may serve as a pivotal strategy to surmount Lenvatinib resistance.
From a lipid metabolism standpoint, hepatocellular carcinoma can induce resistance to Sorafenib and Lenvatinib by upregulating crucial molecules implicated in lipid synthesis. Despite variations in the pathways leading to resistance and lipid synthesis between the two drugs, they primarily accomplish this through direct or indirect activation of the MERK/ERK and PI3K/AKT pathways, which subsequently modulate lipid metabolism. The upregulation of Sterol Regulatory Element Binding Protein 1 expression through activation of the AKT/mTOR signaling pathway plays a significant role in lipid metabolism homeostasis. Sterol Regulatory Element Binding Protein 1, a major regulator in this process, activates key enzymes involved in lipid synthesis, thereby promoting the growth of hepatocellular carcinoma cells.66 Additionally, the phosphorylation of PPARγ by MERK/ERK enhances its transcriptional activity, contributing to the regulation of lipid metabolism.67 It is evident that dysregulation of lipid metabolism holds significant implications for the emergence of resistance to sorafenib and lenvatinib.
Through an analysis of the unique resistance mechanisms to lenvatinib and sorafenib, it is possible to enhance effectiveness by specifically targeting their advantageous pathways. When deciding between sorafenib and lenvatinib, various factors including clinical context, cost-effectiveness, safety, and patient preferences should be carefully considered. Despite the favorable results observed with the integration of molecular targeted therapy and immunotherapy in hepatocellular carcinoma management, the clinical advantages of this combination approach warrant further investigation.
Research Progress in Targeting Lipid Metabolism Pathways to Overcome Hepatocellular Carcinoma Targeted Drug Resistance
In the realm of practical clinical treatment, the primary approach to mitigate the development of resistance to Sorafenib and Lenvatinib is through the implementation of combination therapy. This strategy commonly involves the utilization of various drugs, including EGFR-TKIs, anti-angiogenic agents (such as Bevacizumab), cytotoxic chemotherapeutic drugs (such as Gemcitabine, Cisplatin, etc.), and immune checkpoint inhibitors (ICIs).41,68,69 Nevertheless, the efficacy of combination therapy is hindered by the intricate nature of drug side effects. The dysregulation of lipid metabolism has emerged as a prominent subject in the investigation of hepatocellular carcinoma pathogenesis. Consequently, targeting the diverse pathways implicated in hepatocellular carcinoma lipid synthesis presents a potential avenue for effectively counteracting resistance to Sorafenib and Lenvatinib.
Focusing on the Pathways of Fatty Acid and Cholesterol Synthesis
Although previous research has demonstrated the inhibitory effect of fatty acid synthase on the growth of hepatocellular carcinoma cell lines in vitro,21 limited investigation has been conducted on fatty acid synthase in vivo in hepatocellular carcinoma. Notably, fatty acid synthase expression is upregulated in hepatocellular carcinoma cells resistant to Sorafenib, while Orlistat, an orally administered fatty acid synthase inhibitor, has shown manageable safety and tolerability.70 The targeting of lipid metabolism pathways by Orlistat has been found to induce cell apoptosis and increase the sensitivity of hepatocellular carcinoma cells to Sorafenib, as reported in a study.71 Furthermore, TVB-2640, an orally administered fatty acid synthase inhibitor, has been further optimized for safety and tolerability compared to Orlistat.72 Another novel fatty acid synthase inhibitor, TVB3664, has demonstrated the ability to enhance the therapeutic efficacy of Cabozantinib and Sorafenib by suppressing fatty acid synthase expression, as indicated in a recent study.73
Furthermore, SSI-4, a novel stearoyl-CoA desaturase 1 inhibitor, has exhibited favorable bioavailability and anti-tumor effects in in vitro experiments involving renal clear cell carcinoma74 and Sorafenib-resistant hepatocellular carcinoma cell lines.45 Sorafenib exerts its effects on hepatocellular carcinoma cells by down regulating the expression of stearoyl-CoA desaturase 1 via the AMPK/mTOR/SREBP1 pathway, thereby reducing the synthesis of monounsaturated fatty acids mediated by stearoyl-CoA desaturase 1 and inducing cell death.75 However, the current body of research on the in vivo clinical trials of stearoyl-CoA desaturase 1 inhibitors in hepatocellular carcinoma is limited, which consequently hinders the clinical application of stearoyl-CoA desaturase 1 in the treatment of hepatocellular carcinoma.
The fatty acid translocase CD36 is known to have significant involvement in lipid uptake, immune recognition, and tumor development, making it a promising target for tumor treatment. Several anti-tumor drugs that specifically target CD36 have been subjected to clinical trials, but their clinical translation has been hindered by the occurrence of severe adverse events. The majority of CD36 inhibitors that have been developed are categorized as small molecule inhibitors or fatty acid analogs.76 One such analog, known as SSO, competitively binds to CD36 and effectively inhibits the uptake of long-chain fatty acids and oxLDL by CD36.77 Currently, VT1021 stands as the sole pharmaceutical agent under investigation in clinical trials for its CD36-targeting properties.78 Furthermore, in preclinical investigations, humanized CD36 antibodies, which are specifically designed to impede CD36 activity,79 are frequently employed. Within the realm of tumor therapy, our outlook remains sanguine regarding the potential of VT1021 and humanized CD36 antibodies, as we anticipate that these therapeutic interventions targeting CD36 will offer substantial benefits in the management of tumor patients afflicted with concurrent lipid metabolism disorders.
ATP citrate lyase serves as a pivotal enzyme that establishes a connection between glucose metabolism and lipid metabolism, thereby significantly augmenting the potential for drug development associated with ATP citrate lyase. Molecular inhibitors that specifically target ATP citrate lyase, such as SB-204990 and ECT-1002, have demonstrated the ability to impede the proliferation of hepatocellular carcinoma cells.80,81 Notably, targeted inhibition of ATP citrate lyase has shown promise in partially reversing Sorafenib resistance.44 The combination of the ATP citrate lyase inhibitor BMS-303141 and Sorafenib demonstrates a synergistic impact on restraining the proliferation of mouse hepatocellular carcinoma cell xenograft models.82 Given the intricate nature of the downstream metabolic pathways associated with ATP citrate lyase, our future endeavors will focus on the development of enhanced and specific ATP citrate lyase inhibitors.
Simvastatin, a member of the statin drug class, is utilized for the purpose of reducing cholesterol levels. Its mechanism of action involves the inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) activity, leading to the suppression of cholesterol synthesis. Notably, simvastatin has demonstrated a preventive effect on the development of hepatocellular carcinoma.83 Additionally, when combined with Sorafenib, simvastatin has been found to enhance the efficacy of Sorafenib in hepatocellular carcinoma cells.84 Recent investigations have further revealed that simvastatin effectively hinders the HIF-1α/PAR-γ/PKM2 axis and PKM2-mediated glycolysis, thereby impeding the proliferation of hepatocellular carcinoma cells and rendering them more susceptible to Sorafenib treatment once again.84 Additionally, the use of organoid models derived from hepatocellular carcinoma patients has demonstrated that simvastatin can effectively suppress hepatocellular carcinoma proliferation and increase the sensitivity of hepatocellular carcinoma cells towards Sorafenib and Lenvatinib. However, a recent Phase III clinical trial has shown that the combination of Sorafenib and pravastatin does not lead to an improvement in the survival rate of advanced hepatocellular carcinoma patients.85 Consequently, variations in the pharmacological properties and cholesterol-lowering effects of different statin drugs may lead to divergent outcomes in terms of their potential to enhance the prognosis of patients with hepatocellular carcinoma.
The relationship between lipid metabolism and targeted therapy is highly interconnected. The alterations in lipid metabolism can affect the effectiveness of current targeted treatments. In this context, we have summarized the impact of various drugs targeting lipid metabolic pathways on the treatment of hepatocellular carcinoma (Table 1).
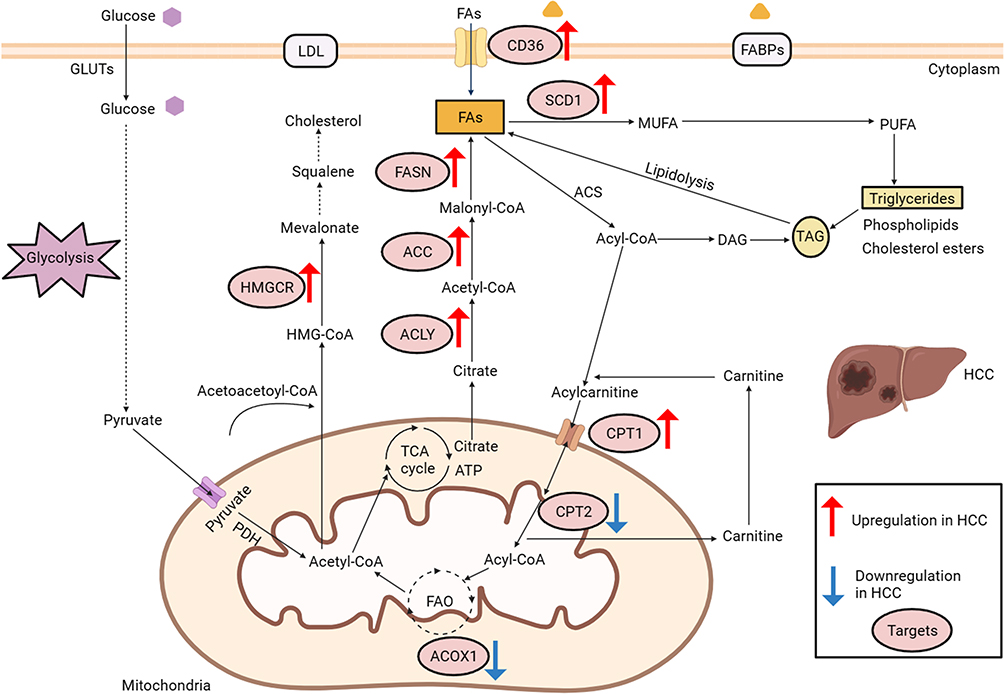 |
Figure 1 Deregulated alterations of fatty acid and cholesterol metabolism in hepatocellular carcinoma. The glycolysis pathway: GLUTs, glucose transporters; PDH, pyruvate dehydrogenase. Cholesterol metabolism: HMG-CoA, 3-hydroxy-3-methylglutaryl coenzymeA; LDL, low density lipoprotein. Abbreviations: HMGCR, 3-hydroxy-3-methylglutaryl coenzyme A reductase; The synthesis and uptake of fatty acids: FABPs, fatty acid binding protein; CD36, fatty acid translocase; ACLY, ATPcitrate lyase; ACC, acetyl-CoA carboxylase; FASN, fatty acid synthase; FAs, fatty acids; SCD1, Stearoyl-CoA desaturase 1; MUFA, monounsaturated fatty acid; PUFA, polyunsaturated fatty acids; TAG, triacylglycerol; DAG, diacylglycerol; ACS, Acyl-CoA Synthetase. Fatty acid β-oxidation: CPT1, carnitine palmitoyltransferase 1; CPT2, carnitine palmitoyltransferase 2; ACOX1, acyl-coenzyme A oxidase 1. HCC, hepatocellular carcinoma. |
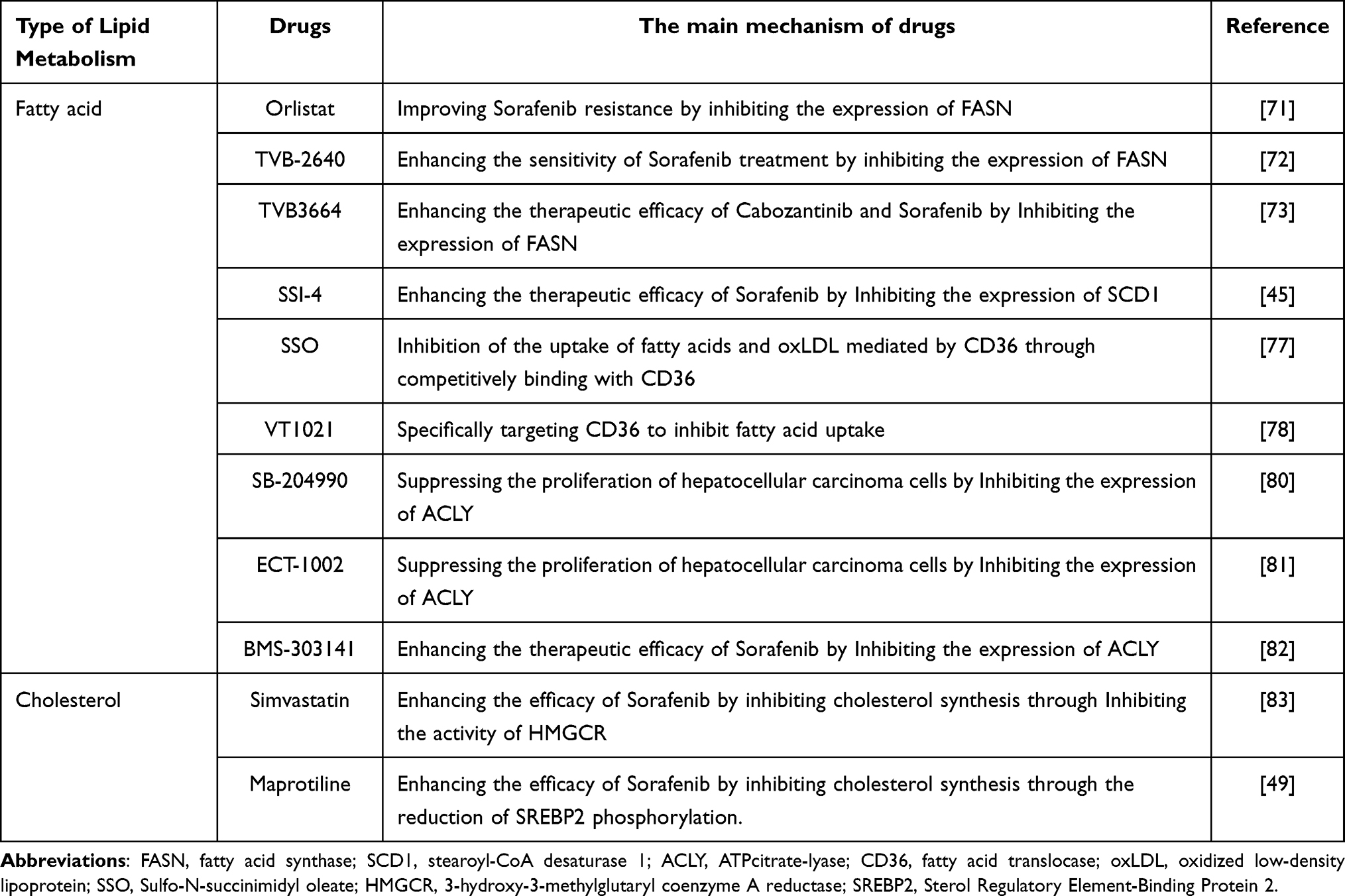 |
Table 1 The Impact of Therapeutic Drugs Targeting the Lipid Metabolism Pathway on the Treatment of Hepatocellular Carcinoma |
Proteolysis-Targeting Chimeras and Lipid Nanoparticles
The proteolysis-targeting chimera (PROTAC) technology has proposed a promising and innovative anti-tumor treatment approach. Proteolysis-targeting chimera achieves the degradation of target proteins through the ubiquitin-proteasome system (UPS). Proteolysis-targeting chimeras are composed of hybrid molecules, wherein one end is linked to a ligand that specifically binds to the target protein, while the other end is connected to a ligand that binds to the E3 ligase. These hybrid molecules possess the capability to establish a connection between the target protein and the E3 ligase, consequently prompting proteasomal degradation.86 Moreover, they exhibit a specific affinity towards receptors located on the surface of target cells, thereby facilitating the precise transportation and subsequent release of functional molecules or pharmaceutical agents. In contrast to conventional small molecule drugs, this approach offers a significant benefit in terms of its potent effectiveness against non-druggable protein targets. The utilization of proteolysis-targeting chimera enables the degradation of the entire target protein, thereby potentially mitigating resistance issues in solid tumors, including hepatocellular carcinoma.87
Nanoparticles possess the capability to augment the stability of drug effectiveness through the enhancement of cellular uptake and intracellular drug concentration. Lipid nanoparticles (LNPs) exhibit commendable biocompatibility and minimal toxicity, rendering them one of the delivery systems for hepatocellular carcinoma treatment that has been acknowledged in clinical settings.88 In recent times, scholars have directed their attention towards the transportation of lipid nanoparticles alongside reactive oxygen species (ROS) probes, employing lipid nanotechnology for the conveyance of fluorescent probes that facilitate the detection of ROS in hepatocellular carcinoma.89 Reactive oxygen species (ROS) has been observed to play a significant role in the progression of hepatocellular carcinoma by stimulating the growth of hepatocellular carcinoma cells.90 The visualization studies of ROS in hepatocellular carcinoma models have proven valuable in facilitating early diagnosis of hepatocellular carcinoma and aiding in the development of innovative drug delivery systems. In the context of hepatocellular carcinoma treatment, the utilization of lipid nanoparticles in conjunction with Sorafenib, Lenvatinib, and other emerging anti-tumor drugs has shown promise in achieving precise targeted drug delivery to cancer cells. Nevertheless, there remain challenges in enhancing the efficiency and stability of drug delivery while minimizing harm to normal cells, which necessitate further attention and resolution.
Despite some progress in research on targeting lipid metabolism pathways in hepatocellular carcinoma to overcome drug resistance, the intricate regulatory mechanisms of lipid metabolism in hepatocellular carcinoma pathophysiology present considerable challenges for its clinical application. Moreover, it is imperative to enhance the customization and precision of lipid metabolism targeting in hepatocellular carcinoma treatment to optimize the survival benefits for patients with hepatocellular carcinoma.
Palmitoylation Modification and Targeted Drug Resistance in Hepatocellular Carcinoma
Protein lipid modification is a significant post-translational alteration of proteins. Notably, palmitoylation has emerged as the most extensively investigated reversible protein lipid modification in recent years, exerting considerable influence on the physiological functions of target proteins, including their stability, membrane localization, and transport.91,92 Palmitoyl acyltransferases (PATs) represent pivotal enzymes responsible for catalyzing palmitoylation reactions.93 Proteins containing a specific Asp-His-His-Cys (DHHC) motif and zinc finger-like structure, commonly referred to as zinc finger DHHC-containing proteins (ZDHHCs), are also known as PATs.94 In mammals, a total of 23 ZDHHCs have been identified.95 The addition of palmitic acid ester to the cysteine (Cys) residue of substrate proteins is facilitated by PATs through the formation of a thioester bond.96 The palmitic acid required for this process is produced through fatty acid synthesis, which is catalyzed by fatty acid synthase. S-palmitoylation represents the primary mode of palmitoylation modification, and its reversibility is attributed to the inherent instability of thioester bonds. Nevertheless, the precise mechanism underlying the recognition and regulatory role of substrate proteins by ZDHHCs remains elusive.
The regulation of palmitoylation, a protein lipid modification, is intricately linked to lipid metabolism. Acetyl-CoA and malonyl-CoA, produced through the enzymatic action of fatty acid synthase, can generate palmitic acid esters that act as substrates for the palmitoylation process. Elevated levels of palmitic acid esters can activate ZDHHCs, thereby enhancing palmitoylation. Consequently, fatty acid synthase may indirectly govern the regulation of palmitoylation. It is established that the internalization of membranes facilitated by CD36 is closely associated with the uptake of fatty acids. Research findings have substantiated that ZDHHC4 and ZDHHC5 play a role in facilitating the palmitoylation process of CD36, thereby governing its positioning within the cellular membrane and influencing the uptake of fatty acids via distinct pathways.97 This suggests that CD36 may be one of the substrates of palmitoylation, and the palmitoylation of CD36 may be an important mechanism underlying lipid metabolism disorders.
The recent studies have shown that Astrocyte Elevated Gene-1 (AEG-1) is overexpressed in hepatocellular carcinoma. Additionally, ZDHHC6 has been identified as a mediator of Astrocyte Elevated Gene-1 (AEG-1) palmitoylation, thereby exerting a negative regulatory influence on hepatocellular carcinoma growth.98 This discovery further underscores the connection between protein lipid modification and the progression of hepatocellular carcinoma. Furthermore, PCSK9, a serine protease secreted by the liver, primarily facilitates the degradation of low-density lipoprotein receptor by binding to it.99 The expression of PCSK9 in hepatocellular carcinoma has been observed to be significantly high, while ZDHHC16 has been identified as the mediator of PCSK9 palmitoylation. This process, in turn, facilitates cell proliferation and confers resistance to Sorafenib in hepatocellular carcinoma by inducing AKT phosphorylation.100 These findings underscore the significance of palmitoylation modification in the development of Sorafenib resistance in hepatocellular carcinoma cells. Consequently, it can be concluded that the investigation of palmitoylation in hepatocellular carcinoma is relatively limited compared to other types of tumors. Hence, it is imperative to gain deeper insights into the correlation between palmitoylation modification in hepatocellular carcinoma and additional lipid metabolic pathways, as this will hold immense importance in identifying novel targets for palmitoylation therapy.
Currently, there is a lack of specific inhibitors for ZDHHC that can be used for targeted intervention in palmitoylation processes. In preclinical studies, the broad-spectrum protein palmitoylation inhibitor, 2-bromopalmitate (2-BP), has been utilized. However, its clinical translation has been restricted due to its high toxicity and lack of specificity.101 The uncertainty surrounding the selection mechanism of palmitoylation substrates allows for the possibility of a single target protein being modified by multiple ZDHHCs, and a ZDHHC can also simultaneously act on multiple target proteins.102 The challenge in developing targeted small molecule inhibitors of palmitoylation is further compounded. Studies have demonstrated that a vaccine consisting of a monopalmitoyl antigen peptide can augment the immune response against tumors in melanoma.103 However, the scarcity of antibodies specific to palmitoylation persists, primarily attributed to challenges in antigen preparation.
Ferroptosis and Hepatocellular Carcinoma Targeted Drug Resistance
Ferroptosis, a recently identified mode of non-apoptotic cell demise, distinguishes itself from necrosis, apoptosis, and autophagy through the excessive buildup of iron-dependent lipid reactive oxygen species within cells. This process is accompanied by the rupture and contraction of the mitochondrial outer membrane, as well as the reduction or vanishing of mitochondrial cristae.104,105 Notably, lipid metabolism exhibits a close association with the occurrence of ferroptosis, as evidenced by relevant studies.106 Specifically, polyunsaturated fatty acids have been identified as the primary substrates for lipid peroxidation in ferroptosis. This process selectively oxidizes long-chain polyunsaturated fatty acids, with the synthesis of these fatty acids being regulated by fatty acid synthase.107,108 In contrast, exogenous monounsaturated fatty acids have been found to possess the ability to resist ferroptosis. This resistance is achieved through the displacement of polyunsaturated fatty acids within the cellular membrane and the subsequent inhibition of reactive oxygen species generation.109 Furthermore, several studies have demonstrated that Sorafenib can also induce ferroptosis by inhibiting cystine/glutamate antiporter (xCT), leading to a decrease in glutathione peroxidase production and an increase in reactive oxygen species levels in hepatocellular carcinoma cells.110 In recent years, a growing body of evidence has substantiated the strong correlation between ferroptosis and the progression of hepatocellular carcinoma as well as the emergence of resistance to targeted therapeutics. Various potential mechanisms can be succinctly outlined as follows.
The involvement of the cystine/glutamate antiporter (xCT) and glutathione peroxidase 4 (GPX4) in the defense against ferroptosis is well-established. The cystine/glutamate antiporter protein, which is encoded by the SLC7A11 gene, plays a crucial role in facilitating the uptake of extracellular cysteine and converting it to cystine. This conversion is necessary for the synthesis of glutathione (GSH), a vital component in the defense against ferroptosis.111 Glutathione peroxidase 4, in turn, relies on glutathione as a substrate to effectively inhibit lipid peroxidation. Notably, certain drugs, including Orlistat and Lenvatinib, have been observed to modulate the xCT/GSH/GPX4 axis, thereby impacting the cellular response to ferroptosis. Orlistat, a fatty acid synthase inhibitor, has been shown to increase the sensitivity of Sorafenib-resistant hepatocellular carcinoma cell lines to Sorafenib treatment by downregulating SLC7A11.112 Lenvatinib, on the other hand, targets FGFR4 to inhibit the expression of cystine/glutamate antiporter and glutathione peroxidase 4, resulting in the accumulation of reactive oxygen species and the induction of ferroptosis.113 Additionally, Sorafenib has been found to enhance the cytotoxicity against liver cancer cells by promoting ferroptosis.114,115 In summary, the xCT/GSH/GPX4 axis plays a crucial role in defending against ferroptosis in hepatocellular carcinoma cells.
The Keap1/Nrf2 pathway is a significant regulatory mechanism implicated in the cellular response to oxidative stress, playing a crucial role in the regulation of cellular antioxidant stress and ferroptosis. Historically, Nrf2 has been acknowledged as a safeguarding transcription factor implicated in cellular antioxidant stress. However, recent findings have revealed that persistent activation of Nrf2 can facilitate the proliferation, invasion, and resistance to drugs in different tumor types, including hepatocellular carcinoma.116 In the context of hepatocellular carcinoma, Sorafenib directly triggers the p62/Keap1/Nrf2 antioxidant stress pathway and mitigates the occurrence of Sorafenib-induced ferroptosis. The Nrf2 inhibitor ML385 has the ability to induce ferroptosis in hepatocellular carcinoma and effectively counteract the resistance to Sorafenib treatment. The deactivation of Keap1 leads to the development of resistance in human hepatocellular carcinoma cells against Sorafenib, Lenvatinib, and regorafenib by upregulating the expression of downstream antioxidant stress genes and reducing levels of reactive oxygen species.117
Metformin, a frequently prescribed antidiabetic medication, has demonstrated specific therapeutic effects in the treatment of hepatocellular carcinoma. These effects are potentially attributed to the activation of the AMPK pathway and phosphorylation of FOXO3, as supported by various studies.118–120 Furthermore, it has been observed that metformin exerts a specific impact on enhancing hepatocellular carcinoma resistance to Lenvatinib, potentially influencing the progression of hepatocellular carcinoma through the regulation of FOXO3 phosphorylation.121 Recent investigations propose that the combined administration of Lenvatinib and metformin may synergistically impede the AKT/FOXO3 signaling pathway, thereby reversing Lenvatinib resistance.120 In summary, the activation of the p62/Keap1/Nrf2 pathway holds the potential to hinder ferroptosis in tumor cells and presents a promising therapeutic target for resistant tumors.
It should be noted that YAP/TAZ functions as a downstream transcription factor of the Hippo pathway, exerting a significant influence on the suppression of hepatocellular carcinoma proliferation and survival.122 It has been observed that YAP/TAZ is subject to negative regulation in cellular ferroptosis. The recently discovered YAP inhibitor CA3 exhibits selective inhibition of YAP/TAZ and enhances the susceptibility of hepatocellular carcinoma to Sorafenib, particularly in hepatocellular carcinoma cells characterized by elevated YAP/TAZ expression levels.123 The NRF2/ABCC5 pathway is known to have a significant impact on the occurrence of ferroptosis. ABCC5 is a member of the ATP-binding cassette (ABC) family. ABCC5, a drug efflux transporter protein, is closely linked to both tumor progression and drug resistance, and it serves as a crucial regulatory factor in the process of ferroptosis.124 Inhibition of ABCC5 expression has the potential to induce ferroptosis, which is associated with resistance to Sorafenib in hepatocellular carcinoma, and it can also enhance the efficacy of molecular targeted drugs.125
Ferroptosis, being morphologically and genetically distinct from apoptosis, presents a novel therapeutic approach to combat drug resistance in tumor cells. Erastin, an inducer of ferroptosis, effectively inhibits voltage-dependent anion channels (VDAC2/VDAC3) in a Ras/Raf/MEK-dependent manner, thereby inducing ferroptosis.126 RSL3 functions as a glutathione peroxidase 4 inhibitor, leading to the deactivation of glutathione and the initiation of lipid peroxidation within cellular systems.107 Furthermore, the concurrent administration of artemisinin and Sorafenib demonstrates a synergistic effect in promoting ferroptosis within hepatocellular carcinoma cells. More specifically, artemisinin exhibits the potential to sensitize hepatocellular carcinoma cells to Sorafenib by inducing apoptosis through the inhibition of the PI3K/AKT/mTOR pathway.127
Considering that lipid metabolism has been recognized as a significant contributor to the occurrence of ferroptosis, the precise regulatory mechanism underlying ferroptosis in hepatocellular carcinoma remains uncertain. Certain scholars posit that specific pivotal molecules implicated in ferroptosis and iron metabolism may serve as prognostic markers for the identification and management of liver cancer.128 Despite the increasing appeal of ferroptosis as a promising anti-tumor strategy, tumor cells can still exploit various adaptive and evasive mechanisms to counteract the effects of ferroptosis. Finally, it is crucial to acknowledge the absence of identifiable biomarkers for monitoring hepatocellular carcinoma ferroptosis in clinical settings. This limitation further hinders the advancement of in vivo studies on ferroptosis in hepatocellular carcinoma and its potential clinical application.
Summary and Outlook
Hepatocellular carcinoma is a prevalent and aggressive solid tumor that exhibits drug resistance, limited treatment options for late-stage patients, and an overall poor prognosis. Consequently, hepatocellular carcinoma represents a formidable challenge with substantial implications for global human health. Given the recent progress in lipidomics analysis, it is crucial to employ a comprehensive approach to investigate changes in hepatic lipid metabolism and identify biomarkers associated with lipid metabolism in hepatocellular carcinoma. Despite previous studies investigating the dysregulation of lipid metabolism in hepatocellular carcinoma, our comprehension of the regulatory network of lipid metabolism in hepatocellular carcinoma remains constrained when compared to the established “Warburg effect” theory in tumor metabolism. Additionally, the metabolic reprogramming of tumor cells is a synchronized process, and alterations in lipid metabolism will inevitably induce adaptive modifications in other metabolic pathways. The investigation of the contribution of these changes to the regulation of the hepatocellular carcinoma microenvironment as a holistic system, as well as the identification of appropriate beneficiaries from these alterations, remains an open area of research. Consequently, additional exploration is imperative to acquire a comprehensive comprehension of the network governing lipid metabolism regulation in hepatocellular carcinoma and its interaction with other metabolic pathways. Such endeavors will facilitate the identification of potential therapeutic targets and the formulation of personalized treatment approaches for patients with hepatocellular carcinoma.
Despite the diligent endeavors of researchers to devise additional combination therapy strategies for surmounting resistance to Sorafenib and Lenvatinib, the overall efficacy of hepatocellular carcinoma treatment remains unsatisfactory. Presently, investigations have been conducted on lipid metabolism inhibitors in both in vitro and in vivo contexts for hepatocellular carcinoma. The advancement of innovative drug delivery systems will enhance the accuracy and consistency of targeted therapy for hepatocellular carcinoma. Additionally, it is imperative to investigate the potential synergistic effects of combining lipid metabolism inhibitors with FDA-approved multi-kinase inhibitors (eg, Sorafenib, Lenvatinib, Regorafenib), immune checkpoint inhibitors, and conventional drugs in the clinical management of hepatocellular carcinoma. This review presents an initial overview of the advancements made in studying targeted resistance related to lipid metabolism in hepatocellular carcinoma. Despite ongoing research efforts, our comprehension of lipid metabolism modifications in hepatocellular carcinoma remains inadequate, and the practical application of targeting lipid metabolism pathways to enhance targeted resistance in hepatocellular carcinoma remains significantly constrained. In the forthcoming years, enhanced comprehension and characterization of pivotal molecules and associated signaling pathways implicated in hepatocellular carcinoma lipid metabolism will expedite the advancement of novel pharmaceuticals. To summarize, the pursuit of hepatocellular carcinoma lipid metabolism pathways presents novel perspectives for the identification of fresh targets for hepatocellular carcinoma resistance and the formulation of precise treatment strategies.
Funding
This research received no external funding.
Disclosure
The authors declare no conflict of interest.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Stavraka C, Rush H, Ross P. Combined hepatocellular cholangiocarcinoma (cHCC-CC): an update of genetics, molecular biology, and therapeutic interventions. J Hepatocell Carcinoma. 2019;6:11–21. doi:10.2147/JHC.S159805
3. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi:10.1038/nrdp.2016.18
4. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857
5. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised Phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–1173. doi:10.1016/S0140-6736(18)30207-1
6. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66. doi:10.1016/S0140-6736(16)32453-9
7. Zhu AX, Kang YK, Yen CJ, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(2):282–296. doi:10.1016/S1470-2045(18)30937-9
8. Abou-Alfa GK, Meyer T, Cheng AL, et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med. 2018;379(1):54–63. doi:10.1056/NEJMoa1717002
9. Cheng AL, Hsu C, Chan SL, Choo SP, Kudo M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J Hepatol. 2020;72(2):307–319. doi:10.1016/j.jhep.2019.09.025
10. Yao C, Wu S, Kong J, et al. Angiogenesis in hepatocellular carcinoma: mechanisms and anti-angiogenic therapies. Cancer Biol Med. 2023;20(1):25–43. doi:10.20892/j.issn.2095-3941.2022.0449
11. Thomas MB, Garrett-Mayer E, Anis M, et al. A Randomized Phase II Open-Label Multi-Institution Study of the Combination of Bevacizumab and Erlotinib Compared to Sorafenib in the First-Line Treatment of Patients with Advanced Hepatocellular Carcinoma. Oncology. 2018;94(6):329–339. doi:10.1159/000485384
12. Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. doi:10.1016/S1470-2045(08)70285-7
13. Bao MH, Wong CC. Hypoxia, Metabolic Reprogramming, and Drug Resistance in Liver Cancer. Cells. 2021;10(7). doi:10.3390/cells10071715
14. Du D, Liu C, Qin M, et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B. 2022;12(2):558–580. doi:10.1016/j.apsb.2021.09.019
15. Foglia B, Beltra M, Sutti S, Cannito S. Metabolic Reprogramming of HCC: a New Microenvironment for Immune Responses. Int J Mol Sci. 2023;24(8). doi:10.3390/ijms24087463
16. Siegel AB, Zhu AX. Metabolic syndrome and hepatocellular carcinoma: two growing epidemics with a potential link. Cancer. 2009;115(24):5651–5661. doi:10.1002/cncr.24687
17. Che L, Paliogiannis P, Cigliano A, Pilo MG, Chen X, Calvisi DF. Pathogenetic, Prognostic, and Therapeutic Role of Fatty Acid Synthase in Human Hepatocellular Carcinoma. Front Oncol. 2019;9:1412. doi:10.3389/fonc.2019.01412
18. Feng XC, Liu FC, Chen WY, Du J, Liu H. Lipid metabolism of hepatocellular carcinoma impacts targeted therapy and immunotherapy. World J Gastrointest Oncol. 2023;15(4):617–631. doi:10.4251/wjgo.v15.i4.617
19. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–777. doi:10.1038/nrc2222
20. Vanauberg D, Schulz C, Lefebvre T. Involvement of the pro-oncogenic enzyme fatty acid synthase in the hallmarks of cancer: a promising target in anti-cancer therapies. Oncogenesis. 2023;12(1):16. doi:10.1038/s41389-023-00460-8
21. Hao Q, Li T, Zhang X, et al. Expression and roles of fatty acid synthase in hepatocellular carcinoma. Oncol Rep. 2014;32(6):2471–2476. doi:10.3892/or.2014.3484
22. Sun H, Wang F, Huang Y, et al. Targeted inhibition of ACLY expression to reverse the resistance of sorafenib in hepatocellular carcinoma. J Cancer. 2022;13(3):951–964. doi:10.7150/jca.52778
23. Budhu A, Roessler S, Zhao X, et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144(5):1066–1075 e1. doi:10.1053/j.gastro.2013.01.054
24. Hac-Wydro K, Jedrzejek K, Dynarowicz-Latka P. Effect of saturation degree on the interactions between fatty acids and phosphatidylcholines in binary and ternary Langmuir monolayers. Colloids Surf B Biointerfaces. 2009;72(1):101–111. doi:10.1016/j.colsurfb.2009.03.019
25. Liu HH, Xu Y, Li CJ, et al. An SCD1-dependent mechanoresponsive pathway promotes HCC invasion and metastasis through lipid metabolic reprogramming. Mol Ther. 2022;30(7):2554–2567. doi:10.1016/j.ymthe.2022.03.015
26. Luo X, Zheng E, Wei L, et al. The fatty acid receptor CD36 promotes HCC progression through activating Src/PI3K/AKT axis-dependent aerobic glycolysis. Cell Death Dis. 2021;12(4):328. doi:10.1038/s41419-021-03596-w
27. Hao JW, Wang J, Guo H, et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat Commun. 2020;11(1):4765. doi:10.1038/s41467-020-18565-8
28. Helms JB, Zurzolo C. Lipids as targeting signals: lipid rafts and intracellular trafficking. Traffic. 2004;5(4):247–254. doi:10.1111/j.1600-0854.2004.0181.x
29. Donatello S, Babina IS, Hazelwood LD, Hill AD, Nabi IR, Hopkins AM. Lipid raft association restricts CD44-ezrin interaction and promotion of breast cancer cell migration. Am J Pathol. 2012;181(6):2172–2187. doi:10.1016/j.ajpath.2012.08.025
30. Ioannou GN, Morrow OB, Connole ML, Lee SP. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology. 2009;50(1):175–184. doi:10.1002/hep.22941
31. Kawata S, Takaishi K, Nagase T, et al. Increase in the active form of 3-hydroxy-3-methylglutaryl coenzyme A reductase in human hepatocellular carcinoma: possible mechanism for alteration of cholesterol biosynthesis. Cancer Res. 1990;50(11):3270–3273.
32. Che L, Chi W, Qiao Y, et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut. 2020;69(1):177–186. doi:10.1136/gutjnl-2018-317581
33. Chen Z, Chen L, Sun B, et al. LDLR inhibition promotes hepatocellular carcinoma proliferation and metastasis by elevating intracellular cholesterol synthesis through the MEK/ERK signaling pathway. Mol Metab. 2021;51:101230. doi:10.1016/j.molmet.2021.101230
34. Cui Y, Liang S, Zhang S, et al. ABCA8 is regulated by miR-374b-5p and inhibits proliferation and metastasis of hepatocellular carcinoma through the ERK/ZEB1 pathway. J Exp Clin Cancer Res. 2020;39(1):90. doi:10.1186/s13046-020-01591-1
35. Chang TY, Chang CC, Lin S, Yu C, Li BL, Miyazaki A. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and −2. Curr Opin Lipidol. 2001;12(3):289–296. doi:10.1097/00041433-200106000-00008
36. Schlaepfer IR, Joshi M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology. 2020;161(2). doi:10.1210/endocr/bqz046
37. Fujiwara N, Nakagawa H, Enooku K, et al. CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut. 2018;67(8):1493–1504. doi:10.1136/gutjnl-2017-315193
38. Huang J, Viswakarma N, Yu S, et al. Progressive endoplasmic reticulum stress contributes to hepatocarcinogenesis in fatty acyl-CoA oxidase 1-deficient mice. Am J Pathol. 2011;179(2):703–713. doi:10.1016/j.ajpath.2011.04.030
39. Li X, Li C, Zhang L, et al. The significance of exosomes in the development and treatment of hepatocellular carcinoma. Mol Cancer. 2020;19(1):1. doi:10.1186/s12943-019-1085-0
40. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7(10):3129–3140. doi:10.1158/1535-7163.MCT-08-0013
41. Chen J, Jin R, Zhao J, et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015;367(1):1–11. doi:10.1016/j.canlet.2015.06.019
42. Tang W, Chen Z, Zhang W, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5(1):87. doi:10.1038/s41392-020-0187-x
43. Mendez-Blanco C, Fondevila F, Garcia-Palomo A, Gonzalez-Gallego J, Mauriz JL. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018;50(10):1–9. doi:10.1038/s12276-018-0159-1
44. Bort A, Sanchez BG, de Miguel I, Mateos-Gomez PA, Diaz-Laviada I. Dysregulated lipid metabolism in hepatocellular carcinoma cancer stem cells. Mol Biol Rep. 2020;47(4):2635–2647. doi:10.1007/s11033-020-05352-3
45. MKF M, Lau EYT, Leung DHW, et al. Stearoyl-CoA desaturase regulates sorafenib resistance via modulation of ER stress-induced differentiation. J Hepatol. 2017;67(5):979–990. doi:10.1016/j.jhep.2017.06.015
46. Gao Q, Zhang G, Zheng Y, et al. SLC27A5 deficiency activates NRF2/TXNRD1 pathway by increased lipid peroxidation in HCC. Cell Death Differ. 2020;27(3):1086–1104. doi:10.1038/s41418-019-0399-1
47. Suk FM, Wang YH, Chiu WC, et al. Secretory NPC2 Protein-Mediated Free Cholesterol Levels Were Correlated with the Sorafenib Response in Hepatocellular Carcinoma. Int J Mol Sci. 2021;22(16). doi:10.3390/ijms22168567
48. Yue X, Kong Y, Zhang Y, et al. SREBF2-STARD4 axis confers sorafenib resistance in hepatocellular carcinoma by regulating mitochondrial cholesterol homeostasis. Cancer Sci. 2023;114(2):477–489. doi:10.1111/cas.15449
49. Zheng C, Zhu Y, Liu Q, Luo T, Xu W. Maprotiline Suppresses Cholesterol Biosynthesis and Hepatocellular Carcinoma Progression Through Direct Targeting of CRABP1. Front Pharmacol. 2021;12:689767. doi:10.3389/fphar.2021.689767
50. Fu R, Jiang S, Li J, Chen H, Zhang X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med Oncol. 2020;37(4):24. doi:10.1007/s12032-020-01350-4
51. Fang T, Jiao Z, You Y, et al. Lenvatinib inhibited HCC cell migration and invasion through regulating the transcription and ubiquitination of UHRF1 and DNMT1. Biochem Pharmacol. 2023;210:115489. doi:10.1016/j.bcp.2023.115489
52. Guo Y, Xu J, Du Q, Yan Y, Geller DA. IRF2 regulates cellular survival and Lenvatinib-sensitivity of hepatocellular carcinoma (HCC) through regulating beta-catenin. Transl Oncol. 2021;14(6):101059. doi:10.1016/j.tranon.2021.101059
53. Zhao Z, Song J, Zhang D, Wu F, Tu J, Ji J. Oxysophocarpine suppresses FGFR1-overexpressed hepatocellular carcinoma growth and sensitizes the therapeutic effect of lenvatinib. Life Sci. 2021;264:118642. doi:10.1016/j.lfs.2020.118642
54. Zhao Z, Zhang D, Wu F, et al. Sophoridine suppresses lenvatinib-resistant hepatocellular carcinoma growth by inhibiting RAS/MEK/ERK axis via decreasing VEGFR2 expression. J Cell Mol Med. 2021;25(1):549–560. doi:10.1111/jcmm.16108
55. Sun D, Liu J, Wang Y, Dong J. Co-administration of MDR1 and BCRP or EGFR/PI3K inhibitors overcomes lenvatinib resistance in hepatocellular carcinoma. Front Oncol. 2022;12:944537. doi:10.3389/fonc.2022.944537
56. Li B, Qin Y, Yu X, Xu X, Yu W. Lipid raft involvement in signal transduction in cancer cell survival, cell death and metastasis. Cell Prolif. 2022;55(1):e13167. doi:10.1111/cpr.13167
57. Wu S, Fu L. Tyrosine kinase inhibitors enhanced the efficacy of conventional chemotherapeutic agent in multidrug resistant cancer cells. Mol Cancer. 2018;17(1):25. doi:10.1186/s12943-018-0775-3
58. Hu B, Zou T, Qin W, et al. Inhibition of EGFR Overcomes Acquired Lenvatinib Resistance Driven by STAT3-ABCB1 Signaling in Hepatocellular Carcinoma. Cancer Res. 2022;82(20):3845–3857. doi:10.1158/0008-5472.CAN-21-4140
59. Jin H, Shi Y, Lv Y, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature. 2021;595(7869):730–734. doi:10.1038/s41586-021-03741-7
60. Mok EHK, Leung CON, Zhou L, et al. Caspase-3-Induced Activation of SREBP2 Drives Drug Resistance via Promotion of Cholesterol Biosynthesis in Hepatocellular Carcinoma. Cancer Res. 2022;82(17):3102–3115. doi:10.1158/0008-5472.CAN-21-2934
61. Mendelson K, Evans T, Hla T. Sphingosine 1-phosphate signalling. Development. 2014;141(1):5–9. doi:10.1242/dev.094805
62. Wang X, Qiu Z, Dong W, et al. S1PR1 induces metabolic reprogramming of ceramide in vascular endothelial cells, affecting hepatocellular carcinoma angiogenesis and progression. Cell Death Dis. 2022;13(9):768. doi:10.1038/s41419-022-05210-z
63. Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24(26):4293–4300. doi:10.1200/JCO.2005.01.3441
64. Zhang Z, Zhou X, Shen H, Wang D, Wang Y. Phosphorylated ERK is a potential predictor of sensitivity to sorafenib when treating hepatocellular carcinoma: evidence from an in vitro study. BMC Med. 2009;7:41. doi:10.1186/1741-7015-7-41
65. Shi T, Iwama H, Fujita K, et al. Evaluating the Effect of Lenvatinib on Sorafenib-Resistant Hepatocellular Carcinoma Cells. Int J Mol Sci. 2021;22(23). doi:10.3390/ijms222313071
66. Calvisi DF, Wang C, Ho C, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140(3):1071–1083. doi:10.1053/j.gastro.2010.12.006
67. Jiang X, Huang L, Xing D. Photoactivation of Dok1/ERK/PPARgamma signaling axis inhibits excessive lipolysis in insulin-resistant adipocytes. Cell Signal. 2015;27(7):1265–1275. doi:10.1016/j.cellsig.2015.03.010
68. Srimuninnimit V, Sriuranpong V, Suwanvecho S. Efficacy and safety of sorafenib in combination with gemcitabine in patients with advanced hepatocellular carcinoma: a multicenter, open-label, single-arm phase II study. Asia Pac J Clin Oncol. 2014;10(3):255–260. doi:10.1111/ajco.12191
69. Ouyang T, Kan X, Zheng C. Immune Checkpoint Inhibitors for Advanced Hepatocellular Carcinoma: monotherapies and Combined Therapies. Front Oncol. 2022;12:898964. doi:10.3389/fonc.2022.898964
70. Angeles TS, Hudkins RL. Recent advances in targeting the fatty acid biosynthetic pathway using fatty acid synthase inhibitors. Expert Opin Drug Discov. 2016;11(12):1187–1199. doi:10.1080/17460441.2016.1245286
71. Shueng PW, Chan HW, Lin WC, Kuo DY, Chuang HY. Orlistat Resensitizes Sorafenib-Resistance in Hepatocellular Carcinoma Cells through Modulating Metabolism. Int J Mol Sci. 2022;23(12). doi:10.3390/ijms23126501
72. Jones SF, Infante JR. Molecular Pathways: fatty Acid Synthase. Clin Cancer Res. 2015;21(24):5434–5438. doi:10.1158/1078-0432.CCR-15-0126
73. Wang H, Zhou Y, Xu H, et al. Therapeutic efficacy of FASN inhibition in preclinical models of HCC. Hepatology. 2022;76(4):951–966. doi:10.1002/hep.32359
74. von Roemeling CA, Caulfield TR, Marlow L, et al. Accelerated bottom-up drug design platform enables the discovery of novel stearoyl-CoA desaturase 1 inhibitors for cancer therapy. Oncotarget. 2018;9(1):3–20. doi:10.18632/oncotarget.21545
75. Liu G, Kuang S, Cao R, Wang J, Peng Q, Sun C. Sorafenib kills liver cancer cells by disrupting SCD1-mediated synthesis of monounsaturated fatty acids via the ATP-AMPK-mTOR-SREBP1 signaling pathway. FASEB J. 2019;33(9):10089–10103. doi:10.1096/fj.201802619RR
76. Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. 2019;9(17):4893–4908. doi:10.7150/thno.36037
77. Kuda O, Pietka TA, Demianova Z, et al. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J Biol Chem. 2013;288(22):15547–15555. doi:10.1074/jbc.M113.473298
78. Mahalingam D, Harb W, Patnaik A, et al. 374 A first-in-human Phase 1/2 open label trial evaluating the safety, pharmacology, and preliminary efficacy of VT1021 in subjects with advanced solid tumors. Journal for ImmunoTherapy of Cancer. 2020;8(Suppl 3):A228–A228. doi:10.1136/jitc-2020-SITC2020.0374
79. Feng WW, Zuppe HT, Kurokawa M. The Role of CD36 in Cancer Progression and Its Value as a Therapeutic Target. Cells. 2023;12(12). doi:10.3390/cells12121605
80. Wei J, Leit S, Kuai J, et al. An allosteric mechanism for potent inhibition of human ATP-citrate lyase. Nature. 2019;568(7753):566–570. doi:10.1038/s41586-019-1094-6
81. Granchi C. ATP citrate lyase (ACLY) inhibitors: an anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur J Med Chem. 2018;157:1276–1291. doi:10.1016/j.ejmech.2018.09.001
82. Zheng Y, Zhou Q, Zhao C, Li J, Yu Z, Zhu Q. ATP citrate lyase inhibitor triggers endoplasmic reticulum stress to induce hepatocellular carcinoma cell apoptosis via p-eIF2alpha/ATF4/CHOP axis. J Cell Mol Med. 2021;25(3):1468–1479. doi:10.1111/jcmm.16235
83. Lai SW, Liao KF, Lai HC, Muo CH, Sung FC, Chen PC. Statin use and risk of hepatocellular carcinoma. Eur J Epidemiol. 2013;28(6):485–492. doi:10.1007/s10654-013-9806-y
84. Feng J, Dai W, Mao Y, et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1alpha/PPAR-gamma/PKM2-mediated glycolysis. J Exp Clin Cancer Res. 2020;39(1):24. doi:10.1186/s13046-020-1528-x
85. Jouve JL, Lecomte T, Bouche O, et al. Pravastatin combination with sorafenib does not improve survival in advanced hepatocellular carcinoma. J Hepatol. 2019;71(3):516–522. doi:10.1016/j.jhep.2019.04.021
86. Mullard A. Targeted protein degraders crowd into the clinic. Nat Rev Drug Discov. 2021;20(4):247–250. doi:10.1038/d41573-021-00052-4
87. Burslem GM, Crews CM. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell. 2020;181(1):102–114. doi:10.1016/j.cell.2019.11.031
88. Bayon-Cordero L, Alkorta I, Arana L. Application of Solid Lipid Nanoparticles to Improve the Efficiency of Anticancer Drugs. Nanomaterials (Basel). 2019;9(3). doi:10.3390/nano9030474
89. Shashkovskaya VS, Vetosheva PI, Shokhina AG, et al. Delivery of Lipid Nanoparticles with ROS Probes for Improved Visualization of Hepatocellular Carcinoma. Biomedicines. 2023;11(7). doi:10.3390/biomedicines11071783
90. Cichoz-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol. 2014;20(25):8082–8091. doi:10.3748/wjg.v20.i25.8082
91. Ren W, Jhala US, Du K. Proteomic analysis of protein palmitoylation in adipocytes. Adipocyte. 2013;2(1):17–28. doi:10.4161/adip.22117
92. Chamberlain LH, Shipston MJ. The physiology of protein S-acylation. Physiol Rev. 2015;95(2):341–376. doi:10.1152/physrev.00032.2014
93. Gottlieb CD, Linder ME. Structure and function of DHHC protein S-acyltransferases. Biochem Soc Trans. 2017;45(4):923–928. doi:10.1042/BST20160304
94. Lawrence DS, Zilfou JT, Smith CD. Structure-activity studies of cerulenin analogues as protein palmitoylation inhibitors. J Med Chem. 1999;42(24):4932–4941. doi:10.1021/jm980591s
95. Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. Protein palmitoylation by a family of DHHC protein S-acyltransferases. J Lipid Res. 2006;47(6):1118–1127. doi:10.1194/jlr.R600007-JLR200
96. Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8(1):74–84. doi:10.1038/nrm2084
97. Wang J, Hao JW, Wang X, et al. DHHC4 and DHHC5 Facilitate Fatty Acid Uptake by Palmitoylating and Targeting CD36 to the Plasma Membrane. Cell Rep. 2019;26(1):209–221 e5. doi:10.1016/j.celrep.2018.12.022
98. Zhou B, Wang Y, Zhang L, et al. The palmitoylation of AEG-1 dynamically modulates the progression of hepatocellular carcinoma. Theranostics. 2022;12(16):6898–6914. doi:10.7150/thno.78377
99. Grzesk G, Dorota B, Wolowiec Ł, et al. Safety of PCSK9 inhibitors. Biomed Pharmacother. 2022;156:113957. doi:10.1016/j.biopha.2022.113957
100. Sun Y, Zhang H, Meng J, et al. S-palmitoylation of PCSK9 induces sorafenib resistance in liver cancer by activating the PI3K/AKT pathway. Cell Rep. 2022;40(7):111194. doi:10.1016/j.celrep.2022.111194
101. Davda D, El Azzouny MA, Tom CTMB, et al. Profiling targets of the irreversible palmitoylation inhibitor 2-bromopalmitate. ACS Chem Biol. 2013;8(9):1912–1917. doi:10.1021/cb400380s
102. Lemonidis K, Salaun C, Kouskou M, Diez-Ardanuy C, Chamberlain LH, Greaves J. Substrate selectivity in the zDHHC family of S-acyltransferases. Biochem Soc Trans. 2017;45(3):751–758. doi:10.1042/BST20160309
103. Stolk DA, Horrevorts SK, Schetters STT, et al. Palmitoylated antigens for the induction of anti-tumor CD8(+) T cells and enhanced tumor recognition. Mol Ther Oncolytics. 2021;21:315–328. doi:10.1016/j.omto.2021.04.009
104. Li J, Cao F, Yin HL, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. doi:10.1038/s41419-020-2298-2
105. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–2081. doi:10.1080/15548627.2020.1810918
106. Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508(4):997–1003. doi:10.1016/j.bbrc.2018.12.039
107. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
108. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
109. Magtanong L, Ko PJ, To M, et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol. 2019;26(3):420–432 e9. doi:10.1016/j.chembiol.2018.11.016
110. Louandre C, Ezzoukhry Z, Godin C, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int, J, Cancer. 2013;133(7):1732–1742. doi:10.1002/ijc.28159
111. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12(8):599–620. doi:10.1007/s13238-020-00789-5
112. Li Y, Yang W, Zheng Y, et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J Exp Clin Cancer Res. 2023;42(1):6. doi:10.1186/s13046-022-02567-z
113. Iseda N, Itoh S, Toshida K, et al. Ferroptosis is induced by lenvatinib through fibroblast growth factor receptor-4 inhibition in hepatocellular carcinoma. Cancer Sci. 2022;113(7):2272–2287. doi:10.1111/cas.15378
114. Sun X, Niu X, Chen R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488–500. doi:10.1002/hep.28574
115. Lai Y, Lu N, Luo S, Wang H, Zhang P. A Photoactivated Sorafenib-Ruthenium(II) Prodrug for Resistant Hepatocellular Carcinoma Therapy through Ferroptosis and Purine Metabolism Disruption. J Med Chem. 2022;65(19):13041–13051. doi:10.1021/acs.jmedchem.2c00880
116. Ganan-Gomez I, Wei Y, Yang H, Boyano-Adanez MC, Garcia-Manero G. Oncogenic functions of the transcription factor Nrf2. Free Radic Biol Med. 2013;65:750–764. doi:10.1016/j.freeradbiomed.2013.06.041
117. Zheng A, Chevalier N, Calderoni M, et al. CRISPR/Cas9 genome-wide screening identifies KEAP1 as a sorafenib, lenvatinib, and regorafenib sensitivity gene in hepatocellular carcinoma. Oncotarget. 2019;10(66):7058–7070. doi:10.18632/oncotarget.27361
118. Lai HY, Tsai HH, Yen CJ, et al. Metformin Resensitizes Sorafenib-Resistant HCC Cells Through AMPK-Dependent Autophagy Activation. Front Cell Dev Biol. 2020;8:596655. doi:10.3389/fcell.2020.596655
119. Feng J, Lu H, Ma W, et al. Genome-wide CRISPR screen identifies synthetic lethality between DOCK1 inhibition and metformin in liver cancer. Protein Cell. 2022;13(11):825–841. doi:10.1007/s13238-022-00906-6
120. Cheng Y, Zhan P, Lu J, et al. Metformin synergistically enhances the antitumour activity of Lenvatinib in hepatocellular carcinoma by altering AKT-FOXO3 signalling pathway. Liver Int. 2023;43(7):1577–1592. doi:10.1111/liv.15611
121. Shen Z, Zhou H, Li A, et al. Metformin inhibits hepatocellular carcinoma development by inducing apoptosis and pyroptosis through regulating FOXO3. Aging. 2021;13(18):22120–22133. doi:10.18632/aging.203464
122. Misra JR, Irvine KD. The Hippo Signaling Network and Its Biological Functions. Annu Rev Genet. 2018;52:65–87. doi:10.1146/annurev-genet-120417-031621
123. Han S, Lim JY, Cho K, et al. Anti-Cancer Effects of YAP Inhibitor (CA3) in Combination with Sorafenib against Hepatocellular Carcinoma (HCC) in Patient-Derived Multicellular Tumor Spheroid Models (MCTS). Cancers (Basel). 2022;14(11). doi:10.3390/cancers14112733
124. Chen L, Yang Z, Cao Y, et al. Pan-cancer analysis and single-cell analysis revealed the role of ABCC5 transporter in hepatocellular carcinoma. Channels. 2021;15(1):541–554. doi:10.1080/19336950.2021.1968592
125. Huang W, Chen K, Lu Y, et al. ABCC5 facilitates the acquired resistance of sorafenib through the inhibition of SLC7A11-induced ferroptosis in hepatocellular carcinoma. Neoplasia. 2021;23(12):1227–1239. doi:10.1016/j.neo.2021.11.002
126. Yagoda N, von Rechenberg M, Zaganjor E, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–868. doi:10.1038/nature05859
127. Jing W, Shuo L, Yingru X, et al. Artesunate promotes sensitivity to sorafenib in hepatocellular carcinoma. Biochem Biophys Res Commun. 2019;519(1):41–45. doi:10.1016/j.bbrc.2019.08.115
128. Tang B, Zhu J, Li J, et al. The ferroptosis and iron-metabolism signature robustly predicts clinical diagnosis, prognosis and immune microenvironment for hepatocellular carcinoma. Cell Commun Signal. 2020;18(1):174. doi:10.1186/s12964-020-00663-1
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Lenvatinib Plus PD-1 Inhibitors versus Regorafenib in Patients with Advanced Hepatocellular Carcinoma After the Failure of Sorafenib: A Retrospective Study
Xu Y, Fu S, Liu K, Mao Y, Wu J
Therapeutics and Clinical Risk Management 2023, 19:853-863
Published Date: 24 October 2023
Mechanisms of Sorafenib Resistance in HCC Culture Relate to the Impaired Membrane Expression of Organic Cation Transporter 1 (OCT1)
Chava S, Ekmen N, Ferraris P, Aydin Y, Moroz K, Wu T, Thung SN, Dash S
Journal of Hepatocellular Carcinoma 2024, 11:839-855
Published Date: 9 May 2024
Efficacy of Lenvatinib Combined with PD-1 Inhibitor versus Sorafenib and PD-1 Inhibitor with or Without TACE for Hepatocellular Carcinoma with Extrahepatic Metastasis
Duan WB, Wang XH, Zhang GC, He Z, Li SQ, Zhou J
ImmunoTargets and Therapy 2024, 13:247-258
Published Date: 16 May 2024
Real-World Comparison of Lenvatinib and Sorafenib as First-Line Treatments for Hepatocellular Carcinoma: A Multicenter Study
Kang M, Cha WC, Sinn DH, Jeong WK, Kim DY, Lee MJ, Lim S, Kim D, Kim KP, Ryoo BY, Choi WM, Kim KM, Kim KH, Lee D, Choi EJ, Jung C, Kim J, Hong JY
Journal of Hepatocellular Carcinoma 2025, 12:2611-2623
Published Date: 25 November 2025