")
Back to Journals » OncoTargets and Therapy » Volume 9
Advances in targeted and immunobased therapies for colorectal cancer in the genomic era
Authors Seow H, Yip WK, Fifis T
Received 25 August 2015
Accepted for publication 22 January 2016
Published 31 March 2016 Volume 2016:9 Pages 1899—1920
DOI https://doi.org/10.2147/OTT.S95101
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Heng Fong Seow,1 Wai Kien Yip,1 Theodora Fifis2
1Immunology Unit, Department of Pathology, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, Serdang, Malaysia; 2Department of Surgery, University of Melbourne, Melbourne, Australia
Abstract: Targeted therapies require information on specific defective signaling pathways or mutations. Advances in genomic technologies and cell biology have led to identification of new therapeutic targets associated with signal-transduction pathways. Survival times of patients with colorectal cancer (CRC) can be extended with combinations of conventional cytotoxic agents and targeted therapies. Targeting EGFR- and VEGFR-signaling systems has been the major focus for treatment of metastatic CRC. However, there are still limitations in their clinical application, and new and better drug combinations are needed. This review provides information on EGFR and VEGF inhibitors, new therapeutic agents in the pipeline targeting EGFR and VEGFR pathways, and those targeting other signal-transduction pathways, such as MET, IGF1R, MEK, PI3K, Wnt, Notch, Hedgehog, and death-receptor signaling pathways for treatment of metastatic CRC. Additionally, multitargeted approaches in combination therapies targeting negative-feedback loops, compensatory networks, and cross talk between pathways are highlighted. Then, immunobased strategies to enhance antitumor immunity using specific monoclonal antibodies, such as the immune-checkpoint inhibitors anti-CTLA4 and anti-PD1, as well as the challenges that need to be overcome for increased efficacy of targeted therapies, including drug resistance, predictive markers of response, tumor subtypes, and cancer stem cells, are covered. The review concludes with a brief insight into the applications of next-generation sequencing, expression profiling for tumor subtyping, and the exciting progress made in in silico predictive analysis in the development of a prescription strategy for cancer therapy.
Keywords: targeted therapies, colorectal cancer, signaling pathways, immune cells, personalized medicine
Introduction
Colorectal cancer (CRC) is one of the most common cancers in the world and the second-most common cause of cancer-related deaths worldwide.1 Approximately 1.4 million cases and 694,000 deaths were reported for the year 2012.2 The increased incidence of CRC in developing nations, eg, countries in Asia, has been associated with smoking, obesity, and changes in lifestyle and diet.3
Over the past 10 years, survival in metastatic CRC has improved significantly; this is due to major advances in chemotherapy and targeted drugs. A number of targeted biologic therapies have been approved by the US Food and Drug Administration (FDA), and they are summarized in Table 1. A systematic review on anti-EGFR and anti-VEGF targeted therapies in combination with chemotherapy was recently published.4 The combination of fluorouracil and leucovorin (FL) resulted in median overall survival durations of approximately 12 months in a Phase III trial first reported in 1994.5,6 Survival was further improved by another 3 months with irinotecan treatment in combination with FL,7,8 and 16–20 months with oxaliplatin treatment in combination with FL.9–11
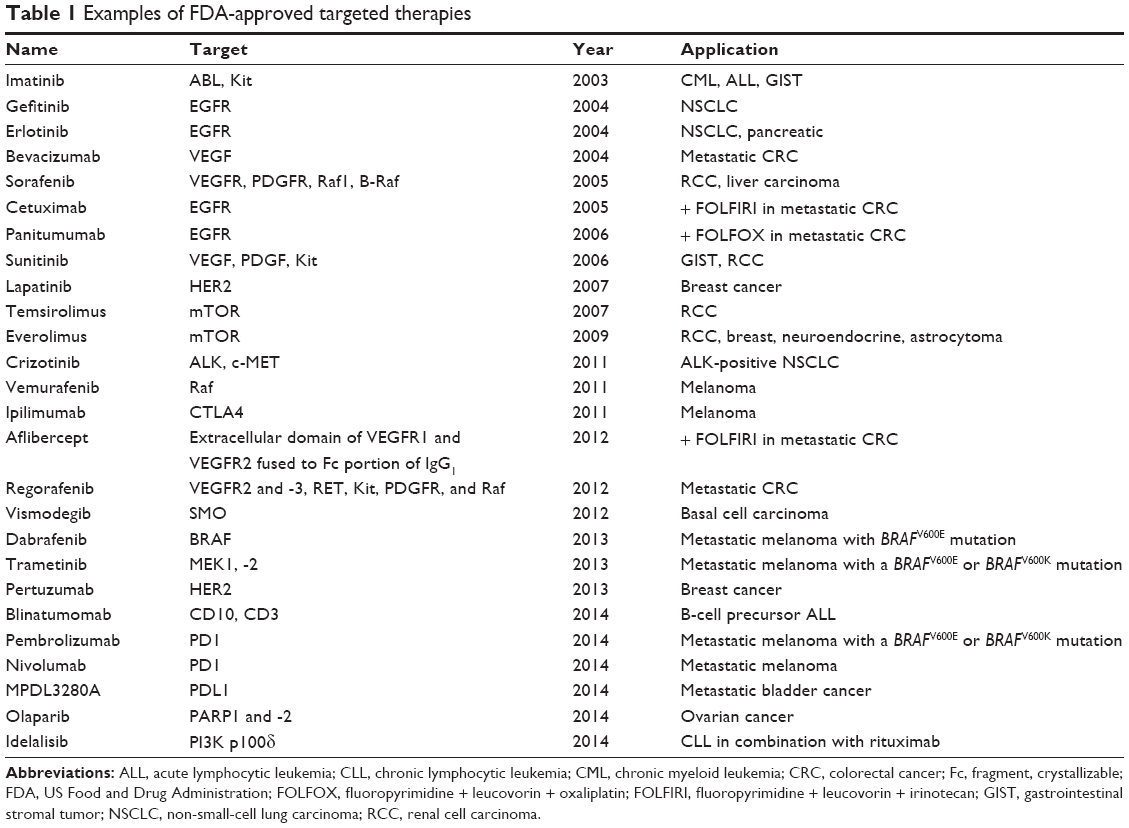 | Table 1 Examples of FDA-approved targeted therapies |
The major step forward in clinical management of metastatic CRC was the combination of FL, irinotecan, and oxaliplatin, which increased the survival rate from 12 to 20 months.12 In addition to this, the use of these drugs to reduce tumor mass before tumor resection enabled a significant proportion of patients who previously were only treated with chemotherapy to undergo resection of their metastases. The introduction of biologic agents, such as inhibitors of VEGF and EGFR, further increased survival rates to more than 2 years.13,14
The current standard of care for unresectable metastatic CRC combines standard cytotoxic chemotherapy with biologic agents. The biologic agents available for metastatic CRC are categorized into three groups, as shown in Table 2. They are: 1) monoclonal antibodies (mAbs; cetuximab and panitumumab) against EGFR on the surface of tumor cells; 2) antiangiogenic inhibitors targeting tumor vascularization and directed mainly against VEGF or its receptors (the mAbs bevacizumab and aflibercept); and 3) regorafenib, an oral small-molecule inhibitor of intracellular kinases involved in various signaling cascades, such as VEGFR2 and -3, as well as RET, Kit, PDGFR, and Raf kinases.
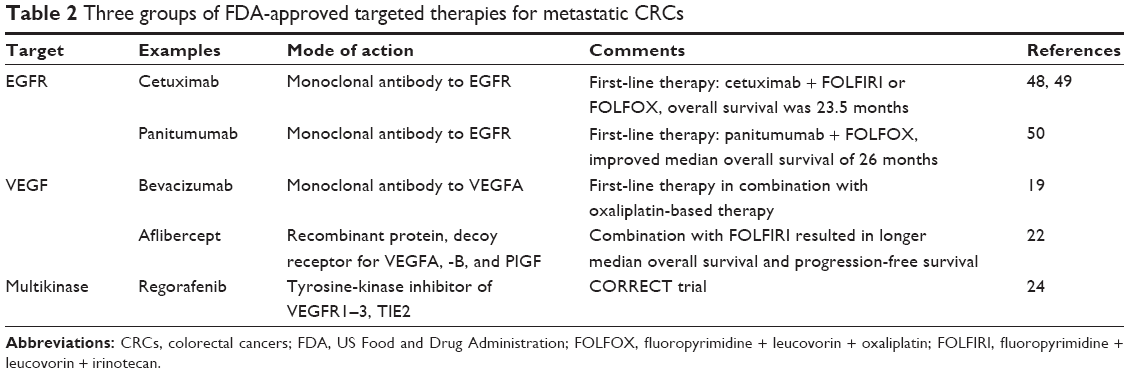 | Table 2 Three groups of FDA-approved targeted therapies for metastatic CRCs |
Many excellent reviews have been written on the application of biological targeted therapies for CRC.15–17 A brief overview is included here to aid readers to understand the rationale behind these applications.
Current status with antiangiogenic agents and EGFR inhibitors in CRC
Targeting the VEGF system
Bevacizumab is a humanized mAb that binds to VEGFA, preventing it from binding to its target receptors. Bevacizumab has been approved by the FDA for use in combination with the fluoropyrimidine analog leucovorin and topoisomerase I inhibitor irinotecan (FOLFIRI) for the treatment of patients with metastatic CRC who have progressed following an oxaliplatin-containing regimen.18 Major limitations of anti-VEGF therapy include the need for expensive maintenance therapy and the onset of secondary resistance, as highlighted in several randomized trials.19–21
Besides bevacizumab, some progress has been made with new antiangiogenic agents, such as aflibercept and regorafenib. Several other antiangiogenic agents are in clinical development, including ramucirumab, famitinib, vatalanib, SU11248, and vascular targeting agents (VTAs).
Aflibercept is a recombinant fusion protein consisting of the extracellular domain of human VEGFR1 and VEGFR2 fused to the Fc (fragment, crystallizable) portion of human IgG1. The VELOUR trial showed that regardless of prior bevacizumab exposure, treatment with FOLFIRI and aflibercept resulted in longer median overall survival and progression-free survival compared to FOLFIRI with placebo. These patients had previously progressed within 6 months of receiving oxaliplatin-containing chemotherapy (with or without bevacizumab).22
Regorafenib is a small-molecule multikinase inhibitor. It blocks the ATP-binding site of tyrosine kinases involved in various signaling cascades, such as VEGFR2 and -3, RET, Kit, PDGFR, and Raf kinases, thus preventing downstream signaling.23 In the CORRECT trial, the median overall survival for patients with metastatic CRC refractory to standard chemotherapy in the regorafenib-treated arm was 6.4 months compared with 5 months in the placebo-treated arm,24 and no benefit was observed in 50% of patients. Since there are no known predictive biomarkers for regorafenib, it is unclear why this group of patients did not benefit from this novel multikinase-inhibitor treatment.
Promising results were also demonstrated with ramucirumab, a VEGFR2 inhibitor combined with modified FOLFOX (fluoropyrimidine + leucovorin + oxaliplatin)-6 (mFOLFOX6) as a first-line therapy for metastatic CRC.25 Results from the Phase III RAISE trial reported at the 2015 American Society of Clinical Oncology Gastrointestinal Symposium showed second-line treatment with the VEGFR2 inhibitor ramucirumab combined with standard FOLFIRI extended survival by 1.6 months versus FOLFIRI alone in patients with metastatic CRC.26
Famitinib is another small-molecule tyrosine-kinase inhibitor that primarily targets VEGFR2, c-Kit, and PDGFR. Its safety and efficacy in CRC treatment was tested. While it resulted in improved progression-free survival by 1.3 months (13.1% vs 5.5%), drug-related adverse events led to its discontinuation.27
Other oral small multitargeted receptor tyrosine-kinase inhibitors include sunitinib, sorafenib, vatalinib (PTK787/ZK222584), axitinib, cediranib, and brivanib.28–33 Vatalanib (PTK787/ZK222584) inhibits all three VEGF receptors, as well as PDGFRβ, c-Kit, and CSF1R. Two Phase II trials in metastatic CRC are ongoing. SU11248 is being evaluated in a number of clinical trials in metastatic breast, renal, and CRCs. Objective responses were observed with SU11248-treated patients with metastatic gastrointestinal stroma tumors resistant to imatinib. This supported further evaluation of this compound in Phase III trials.
Another area of investigation is VTAs, which target the already formed vasculature of tumors rather than inhibiting the growth of new blood vessels.34 Antitumor activities of VTAs in preclinical models and clinical trials have been comprehensively reviewed.35
The first class of VTAs consists of flavone-8-acetic acid and its derivatives, with 5,6-dimethylxanthenone-4 acetic acid being the most promising. In preclinical studies, this acid demonstrated antitumor activity in a wide variety of murine tumors, induced extensive tumor necrosis in patient-derived xenografts, and blocked angiogenesis.36 However, Phase III trials with non-small-cell lung cancer patients produced negative results.37
The second class of small-molecule VTAs are tubulin-binding agents consisting of combretastatins and their analogs.35 Combretastatin A4 disodium phosphate (CA4DP) has shown potent vascular disruption and antitumor effects in a wide variety of preclinical tumor models.38 However, a robust proangiogenic response was observed once treatment stopped. Additionally, tumor pain was reported in three Phase I clinical trials of CA4DP involving a total of 96 patients with advanced cancers.39–41 In one of these clinical trials, a patient with anaplastic thyroid cancer had a complete response.39 Several other tubulin-binding agents are also in Phase I clinical development, including AVE8062A, ZD6126, and ABT751.35
EGFR-targeted therapies
EGFR, also known as ErbB1 or HER1, is a member of the ErbB transmembrane tyrosine-kinase receptor family. Other members of the family include HER2 and HER3. Ligands that bind EGFR include EGF, amphiregulin, and epiregulin. As shown in Figure 1, ligand binding to the extracellular domain of EGFR leads to activation of the tyrosine-kinase domain in the cytoplasm, activating several intracellular signal-transduction pathways, including Ras/Raf/MEK/ERK, which is mainly involved in cell proliferation, PI3K/Akt, which is involved in cell survival and tumor invasion, JAK/STAT, and phospholipase C.42 The last two are downstream effectors in a wide range of signal-transduction pathways. Blocking of EGFR with an antibody to EGFR leads to inhibition of these downstream pathways, resulting in reduced cell proliferation, decreased cell survival, and inhibition of invasion/metastasis.43,44 However, point mutations in KRAS result in constitutive activation of the downstream pathways, such as Raf/MEK/ERK and PI3K/Akt.45
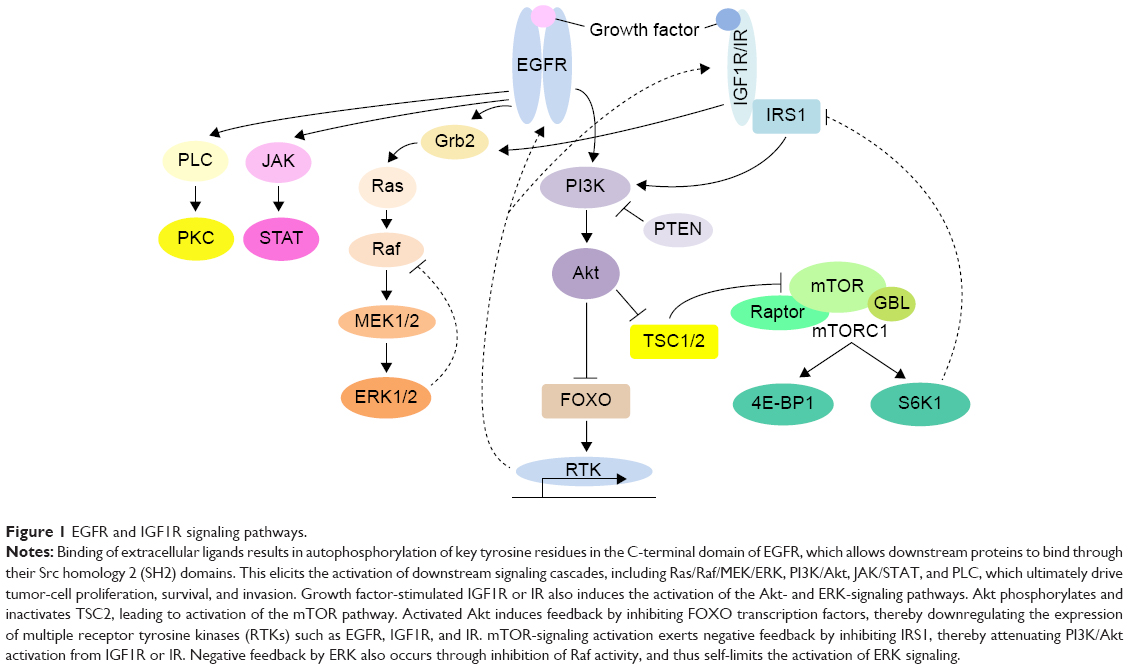 | Figure 1 EGFR and IGF1R signaling pathways. |
Cetuximab (a recombinant chimeric IgG1 anti-EGFR mAb) treatment provides survival benefit in metastatic CRCs that harbor wild-type KRAS,46,47 and was first approved by the FDA in 2005 for use with FOLFIRI chemotherapy in metastatic CRC. Panitumumab (a fully human IgG2 antibody) targeting EGFR was approved for use with FOLFOX in 2006 (Table 1).
A retrospective analysis of the Phase II OPUS and Phase III CRYSTAL clinical trials showed that cetuximab combined with a first-line chemotherapy with either FOLFOX4 or FOLFIRI significantly improved response rate, progression-free survival, and overall survival in patients with KRAS wild-type tumors.48 The CRYSTAL study reported overall survival of 23.5 months in patients treated with FOLFIRI and cetuximab compared to 20 months with FOLFIRI alone in previously untreated KRAS wild-type metastatic CRC.49 In the PRIME study, first-line metastatic CRC patients treated with FOLFOX and panitumumab had a 4.2-month improvement in overall survival compared to FOLFOX alone.50 Cetuximab and panitumumab are currently used in clinical practice in combination with standard combination-chemotherapy regimens or as single agents.
EGFR mutations are rare in CRC, and they are not routinely analyzed in clinical practice. One important finding is that patients with mutation at S492R within the extracellular domain are resistant to cetuximab, but are sensitive to panitumumab.51 EGFR expression is not a useful marker, since its immunohistochemical expression only weakly correlates with treatment response.52–54 In addition, there is no correlation between EGFR-protein expression and EGFR-gene amplification.55 However amplification of EGFR56 or loss of PTEN expression57 may indicate response to cetuximab. It has also been shown that cetuximab provides clinical benefit in patients with high expression of the EGFR ligands amphiregulin and epiregulin.58,59 In contrast, poor prognosis correlated with high expression of TGFα, another EGFR ligand.60 Expression analysis of EGFR and EGFR ligands is not routinely implemented in clinical practice.
KRAS mutations, which are seen in 35%–40% of CRCs, have emerged as the most important predictive biomarkers in selecting patients who will benefit from cetuximab.46,47,61,62 Mutations in KRAS codons 12 or 13 have been reported in 40% of metastatic CRCs, and are predictive for lack of response to treatment with antibodies to EGFR.63 Mutations in BRAF, NRAS, and PI3K are also associated with poor response to cetuximab.64 Recent data show patients with mutations in codons 61 and 146 of KRAS and codons 12, 13, and 61 of NRAS do not benefit from anti-EGFR treatment.63 Therefore, it has been recommended that testing be expanded to include these mutations.65
Resistance mechanisms to cetuximab
As alluded to earlier, one of the major problems in clinical application of anti-EGFR inhibitors is acquired drug resistance. A subset of metastatic CRCs responds to the anti-EGFR drugs cetuximab and panitumumab, but resistance develops within several months of therapy initiation.43 The factors contributing to this acquired resistance are summarized in Table 3.
 | Table 3 Possible reasons for acquired resistance to anti-EGFR inhibitors and strategies |
Mutations in KRAS can emerge during treatment with cetuximab in patients with wild-type KRAS.66,67 Emergence of the EGFR ectodomain mutant S492R has also been reported.68 Other studies have demonstrated that oncogenic activation of effectors downstream of EGFR, such as mutant BRAF,69,70 PIK3CA,71,72 PTEN inactivation,72,73 and PTEN loss,73 are associated with cetuximab resistance. In addition, approximately 25% of CRC patients with wild-type KRAS, BRAF, PIK3CA, and PTEN do not respond to cetuximab, and the resistance mechanism is still unknown.
Other mechanisms that lead to cetuximab resistance include amplification of MET,74 overexpression of IGF1R,75 overexpression of EGFR ligands and receptors, such as ErbB276 and amphiregulin,77 modulation of EGFR by Src-family kinases, transactivation of alternative pathways that bypass the EGFR pathway, such as MET and IGFR, ubiquitination, expression of EGFR variant III, and induction of EGFR translocation.78,79
New-generation anti-EGFR therapies
New developments in anti-EGFR therapies are summarized in Table 4. Sym004 is a mixture of two mAbs that bind to nonoverlapping epitopes of the EGFR extracellular domain. The treatment showed significant clinical activity in KRAS wild-type CRC patients who had acquired resistance to anti-EGFR therapies.80 A Phase II trial comparing Sym004 to 5-fluorouracil, capecitabine, or best-support care is currently in progress (NCT02083653) in metastatic CRC patients who are resistant to anti-EGFR therapies.
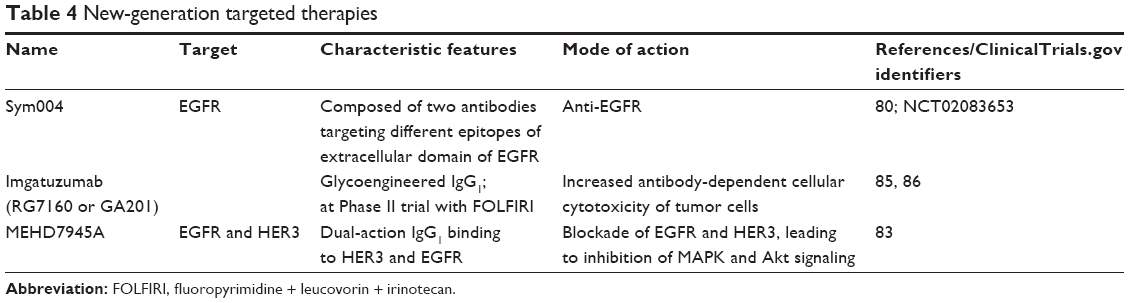 | Table 4 New-generation targeted therapies |
Dual-targeting EGFR- and HER3-mediated signaling by MEHD7945A, an mAb, was found to be effective in blocking MAPK- and Akt-signaling pathways and to elicit antibody-dependent cellular cytotoxicity in colon xenograft models.81 It can also overcome acquired resistance to EGFR inhibition by blocking PI3K/Akt- and ERK-signaling pathways.82 The treatments demonstrated an encouraging safety profile and antitumor activity in a Phase I clinical trial in KRAS wild-type CRC patients.83
GA201 (RG7160), a humanized anti-EGFR mAb with a glycoengineered Fc region for enhanced binding to FcγRIIIA has been shown to be effective in the killing of KRAS wild-type and mutant tumor-cell lines and mouse xenograft tumors.84 Preclinical studies suggest that GA201 is more effective than cetuximab, and it has shown promising results in two clinical trials with KRAS-mutant metastatic CRC patients.85,86
HER2 amplification has been found in 2%–3% of metastatic CRC cases, and is associated with up to 36% of cetuximab resistance in KRAS wild-type patients. Investigations with patient-derived xenografts have identified HER2 as a therapeutic target in cetuximab-resistant CRCs.87 An ongoing Phase II trial (NCT01960023) with cetuximab in combination with neratinib, an HER2 tyrosine-kinase inhibitor in metastatic CRC, is in progress.
New opportunities by targeting other signal-transduction pathways
Examples of clinical trials with a number of targeted agents are summarized in Table 5 and further elaborated in the following sections.
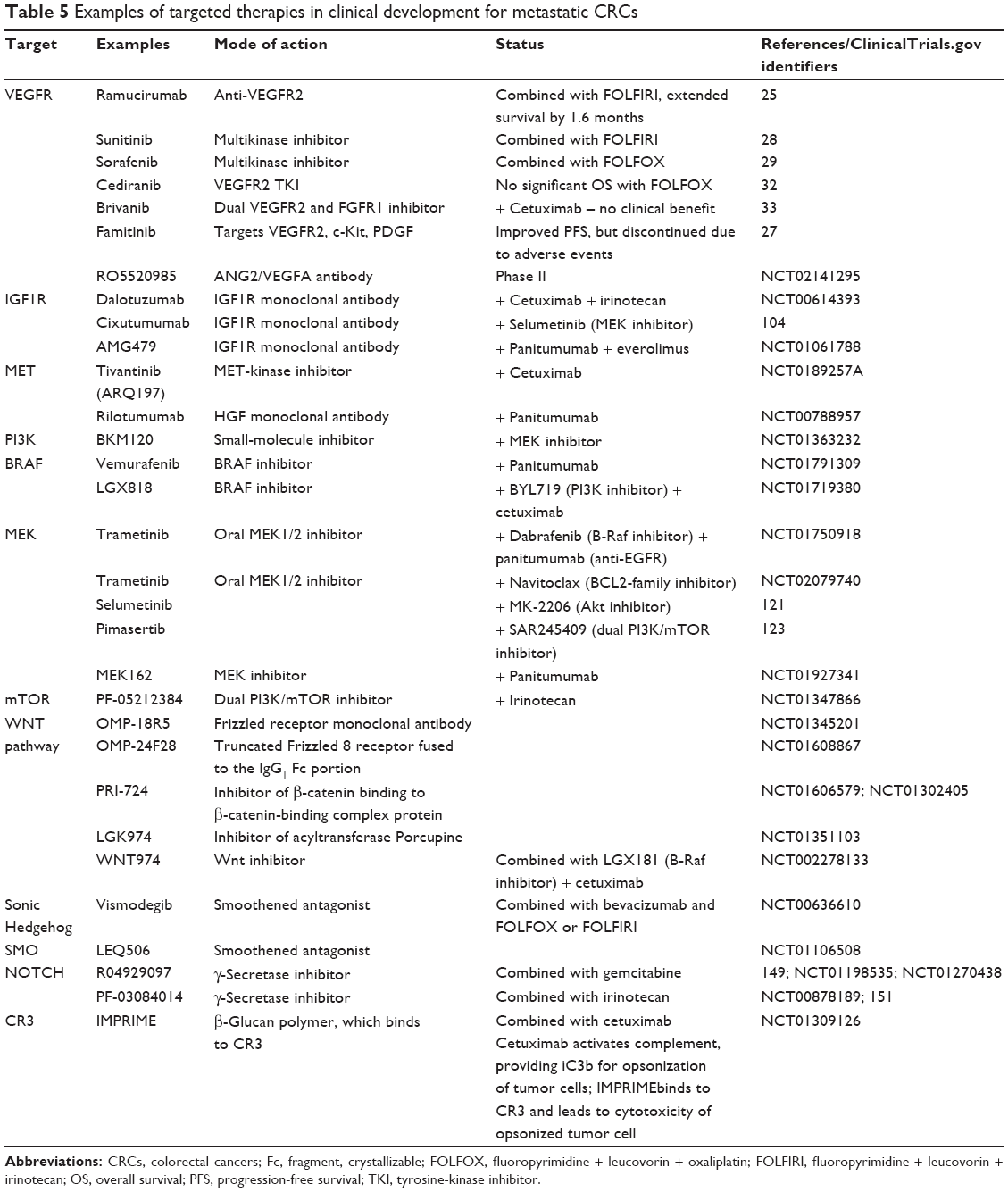 | Table 5 Examples of targeted therapies in clinical development for metastatic CRCs |
Targeting BRAF mutants
Mutations in genes related to EGFR signaling, including KRAS, BRAF, and NRAS, account for about 60% of cases that develop resistance. B-Raf is downstream of Ras and mutations in BRAF have been found in 5%–10% of advanced CRCs and are associated with a very poor prognosis.
Clinical results with vemurafenib (a B-Raf inhibitor) as a single agent in CRC have been disappointing, although the inhibitor is very effective for melanoma patients harboring this mutation.88 Cell-culture studies have shown that colon cancer cells treated with an inhibitor of BRAFV600E mutation were less sensitive to vemurafenib compared to melanoma cell lines with the same mutation.89,90 Studies in melanoma patients treated with vemurafenib suggest that the inhibition of ERK is necessary to effectively inhibit tumor growth.91
Analysis of the phosphorylation status of ERK, EGFR, and Akt in vemurafenib-treated colon cancer cell lines indicated that EGFR and ERK signaling were switched on and that this could be counteracted by cotargeting EGFR.92 These studies concurred with another study that showed that vemurafenib in CRC caused a rapid feedback activation of EGFR, which supported continued tumor growth.89 Therefore, the use of B-Raf- and EGFR-inhibitor combinations appears to be a rational strategy. A pilot study using dual B-Raf and EGFR inhibition in BRAF-mutated CRC showed a response rate of 13%, but in a subset of patients the response was not durable, lasting less than 1 year.93
Another Raf inhibitor, encorafenib, in combination with cetuximab showed a partial response in two of 18 patients.94 In contrast, improvements were seen with the combination of another B-Raf inhibitor, dabrafenib, with trametinib (MEK inhibitor) and panitumumab, where seven of eight patients achieved stable disease.95 Further investigations are warranted to clarify the need for ERK inhibition and the role of compensating pathways in these tumors.
Targeting the MET pathway
MET is the tyrosine-kinase receptor for HGF, and is overexpressed in metastatic colon cancer.96–98 CRC resistance to anti-EGFR therapies can be driven by MET gene amplification in tumor samples. MET amplification was detected in drug-resistant tumor xenografts,74,99 and was also detected in patient blood samples collected at regular intervals during treatment, thus providing a convenient method for monitoring and predicting resistance. Overexpression of the MET gene and MET protein were detected in resistant CRC tumors by fluorescent in situ hybridization and immunohistochemistry, respectively. In addition, antitumor activity was demonstrated using a combination of the clinically approved MET inhibitor crizotinib, and anti-EGFR demonstrated efficacy in two patient-derived xenografts, thus providing proof of concept that combination of MET- and EGFR-inhibitor therapies offers novel opportunities to overcome anti-EGFR resistance. Other studies have also demonstrated that activation of the HGF-dependent MET-signaling pathway contributes to cetuximab resistance in colon cancer.100 In addition, constitutive MET activation was shown to be due to the formation of an MET/SRC/EGFR complex.100 This provides a rationale for combinatorial inhibition of EGFR and MET or EGFR and SRC in therapy targeting colon cancer.
A single-arm Phase II study of tivantinib (ARQ 197; MET-kinase inhibitor) combined with cetuximab is in progress in locally advanced or metastatic CRC patients with wild-type KRAS, MET-amplified and EGFR inhibitor-resistant tumors (NCT01892527). Another clinical trial (NCT00788957) in progress is testing the combination of an anti-HGF mAb (MG 102; rilotumumab) with panitumumab.
MET has been found to be upregulated in response to VEGF-pathway inhibition, implicating it in antiangiogenic therapy resistance. In a preclinical CRC tumor xenograft model, potent antitumor activity was demonstrated in 80% of tumors with cabozantinib, a dual c-MET and VEGFR2 inhibitor.101 The antitumor activity was attributed to the inhibition of angiogenesis, inhibition of Akt, and downregulation of genes involved in the PI3K pathway. A clinical trial of cabozantinib in refractory metastatic CRC is currently being activated.
Insulin-like growth factor-signaling pathway
The IGF-signaling pathway plays a key role in cell growth and metabolism. Overexpression of IGF1 and IGF1R has been found in many cancers, including CRC.102 Binding of IGF1 to its receptor induces the activation of IRS1 and -2, which then activate several intracellular signaling pathways, including the PI3K/AKT/mTOR and Ras/Raf/MAPK pathways. This leads to cell-cycle progression, cell proliferation, and cell survival. Therefore, blockade of this pathway would reduce cell proliferation and growth.
Indeed, it has been demonstrated that a combination of OSI-906 (IGF1R inhibitor) with selumetinib (MEK1/2 inhibitor) exerted synergistic antiproliferative activity in 13 colon cancer cell lines and an in vivo xenograft model.103 A Phase I clinical trial targeting these two pathways with selumetinib (MEK1/2 inhibitor) and cixutumumab (IGF1R mAb) in 30 patients with a variety of tumor types, including 13 patients with gastrointestinal tumors (colorectal, pancreatic, and biliary) and four patients with thyroid cancers, is in progress.104 The combination appears to be well tolerated in these patients.
Activation of IGF1R has been implicated in resistance of lung cancer cells and colon cancer cells to erlotinib, an EGFR tyrosine-kinase inhibitor,103 further supporting the need to block the IGF1R-signaling pathway when administering anti-EGFR-targeted therapies. A clinical trial with a combination of AMG479 (IGF1R mAb) and panitumumab (anti-EGFR) is in progress (NCT01061788). A Phase II clinical trial with dalotuzumab (MK0646; anti-IGF1R) and cetuximab in combination with chemotherapy (irinotecan) is also ongoing (NCT00614393).
Targeting MEK- and PI3K-signaling pathways
The Ras/Raf/MEK/ERK and PI3K/Akt/mTOR signaling pathways are among the best-characterized kinase cascades in cancer cell biology.105,106 Class I PI3K consists of a regulatory p85 subunit and a catalytic p110 α-subunit. The p110 catalytic subunit occurs in four isoforms, designated as p110α, -β, -γ and -δ. Three isoforms consist of the regulatory p85 subunit and p110α/β/δ subunit, while the fourth isoform consists of p110γ and a regulatory subunit – p101. A phase I clinical trial with an oral panclass IPI3K inhibitor, pictilisib, has shown that it is safe and tolerable, so further investigation is warranted.107
Idelalisib is a first-in-class, oral PI3Kδ-specific inhibitor approved for the treatment of chronic lymphocytic leukemia in combination with rituximab. In addition to idelalisib, two other oral PI3Kδ-specific inhibitors in development are TGR-1202 and AMG-319 (NCT01767766, NCT01300026).
The activation of these pathways is triggered by growth factors or activating mutations of oncogenic kinases, such as K-Ras, N-Ras, B-Raf, and PI3K, resulting in enhanced cell survival, proliferation, and motility. Mutations in KRAS, NRAS, or BRAF (all upstream of MEK complexes) have been found in approximately 40%, 1%–3% and 5%–15% of CRC tumors, respectively.108 Mutations of the PI3K catalytic p110α subunit have been found in 10%–20% of CRC tumors. Activating mutations occur in two hotspots located in exon 9 (E542K, E545K) and exon 20 (H1047R) in approximately 15% of CRCs.109 Only exon 20 mutations (about 25% of total), but not exon 9 mutations, are associated with clinical resistance to anti-EGFR mAbs.64,110 A third of CRC tumors have co-occurrence of both KRAS and PI3K mutations.108 Targeting PI3K alone has not been found to be effective for treating solid cancers.111,112 This may be due to negative-feedback loops, compensatory networks, and cross talk between pathways.
Activating PIK3CA mutants can induce phosphorylation of Akt, which then promotes cell growth and suppresses apoptosis in CRCs. A downstream component of the PI3K-signaling pathway is mTORC1, which plays a major role in nutrient uptake and cell growth.113 mTOR is the catalytic component of the mTORC2 and mTORC1 multiprotein-kinase complexes. Akt phosphorylates mTORC1, which then drives protein synthesis. The signaling events in both these pathways intersect, and they regulate each other and their downstream functions. The extent of this cross talk is clinically important, and has provided insights into negative-feedback loops and compensatory pathways. Aberrant hyperactivation of these pathways leads to tumorigenesis. Therefore, these pathways are attractive targets for inhibition, and a number of targeted agents are in clinical development.
Signaling through an mTOR-dependent negative-feedback loop results in the inhibition of PI3K signaling. When mTORC1 is inhibited by rapamycin or its derivatives (rapalogs), this negative-feedback loop is disrupted, leading to activation of PI3K and its effector Akt.114 PI3K can then act on Ras to promote Ras-dependent ERK activation.115 The binding of Ras to PI3K activates the EGFR- and FGF2-signalling pathways. As shown in Figure 1, activated mTOR also phosphorylates the downstream substrate S6K1, which then phosphorylates the IRS protein and induces IRS degradation, resulting in less interaction between IRS with IGF1R and insulin receptors. Disruption of this feedback loop by mTORC1 inhibitors will enhance the stability of IRS, which can then interact with IGF1R and insulin receptors. This illustrates the alternative pathways that bypass mTORC1 inhibition. Therefore, dual blockade of PI3K and mTORC1 appears to be a rational step to inhibit tumor survival.
A number of dual PI3K/mTOR inhibitors have been discovered and are currently in clinical development.116 Some compounds have also been developed to inhibit both class I PI3K isoforms and mTORC1 and mTORC2.117 Combination therapies with dual PI3K/mTOR inhibitors and chemotherapy are also being investigated, whereby a Phase II trial is testing PF-05212384 (dual PI3K/mTOR inhibitor) with or without irinotecan (NCT01347866), and another trial is comparing PF-05212384 plus irinotecan versus cetuximab plus irinotecan (NCT01925274).
Due to multiple points of cross talk and negative-feedback loops, inhibition of the PI3K/Akt/mTOR pathway gives rise to activation of the Ras/Raf/MEK-signaling pathway and vice versa.114,118 Numerous preclinical models in Ras-driven tumors have shown that single-agent inhibitors of downstream Ras/Raf/MEK/ERK and PI3K/Akt/mTOR pathways do not lead to significant antitumor activity, due to multiple points of cross talk, negative-feedback loops, and redundancy.115,119 One example is the finding that selumetinib (MEK inhibitor) treatment upregulated Akt phosphorylation,120 indicating cross talk between these two pathways and suggesting simultaneous inhibition of both pathways may be of clinical benefit. A Phase I clinical trial with MK-2206 (an allosteric inhibitor of Akt) and selumetinib (a non-ATP competitive inhibitor of MEK) demonstrated that this combination had antitumor activity at tolerable doses, and thus further investigation is warranted.121 Another promising potential combination is to cotarget MEK and PI3K pathways. Table 5 shows a summary of ongoing Phase II and III clinical trials with MEK and PI3K inhibitors.116,122 A phase IB study of the MEK inhibitor pimasertib and the PI3K/mTOR inhibitor SAR245409, however, found this combination to be toxic.123
One possible mechanism for the intrinsic anti-EGFR resistance in CRC patients harboring KRAS mutations may be the direct activation of the MAPK-signaling pathway.61,62,124–126 Therefore, one possible strategy is to cotarget the MAPK-signaling pathway. One example is the use of panitumumab and MEK162 in a Phase II clinical trial (NCT01927341).
Resistance to B-Raf inhibitors in BRAF-mutated tumors has been found to be due to PI3K activation.127 However, resistance can be overcome by combining B-Raf inhibitors with chemotherapy120 or with PI3K inhibitors, as shown in a murine model of BRAF-mutation-positive CRC.128 In cultured BRAF-mutated cell lines, resistance to vemurafenib can also be overcome by combining vemurafenib with PI3K inhibitors.129 The importance of the PI3K pathway in BRAF-mutated tumors is now being tested in a clinical trial with a combination of LGX818 (B-Raf inhibitor) and cetuximab with and without the PI3K inhibitor BYL719 (NCT01719380).
Targeting the Wnt-signaling pathway
Wnt is a family of 19 secreted glycoproteins that accumulate in the extracellular matrix and activate cell-surface receptor-mediated signal-transduction pathways to regulate a range of cellular processes, such as cell migration, proliferation, cell polarity, and development. The Wnt/β-catenin pathway is activated when a Wnt ligand binds to the Fz receptor and its coreceptor LRP6 or its close relative LRP5. The formation of a likely Wnt–Fz–LRP6 complex together with the recruitment of the scaffolding protein Dvl results in LRP6 phosphorylation and the recruitment of the Axin complex to the receptors. In the canonical pathway, when Wnt signaling is not active, β-catenin is recruited into the Axin complex, which consists of APC, Axin and GSK3. This complex is then degraded following phosphorylation of β-catenin. In contrast, in Wnt-activated cells, the receptor occupancy by Wnt ligands in cells inhibits the kinase activity needed for the destruction of the complex, resulting in accumulation of β-catenin in the cytoplasm and its translocation to the nucleus. The accumulating β-catenin in the nucleus combines with the T-cell factor (TCF) transcriptional regulator complex to activate the transcription of many target genes, such as MYC, CCND1, BIRC5, and VEGF, and metalloproteinases that promote cell proliferation, survival, and angiogenesis.130 Association of deregulated Wnt/β-catenin signaling with cancer and particularly with CRC has been well documented, and thus blockade of this pathway has been of great interest for cancer therapy.131,132
The tumor-suppressor gene APC has been found to be mutated in approximately 80% of CRCs. Mutational inactivation of APC leads to hyperactivation of the Wnt pathway. In the Cancer Genome Atlas Network study, 16 different altered Wnt-pathway genes were found in CRC.108 The consequence of aberrant Wnt activation is a rapid proliferation of the stem cell compartment in the crypts and the generation of adenomas.
A number of Wnt-signaling pathway inhibitors have been described to date.133 The first class of Wnt-pathway inhibitors are small molecules that block TCF/β-catenin signaling by disrupting TCF–β-catenin interaction134 or β-catenin–co-activator (CBP) interaction, such as ICG-001.135 The disruption of TCF–β-catenin interaction inhibits production of prostaglandin E2, resembling the action of nonsteroidal anti-inflammatory drug, which has been found to be beneficial in CRC prevention and therapy.136,137
A second class of Wnt-pathway inhibitors blocks binding of β-catenin to the Axin-binding complex. One such small-molecule inhibitor is PRI-724, which is currently in clinical trials (NCT01606579, NCT01302405), in combination with a modified FOLFOX6 regimen for refractory colon cancer patients.138
A third class of Wnt-pathway inhibitors targets the activity of Porcupine, a membrane-bound acyltransferase that is responsible for secretion of Wnt ligands.139,140 Clinical trials are ongoing with LGK974 (inhibitor of acyltransferase Porcupine; NCT01351103). A study of WNT974 (Porcupine inhibitor that blocks the palmitoylation and secretion of Wnt ligands) in combination with LGX818 (B-Raf inhibitor) and cetuximab (EGFR inhibitor) in patients with BRAF-mutant metastatic CRC and Wnt-pathway mutations has also been reported.141
A fourth class of inhibitors stabilize the Axin protein, thereby promoting β-catenin degradation even in cancer cells that have lost APC function.139,140 One such inhibitor is JW55,142 a small-molecule tankyrase 1/2 inhibitor that can inhibit the growth of colon cancer cells in both cell cultures and animal models.
Finally, several mAbs have been developed to bind and sequester either Wnt ligands or Fz receptors. OMP-18R5, an mAb that binds to Fz receptors 1, 2, 5, 7, and 8, and blocks β-catenin signaling in response to Wnt3a, inhibited growth of human tumor xenografts in several cancers, including colon, breast, pancreatic, and lung cancers.143 Clinical trials for OMP-18R5 are ongoing (NCT01345201).
OMP-24F28 is a truncated Fr8 receptor fused to the IgG1 Fc portion, which has the ability to sequester the Wnt ligand. Antitumor activity was demonstrated in preclinical models, and Phase I trials in patients with a variety of advanced solid tumors are in progress, including three Phase III trials in combination with chemotherapy for pancreatic, hepatocellular, and ovarian cancers (NCT01608867).144
Targeting the Notch-signaling pathway
The Notch-signaling pathway consists of four receptors (Notch 1, 2, 3, and 4) and at least five ligands: Jagged 1, Jagged 2, Delta 1, Delta 3, and Delta 4.145 In the canonical Notch pathway, the binding of a ligand to the Notch receptor results in a cascade of proteolytic cleavages mediated first by a metalloprotease and then by a γ-secretase,145 resulting in release of a constitutively active intracytoplasmic Notch fragment that is then translocated to the nucleus.146 The Notch-signaling pathway has been shown to be aberrantly activated in colon adenocarcinoma,147 and thus blockade of this pathway appears to be a rational therapeutic strategy. However, single-agent treatment of metastatic CRC patients with the γ-secretase inhibitor RO4929097 showed little to no effect.147
Treatment of colon cancer cell lines with a γ-secretase inhibitor renders the cell lines more sensitive to chemotherapeutic treatments, such as with oxaliplatin, 5-fluorouracil, or SN-38 (irinotecan).148 Whether this in vitro study can be extrapolated to in vivo is still unknown. A total of 18 patients with advanced solid tumors are currently enrolled in a Phase I study of the oral γ-secretase inhibitor R04929097 in combination with gemcitabine.149 In addition, two ongoing trials are testing the efficacy of RO4929097 in CRC in combination with other chemotherapeutic drugs (NCT01198535 and NCT01270438). Another γ-secretase inhibitor, PF-03084014, has been evaluated in a CRC-explant model, where patient tumors were explanted into nude mice.150 A PF-03084014-plus-irinotecan combination was found to be more effective than a γ-secretase inhibitor or irinotecan administered as single agents. PF-03084014 (NCT00878189) has undergone one clinical trial and shown promising results, with durable tumor responses, thus warranting further evaluation.151
Targeting the Hedgehog-signaling pathway
The Hedgehog (Hh)-signaling pathway plays a crucial role in embryonic development, and is generally silenced in adults but reactivated during tissue repair and regeneration.152–154 Aberrant Hh-pathway activation has been found in a variety of cancers, such as glioma, medulloblastoma, and gastric, pancreatic, breast, and basal cell cancers.155–160
Binding of Hh ligands to the receptor Patched leads to derepression of Smoothened (Smo) and modulation of the transcription factors Gli1, Gli2, and Gli3.161,162 It has been shown that sonic Hh (Shh), which is the best-studied ligand of the Hh-signaling pathway, increases growth of xenograft colon tumors163,164 and Shh–Gli1 signaling contributes to the enhancement of tumor stemness.165 Therefore, Hh signaling results in the initiation, progression, and relapse of cancer, including the acquisition of the cancer stem cell phenotype. Gli3 and Shh are expressed in subsets of CRC,166 and thus are attractive therapeutic targets for development of anticancer agents. Efforts toward developing Hh-pathway inhibitors led to the discovery of the orphan G-protein-coupled receptor GPR39.167
Several targeted agents against Hh-signaling pathways have been developed, and most of these agents antagonize Smo. The Smo inhibitors in clinical trials include vismodegib, BMS-833923, IPI-926, LDE-225, PF-04449913, LEQ 506, and TAK-441, and are summarized in Banerjee and Hadden.168
Vismodegib was the first Hh-pathway inhibitor approved in the US (in 2012) for the treatment of metastatic or locally advanced basal cell carcinoma. A Phase II randomized clinical trial of vismodegib in combination with FOLFOX1 or FOXFIRI in 195 patients with previously untreated metastatic CRC found no extension of progression-free survival.169
Another Smo antagonist, IPI-926, an orally bioavailable derivative of cyclopamine, has demonstrated antitumor activity in a mouse pancreatic cancer model, but a Phase II clinical trial in combination with gemcitabine was stopped early, due to adverse side effects. Different combinations of IPI-926 with chemotherapy in clinical trials were summarized in a recent review.170
The Smo antagonist LDE-225 has shown partial response in medulloblastoma and complete response in basal cell carcinoma. GANT61 is a small molecule that inhibits binding of GLI1 and GLI2 to the promoters of target genes, and has been found to kill colon cancer cells.171
Initial preclinical studies with the Smo antagonist vismodegib showed that resistance can develop due to SMO mutations that promote Hh-pathway activation. An SMO mutation at position 1,697, changing codon 473 from Asp to His and conferring resistance to vismodegib, has been described.172 Another mechanism for resistance is the amplification of the GLI2 transcription factor and the Hh-target gene CCND1.173 Lastly, upregulation of the compensatory pathway IGF1R/PI3K was observed in LDE-225-resistant tumor samples. In a mouse medulloblastoma model, addition of the PI3K inhibitor BMK120 or the dual PI3K–mTOR inhibitor BEZ235 to an Smo antagonist delayed or prevented the development of drug resistance.174 Therefore, the combination of an Smo antagonist and dual PI3K–mTOR inhibitor appears to be a rational combination for cancer therapy.
Targeting the TGFβ-signaling pathway
The role of TGFβ as a tumor promoter or tumor suppressor is still debatable, as it has a differential role in early and late-stage cancer. Targeting the tumor microenvironment by inhibiting the TGFβ pathway appears to be a rational approach, since TGFβ functions to generate a favorable microenvironment for tumor growth and metastasis throughout all the steps of carcinogenesis. Many TGFβ-pathway inhibitors have been discovered, some of which are now in clinical development. These inhibitors include fresolimumab, TβM1, and PF-03446962. An excellent review on TGFβ inhibitors has been published.175 To date, reliable predictive biomarkers of response to TGFβ inhibitors and appropriate combinations with chemotherapy have not been identified. As with many of the targeted therapies, until predictive markers of response are available to guide the selection of patients, the efficacy of TGFβ inhibitors will be limited.
Activation of death-receptor 4 and 5 pathways
Death receptors (DRs), including TNFR1, Fas, DR4, and DR5, are attractive targets for cancer therapy. These DRs have an intracellular death domain that transmits a death signal upon binding of their cognate ligands, such as TNFα, FasL, or TRAIL. TRAIL induces apoptosis through engaging DR4 and/or DR5 expressed on the plasma membrane of target cancer cells, and triggers signal transduction via the assembly of a death-inducing signaling complex, activation of a caspase cascade, and cleavage of cellular proteins.176 Multiple clinical trials have been conducted to evaluate the antitumor activity of recombinant human TRAIL (rhTRAIL), which binds DR4 and DR5 and is used in combination with various chemotherapies or targeted therapies177,178 Agonistic antibodies that target DR4/5 in combination with other therapies are also being trialed. Examples include the application of dulanermin (Apo2L/rhTRAIL) in combination with cetuximab + irinotecan or bevacizumab + FOLFIRI (NCT00671372) and bevacizumab + FOLFOX (NCT00873756). In vitro and in vivo studies have demonstrated that conatumumab (AMG-655) a fully human IgG1 mAb that binds to the extracellular domain of DR5, induces apoptosis in CRC and pancreatic cancer cell lines, as well as in xenograft tumors. Preclinical studies also indicated that conatumumab enhances the antitumor activity of agents, such as irinotecan and gemcitabine.179 It has also been tested in Phase II clinical trials in combination with FOLFIRI (NCT00813605), with mFOLFOX6 + bevacizumab (NCT00625651), and with panitumumab (NCT00630786) for metastatic CRC patients.
Immunobased therapies to enhance antitumor immunity
Normally transformed cells are eliminated by the immune system, and cells of both the adaptive (T cells) and the innate (macrophages, natural killer cells) immune system contribute to this. Established tumors, however, subvert the immune system to produce tumor-protective and tumor-promoting factors. Immunotherapy was named the breakthrough of the year in the last 2013 issue of the journal Science. Cancer immunotherapy attempts to rebalance the immune response by inhibiting the immunosuppressive factors in the tumor microenvironment and/or by enhancing the antitumor functions of the immune system.
Enhancing T-cell function
The first approach is to enhance the T-cell response by using immune-checkpoint inhibitors. Immune-checkpoint inhibitors in clinical trials for CRC patients are summarized in Table 6. CTLA4 is a negative costimulatory molecule expressed on both T cells and regulatory T cells. It acts by inhibiting T cells and promoting regulatory T-cell function. Promising results with combination of mAbs against CTL4 and PD1 have been demonstrated in advanced melanoma.180
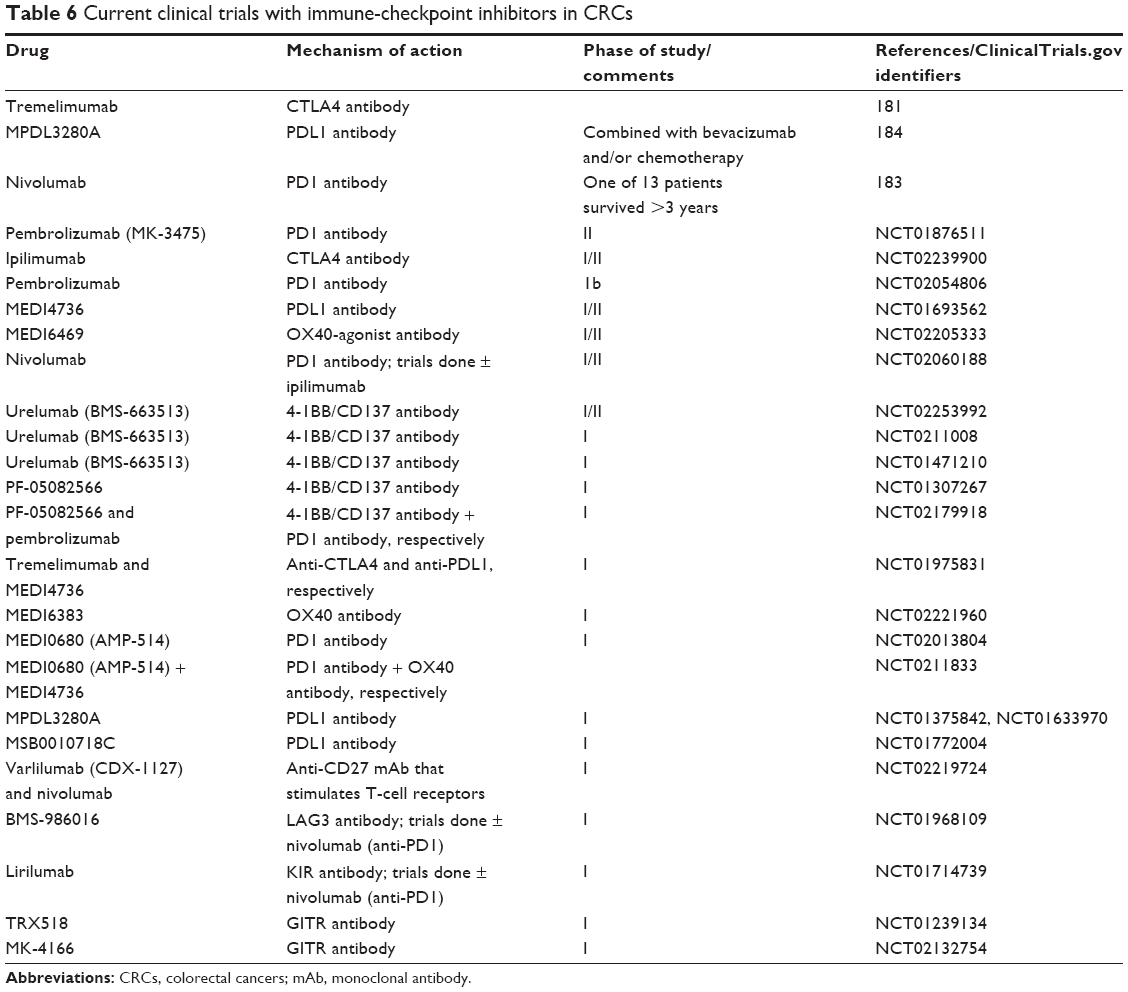 | Table 6 Current clinical trials with immune-checkpoint inhibitors in CRCs |
The first FDA-approved immune-checkpoint inhibitor was ipilimumab (anti-CTLA4; Yervoy) for advanced melanoma. In 2014, the FDA granted accelerated approval to pembrolizumab (PD1 inhibitor) for treatment of advanced melanoma. Since the approval of ipilimumab in 2011, another PD1 inhibitor, Opdivo (nivolumab), has been approved by the FDA. Unfortunately, immunotherapy with these antibodies has been disappointing in unselected CRC patients. It is possible that this treatment may be beneficial in selected CRC patients based on predictive biomarkers. A clinical trial with an anti-PD1 mAb is currently in progress in metastatic CRC patients with high microsatellite-instability tumors (NCT01876511). In the case of tremelimumab (anti-CTLA4), one patient showed a partial response, and 21 of 47 refractory metastatic CRC patients (45%) lived ≥180 days after enrollment.181 In another study, a patient with refractory CRC treated with nivolumab experienced a complete response for more than 3 years. Further analysis found that the tumors were high in microsatellite instability and were associated with infiltrating macrophages and lymphocytes expressing PDL1.182,183
Pembrolizumab (MK-3475) is currently in a Phase II trial for patients with microsatellite-unstable tumors (NCT01876511). Preclinical studies have shown that synergistic antitumor activity can be achieved with a combination of PD1 pathway inhibitors plus inhibitors of other immune checkpoints expressed on T cells, such as LAG3 and TIM3. BMS986016, an antibody against LAG3, is in a Phase I clinical trial with or without nivolumab (anti-PD1) in patients with solid tumors (NCT01968109).
The combination of anti-PDL1 therapy with the anti-CTLA4 therapy tremelimumab across multiple tumor types is also in progress. Other current studies include Phase III clinical trials testing the PDL1 inhibitor MPDL3280A in non-small-cell lung cancer (NCT02366143) and MEDI4736 (anti-PDL1 antibody; NCT02227667), a Phase II trial testing the PDL1 inhibitor MSB0010718 (avelumab) in metastatic Merkel cell carcinoma, and Phase I clinical trials for solid tumors. Encouraging results with MPDL3280 and bevacizumab and/or chemotherapy combination were observed in metastatic CRC.184
Other antibodies to costimulatory molecules used to enhance T-cell function include urelumab or PF-05082566 (anti-41BB/CD137), varlilumab (anti-CD27), and MOXR0916 (anti-OX40). Phase I trials with urelumab (NCT01471210), PF-05082566 (NCT01307267), PF-05082566 and pembrolizumab (NCT02179918), varlilumab (NCT01460134), varlilumab and nivolumab (NCT02335918), and MOXR0916 are underway.
Enhancing the function of cells in the innate immune system
A second approach is to enhance the function of innate cells by either engaging cetuximab in complement-mediated cytotoxicity or antibody-dependent cellular cytotoxicity or inducing phagocytosis of cancer cells with anti-CD47.
A Phase II study of Imprime PGG and cetuximab combination in metastatic CRC patients with KRAS mutation was conducted.185 Imprime is a β-glucan polymer that binds to CR3.186 Cetuximab activates the complement, resulting in production of iC3b, which opsonizes tumor cells. Imprime PGG primes neutrophils and macrophages through a CR3-dependent mechanism to exert killing activity against tumor cells that have been opsonized with complement following targeting with therapeutic mAbs, such as cetuximab.
Preclinical studies have shown that antibody-dependent cellular cytotoxicity by natural killer cells kills EGFR- expressing cancer cell lines following incubation with urelumab (anti-41BB/anti-CD137) in combination with cetuximab.187 This provided the rationale for studying cetuximab in combination with anti-CD137 (NCT02110082).
Phagocytosis of tumor cells by macrophages is prevented, due to the interaction between CD47 on tumor cells interacting with the ligand SIRPα on macrophages. Preclinical studies have shown that blockade of CD47 with anti-CD47 induces macrophage phagocytosis of tumor cells, including breast, colon, prostate and bladder cancers.188 The treatment can also prime an effective antitumor T-cell immune response.189 A Phase I clinical trial with anti-CD47 is underway (NCT02096770).
Targeting tumor-associated macrophages
Cells in the tumor microenvironment communicate with each other through a complex network of cytokines and growth factors. Some of these cells, such as the macrophages and fibroblasts, enhance tumor progression by modulating angiogenesis and immunosuppression by providing growth factors and other signaling molecules that aid in tumor survival and progression to metastasis. Classically activated macrophages, also referred to as M1 macrophages can kill tumors during the early steps of carcinogenesis. Dynamic changes in the tumor microenvironment occur during the transition from early transformation to advanced tumor stages that gradually drive the switch from M1 to M2 macrophages. Tumor-associated macrophages usually exhibit the M2 phenotype, secreting immunosuppressive cytokines, such as IL10, CCL17, and CCL22.190 Tumor-associated macrophages represent a dominant myeloid population in many tumors, and their accumulation correlates with poor prognosis.191 Therefore, they would appear to be rational therapeutic targets in cancer therapy.
One potential strategy is to deplete M2 macrophages. This can be achieved with CSF1R inhibitors. Increased infiltration of cytotoxic T lymphocytes and reduced survival of M2 macrophages were observed in tumors of patients treated with PLX3397 (a multikinase inhibitor targeting CSF1R kinase and c-Kit).192 Other CSF1R inhibitors, such as BLZ945 and RG7155 (humanized anti-CSF1R antibody), have shown antitumor effects in preclinical models.193 However, their efficacy in human trials is yet to be determined.
Another approach to modulate the immune system is to use chemokine-receptor antagonists. Welford et al194 showed that a CXCR4 antagonist (AMD3100) inhibited recruitment of Tie2-expressing macrophages, resulting in increased efficacy of VTAs in a mouse model. A Phase I clinical trial with folinic acid, flurouracil, irinotecan, oxaliplatin (FOLFIRINOX) plus PF-04136309, a CCR2 chemokine-receptor antagonist that blocks macrophage recruitment and is associated with anti-inflammatory responses, had partial responses in 14 of 29 patients: 14 had stable disease, and only one had progressive disease.195
Radiation has been traditionally used to kill tumor cells directly, but has also been shown to have immunomodulatory effects.196 Ionizing radiation results in immunogenic cell death,197 releasing endogenous adjuvants that facilitate dendritic cell maturation, uptake of antigens by dendritic cells, and cross-priming of cytotoxic T lymphocytes.198 In recent years, efforts have been made to augment the antitumor effect of radiation by combining it with immunotherapy.199 Preclinical trials have shown that CTLA4 blockade acts synergistically with radiotherapy to induce an abscopal response to radiotherapy in murine models of breast cancer and colon cancer.200 This approach has great potential, and will be worthy of further investigation.
Challenges in clinical development of targeted and immunobased therapies
Predictive biomarker development
Advances in DNA-sequencing technologies have led to a comprehensive analysis of exome sequences, DNA copy number, and epigenetic or transcriptomic dysregulation in CRC.108 The enormous efforts of many researchers have been summarized in a review.201 These genetic alterations could serve as potential predictive biomarkers of response when selecting patients for targeted therapies. This is exemplified by KRAS mutations in selecting patients for EGFR-inhibitor treatment, such as cetuximab. The mutational profile of tumors, therefore, is important, and they are increasingly incorporated into clinical trials.
In the case of immune-checkpoint inhibitors, predictive biomarker development is still in progress. So far, studies have indicated that patients with tumors high in microsatellite instability and infiltrating macrophages and lymphocytes expressing PDL1 have shown beneficial response to nivolumab.182,183
Existence of CRC subtypes
Molecular classification has categorized CRC into three groups, characterized by chromosomal instability, microsatellite instability, and CpG-island hypermethylation phenotype, respectively. More recently, a new classification of CRC subtypes based on expression profiling has emerged. These new subtypes were found to be associated with treatment outcomes. Patients with goblet and stem-like subtype tumors were found to respond poorly to cetuximab,202 and those with a mesenchymal-like subtype benefited from MET inhibitors. In another study, CRC tumors were classed into five subtypes that displayed significant differences in clinical and molecular characteristics.203 In one of these subtypes, there was upregulation of Aurora kinase A, suggesting that these patients could potentially benefit from treatment with inhibitors of Aurora kinase A.
CRC tumors differ depending on their location along the length of the bowel, and thus rectal and colon cancers not only reside in different anatomic locations but also differ in their embryonic origins and metastatic patterns. The proximal part of the colon up to the splenic flexure originates from the embryonic midgut, while the distal colon and rectum originate from the hindgut. The metastatic site of CRC is primarily dictated by its venous blood drainage: the large bowel drains into the portal system, resulting in liver metastases, while the inferior rectal vein drains into the vena cava, resulting in more frequent lung metastases of rectal tumors than from colon tumors.204
All the molecular subtypes reported herein are found throughout the length of the colorectal tract, but their frequency differs significantly based on location, and thus microsatellite-instability tumors are found mostly in the proximal colon.205 Familial CRCs also show significant association with anatomical locations, so familial adenomatous polyposis syndrome develops in the distal colon in the majority of cases (60%), followed by the rectum (25%), in contrast to the Lynch syndrome (hereditary nonpolyposis CRC), which occurs in the proximal column in 55% of cases and only 15% in the rectum.206 Mutational and epigenetic differences, including gene methylation between the three main locations, are also significant.207 Patients with metastatic CRC arising from distal primary tumors have a better prognosis for first-line chemotherapy alone or in combination with bevacizumab treatment.208
Due to their anatomic location, rectal cancers are treated differently to colon cancers, and invariably require neoadjuvant radiotherapy or radiochemotherapy to reduce the tumor prior to resection. These treatments have the potential to alter recurrent tumor characteristics locally or in metastases, impacting further disease management. As outlined in the immunobased therapies section, radiotherapy is known to kill tumor cells in an immunogenic manner and to induce systemic antitumor immune responses that may target and eliminate distant untreated micrometastases. In the clinic, however, additional enhancement of the immune system is required, such as that covered in the previous section. Chemoradiotherapy of rectal tumors before resection was shown to reduce immune-cell levels in the tumor, peritumoral tissues, and circulation compared to these tissues from patients who underwent surgery alone;209 however, coadministration of PSK, an immunostimulatory protein-bound polysaccharide from the basidiomycete Coriolus versicolor,210 during chemoradiotherapy restored immune-cell levels.211 Among its other immunostimulatory effects, PSK was shown to induce maturation of antigen-presenting dendritic cells.212 Another study found high levels of CXCR4 and CXCL12 expression in primary rectal tumors of patients presenting with metastatic disease. Treatment with radiochemotherapy and bevacizumab further upregulated CXCL12 expression.213 The CXCL12 chemokine attracts immunosuppressive/proangiogenic myeloid-derived suppressor cells and tumor-associated macrophages expressing the CXCR4 receptor into the tumor microenvironment, suggesting that inclusion of inhibitors or receptor antagonists of the CXCL12–CXCR4 axis would improve treatment outcome. A relevant study combining radiotherapy with tadalafil, a small-molecule inhibitor of myeloid-derived suppressor cells function, is ongoing for patients with locally advanced and borderline resectable pancreatic cancer (NCT01903083).
These studies suggest that expression profiling can further enhance our knowledge on the biological reasons for responsiveness or unresponsiveness to targeted therapies. This information can be used for the development of predictive markers using less complex methods, which can be further validated and potentially adopted in routine clinical diagnostics.
Resistance due to cancer stem cells
The cancer stem cell theory states that cancer stem cells are distinct from cells that form the bulk of a tumor in that they can self-renew and differentiate to produce progenitor cells, the way traditional stem cells do. This property enables cancer stem cells to self-renew and generate progenies that can differentiate into the bulk of the proliferating cancer cells within the tumor,214 and like traditional stem cells they are more resistant to chemotherapy. Therefore, therapies specifically targeting cancer stem cells are essential in combination with other treatments to kill the tumor.
Preclinical research with a small molecule, PTC-209, which targets the self-renewal regulator BMI1, appears promising, as it abrogated the tumorigenic capacity of colon cancer stem cells.215 Inhibitors of signaling pathways, such as Notch, Wnt, and Hh, have also been shown to kill these putative cancer stem cells. One example is demonstrated by the γ-secretase inhibitor PF-03084014 plus irinotecan, which resulted in reduced tumor recurrence in a CRC-explant model. The bulk of these effects were seen in ALDH+ tumor cells, which is a subpopulation enriched for cancer-initiating cell activity.150 Selective β-catenin-binding protein antagonists, such as PRI-724, have also been shown to eliminate cancer stem cells by forced differentiation.216
Hh-signaling pathway activation has also been shown to play a role in the maintenance of cancer stem cell phenotype. Preclinical studies have shown that inhibitors of Hh-signaling pathways, such as Smo antagonists, can be used to target cancer stem cells.217 Therefore, elimination of cancer stem cells has been demonstrated with an inhibitor of the self-renewal regulator BMI1 and inhibitors of the Wnt-, Notch-, and Hh-signaling pathways.
Future directions
Recent progress for development of an in silico prescription strategy
As of September 2013, a number of next-generation sequencing (NGS) studies in CRC have been reported.108,218–220 NGS panels have been developed to check tumors for mutations, and currently panels of five to 500 mutations are available for evaluation. Interestingly, a recent report on in silico analysis of genetic mutations for selecting the large pool of previously discovered targeted drugs has potential for prescription strategy beyond targeting EGFR and VEGF systems.221 The in silico prescription strategy requires three steps. Firstly, NGS data need to be analyzed to discover actionable driver events using DriverDB.222 Secondly, information on therapeutic agents needs to be collected from the Drivers Actionability Database. Lastly, the data of both databases will need to be combined to obtain a list of actionable targets with their matching targeted therapeutic agent.
The application of NGS has not only provided a wide spectrum of mutational profiles but also additional information, such as splice variants and novel fusion genes. A novel fusion protein found in three of 97 CRC patients is VTI1A–TCF7L2, which offers a potential new therapeutic target.218 Another example is novel fusion genes encoding R-spondin fusion proteins, which have the ability to potentiate Wnt signaling and thereby function as stem cell growth factors.219
Some of the complicated issues regarding NGS include the fact that the majority of mutations will not provide clinical actionable information, the difficulties of interpreting substantial amounts of genomic data, timing of the tests, and access to treatment options. Integration of the data to enable a better understanding of the biology in order to select appropriate targeted drugs is essential for further progress in this field. This will require a lot of effort and money.
Combination of targeted therapies and silencing of genes by siRNA and microRNAs
RNA-interference strategies appear to be an attractive approach to inhibit expression of oncogenes. Indeed, small interfering RNA (siRNA) has emerged as a major tool to silence gene expression.223 However, clinical trials on the delivery of siRNA in humans are scarce,224 and the development of siRNA as a therapeutic tool has been hindered, due to problems with delivery. A recent publication showed that siRNA against Ras physically coupled to cetuximab exerted antitumor activity on colon cancer mouse xenografts.225 Another potential treatment strategy is to use microRNAs to repress or degrade targeted messenger RNAs.226 It has been demonstrated that miR-4689 delivered into cells was able to induce apoptosis of KRAS-mutant colon cancer cell lines and also exert antitumor activity on mouse xenografts when the microRNA complexes were administered intravenously.227 The miR-4689 targeted both Raf/MEK/ERK and PI3K/Akt pathways through direct inhibition of K-Ras and Akt1. Lastly, genome-editing technologies using zinc-finger nucleases, transcription-like effector nucleases and clustered regularly interspaced short palindromic repeat-associated nuclease 9228 to remove deleterious “driver mutations” or to insert “protective mutations” may present novel strategies for cancer therapy in the future.
Conclusion
Although there are many targeted drugs in clinical trials, many have produced disappointing results. However, with further discovery of novel driver genes through NGS technology, new drug development, and application of in silico analysis, we will be moving toward developing a personalized prescription for cancer treatment. New developments in the use of immune-checkpoint inhibitors and immunotherapy will be seen in the coming years. Further research is needed to elucidate the contribution of the tumor microenvironment, the immune response, and the host–drug response in the development of tumor resistance, in order to achieve an effective treatment strategy. These new discoveries will contribute to our increased understanding of the heterogeneity of most tumors, events in the tumor microenvironment, and new gene and protein signatures that can be translated into clinically meaningful improvements. It is without doubt that application of bioinformatics will be required to link molecular signatures as predictive biomarkers for drug response to overcome the current problems and enable individualized drug therapy for cancer patients. Advances in RNA interference, noncoding RNA, and genome editing will pave the way for an exciting future in gaining new insights into cancer biology and the development of new therapeutic strategies in cancer therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Center MM, Jemal A, Ward E. International trends in colorectal cancer incidence rates. Cancer Epidemiol Biomarkers Prev. 2009;18(6):1688–1694. | ||
Kirstein MM, Lange A, Prenzler A, Manns MP, Kubicka S, Vogel A. Targeted therapies in metastatic colorectal cancer: a systematic review and assessment of currently available data. Oncologist. 2014;19(11):1156–1168. | ||
Buroker TR, O’Connell MJ, Wieand HS, et al. Randomized comparison of two schedules of fluorouracil and leucovorin in the treatment of advanced colorectal cancer. J Clin Oncol. 1994;12(1):14–20. | ||
Poon MA, O’Connell MJ, Moertel CG, et al. Biochemical modulation of fluorouracil: evidence of significant improvement of survival and quality of life in patients with advanced colorectal carcinoma. J Clin Oncol. 1989;7(10):1407–1418. | ||
Douillard JY, Cunningham D, Roth AD, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355(9209):1041–1047. | ||
Saltz LB, Cox JV, Blanke C, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. N Engl J Med. 2000;343(13):905–914. | ||
de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18(16):2938–2947. | ||
Goldberg RM, Sargent DJ, Morton RF, et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol. 2004;22(1):23–30. | ||
Grothey A, Deschler B, Kroening H, et al. Phase III study of bolus 5-fluorouracil (5-FU)/folinic acid (FA) (Mayo) vs weekly high-dose 24h 5-FU infusion/FA + oxaliplatin (OXA) (FUFOX) in advanced colorectal cancer (ACRC). Poster presented at: American Society of Clinical Oncology Annual Meeting; May 18–21, 2002; Orlando, FL. | ||
Grothey A, Sargent D, Goldberg RM, Schmoll HJ. Survival of patients with advanced colorectal cancer improves with the availability of fluorouracil-leucovorin, irinotecan, and oxaliplatin in the course of treatment. J Clin Oncol. 2004;22(7):1209–1214. | ||
Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15(10):1065–1075. | ||
Schwartzberg LS, Rivera F, Karthaus M, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32(21):2240–2247. | ||
Akkad J, Bochum S, Martens UM. Personalized treatment for colorectal cancer: novel developments and putative therapeutic strategies. Langenbecks Arch Surg. 2015;400(2):129–143. | ||
Garrido-Castro AC, Sauri-Nadal T, Macarulla-Mercadé T. New targets and new drug development in colorectal cancer. Curr Colorectal Cancer Rep. 2014;10(3):288–295. | ||
Raj N, Saltz L. Biologic agents in the treatment of colorectal cancer: an update. Colorectal Cancer. 2014;3(4):363–374. | ||
Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30(28):3499–3506. | ||
Saltz LB, Clarke S, Diaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013–2019. | ||
Bennouna J, Sastre J, Arnold D, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 2013;14(1):29–37. | ||
Kuczynski EA, Sargent DJ, Grothey A, Kerbel RS. Drug rechallenge and treatment beyond progression – implications for drug resistance. Nat Rev Clin Oncol. 2013;10(10):571–587. | ||
Tabernero J, Van Cutsem E, Lakomy R, et al. Aflibercept versus placebo in combination with fluorouracil, leucovorin and irinotecan in the treatment of previously treated metastatic colorectal cancer: prespecified subgroup analyses from the VELOUR trial. Eur J Cancer. 2014;50(2):320–331. | ||
Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245–255. | ||
Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–312. | ||
Garcia-Carbonero R, Rivera F, Maurel J, et al. An open-label phase II study evaluating the safety and efficacy of ramucirumab combined with mFOLFOX-6 as first-line therapy for metastatic colorectal cancer. Oncologist. 2014;19(4):350–351. | ||
Tabernero J, Cohn AL, Obermannova R, et al. RAISE: a randomized, double-blind, multicenter phase III study of irinotecan, folinic acid, and 5-fluorouracil (FOLFIRI) plus ramucirumab (RAM) or placebo (PBO) in patients (pts) with metastatic colorectal carcinoma (CRC) progressive during or following first-line combination therapy with bevacizumab (bev), oxaliplatin (ox), and a fluoropyrimidine (fp). J Clin Oncol. 2015;33 Suppl 3:512. | ||
Xu RH, Shen L, Wang KM, et al. A randomized, double-blind, parallel-group, placebo-controlled, multicenter, phase II clinical study of famitinib in the treatment of advanced metastatic colorectal cancer. J Clin Oncol. 2015;33 Suppl 3:513. | ||
Carrato A, Swieboda-Sadlej A, Staszewska-Skurczynska M, et al. Fluorouracil, leucovorin, and irinotecan plus either sunitinib or placebo in metastatic colorectal cancer: a randomized, phase III trial. J Clin Oncol. 2013;31(10):1341–1347. | ||
Tabernero J, Garcia-Carbonero R, Cassidy J, et al. Sorafenib in combination with oxaliplatin, leucovorin, and fluorouracil (modified FOLFOX6) as first-line treatment of metastatic colorectal cancer: the RESPECT trial. Clin Cancer Res. 2013;19(9):2541–2550. | ||
Van Cutsem E, Bajetta E, Valle J, et al. Randomized, placebo-controlled, phase III study of oxaliplatin, fluorouracil, and leucovorin with or without PTK787/ZK 222584 in patients with previously treated metastatic colorectal adenocarcinoma. J Clin Oncol. 2011;29(15):2004–2010. | ||
Infante JR, Reid TR, Cohn AL, et al. Axitinib and/or bevacizumab with modified FOLFOX-6 as first-line therapy for metastatic colorectal cancer: a randomized phase 2 study. Cancer. 2013;119(14):2555–2563. | ||
Hoff PM, Hochhaus A, Pestalozzi BC, et al. Cediranib plus FOLFOX/CAPOX versus placebo plus FOLFOX/CAPOX in patients with previously untreated metastatic colorectal cancer: a randomized, double-blind, phase III study (HORIZON II). J Clin Oncol. 2012;30(29):3596–3603. | ||
Siu LL, Shapiro JD, Jonker DJ, et al. Phase III randomized, placebo-controlled study of cetuximab plus brivanib alaninate versus cetuximab plus placebo in patients with metastatic, chemotherapy-refractory, wild-type K-RAS colorectal carcinoma: the NCIC Clinical Trials Group and AGITG CO.20 trial. J Clin Oncol. 2013;31(19):2477–2484. | ||
Pilat MJ, McCormick J, Lorusso PM. Vascular targeting agents. Curr Oncol Rep. 2004;6(2):103–110. | ||
Kretzschmann VK, Fürst R. Plant-derived vascular disrupting agents: compounds, actions, and clinical trials. Phytochem Rev. 2014;13(1):191–206. | ||
Buchanan CM, Shih JH, Astin JW, et al. DMXAA (Vadimezan, ASA404) is a multi-kinase inhibitor targeting VEGFR2 in particular. Clin Sci (Lond). 2012;122(10):449–457. | ||
Lorusso PM, Boerner SA, Hunsberger S. Clinical development of vascular disrupting agents: what lessons can we learn from ASA404? J Clin Oncol. 2011;29(22):2952–2955. | ||
Holwell SE, Cooper PA, Thompson MJ, et al. Anti-tumor and anti-vascular effects of the novel tubulin-binding agent combretastatin A-1 phosphate. Anticancer Res. 2002;22(6C):3933–3940. | ||
Dowlati A, Robertson K, Cooney M, et al. A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin A-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res. 2002;62(12):3408–3416. | ||
Rustin GJ, Galbraith SM, Anderson H, et al. Phase I clinical trial of weekly combretastatin A4 phosphate: clinical and pharmacokinetic results. J Clin Oncol. 2003;21(15):2815–2822. | ||
Stevenson JP, Rosen M, Sun W, et al. Phase I trial of the antivascular agent combretastatin A4 phosphate on a 5-day schedule to patients with cancer: magnetic resonance imaging evidence for altered tumor blood flow. J Clin Oncol. 2003;21(23):4428–4438. | ||
Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12(18):5268–5272. | ||
Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28(7):1254–1261. | ||
Giehl K. Oncogenic Ras in tumour progression and metastasis. Biol Chem. 2005;386(3):193–205. | ||
Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17(11):1395–1413. | ||
Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–1765. | ||
Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–1417. | ||
Venook A, Niedzwiecki D, Hollis D, et al. Phase III study of irinotecan/5FU/LV (FOLFIRI) or oxaliplatin/5FU/LV (FOLFOX){+/−} cetuximab for patients (pts) with untreated metastatic adenocarcinoma of the colon or rectum (MCRC):CALGB 80203 preliminary results. J Clin Oncol. 2006;24(18 Suppl):3509. | ||
Bokemeyer C, Van Cutsem E, Rougier P, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48(10):1466–1475. | ||
Douillard JY, Siena S, Cassidy J, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28(31):4697–4705. | ||
Montagut C, Dalmases A, Bellosillo B, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18(2):221–223. | ||
Saltz LB, Meropol NJ, Loehrer PJ Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22(7):1201–1208. | ||
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–345. | ||
Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23(9):1803–1810. | ||
Shia J, Klimstra DS, Li AR, et al. Epidermal growth factor receptor expression and gene amplification in colorectal carcinoma: an immunohistochemical and chromogenic in situ hybridization study. Mod Pathol. 2005;18(10):1350–1356. | ||
Moroni M, Sartore-Bianchi A, Veronese S, Siena S. EGFR FISH in colorectal cancer: what is the current reality? Lancet Oncol. 2008;9(5):402–403. | ||
Razis E, Briasoulis E, Vrettou E, et al. Potential value of PTEN in predicting cetuximab response in colorectal cancer: an exploratory study. BMC Cancer. 2008;8:234. | ||
Jacobs B, De Roock W, Piessevaux H, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27(30):5068–5074. | ||
Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25(22):3230–3237. | ||
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. | ||
Di Fiore F, Blanchard F, Charbonnier F, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by cetuximab plus chemotherapy. Br J Cancer. 2007;96(8):1166–1169. | ||
Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–3995. | ||
Morris VK, Lucas FA, Overman MJ, et al. Clinicopathologic characteristics and gene expression analyses of non-KRAS 12/13, RAS-mutated metastatic colorectal cancer. Ann Oncol. 2014;25(10):2008–2014. | ||
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. | ||
Atreya CE, Corcoran RB, Kopetz S. Expanded RAS: refining the patient population. J Clin Oncol. 2015;33(7):682–685. | ||
Diaz LA Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–540. | ||
Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. | ||
Esposito C, Rachiglio AM, La Porta ML, et al. The S492R EGFR ectodomain mutation is never detected in KRAS wild-type colorectal carcinoma before exposure to EGFR monoclonal antibodies. Cancer Biol Ther. 2013;14(12):1143–1146. | ||
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67(6):2643–2648. | ||
Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–5712. | ||
Jhawer M, Goel S, Wilson AJ, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68(6):1953–1961. | ||
Perrone F, Lampis A, Orsenigo M, et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol. 2009;20(1):84–90. | ||
Frattini M, Saletti P, Romagnani E, et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007;97(8):1139–1145. | ||
Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–673. | ||
Huang F, Xu LA, Khambata-Ford S. Correlation between gene expression of IGF-1R pathway markers and cetuximab benefit in metastatic colorectal cancer. Clin Cancer Res. 2012;18(4):1156–1166. | ||
Yonesaka K, Zejnullahu K, Okamoto I, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3(99):99ra86. | ||
Hobor S, Van Emburgh BO, Crowley E, Misale S, Di Nicolantonio F, Bardelli A. TGFα and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin Cancer Res. 2014;20(24):6429–6438. | ||
Corso S, Giordano S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: from basic research to a clinical perspective. Cancer Discov. 2013;3(9):978–992. | ||
Shaib W, Mahajan R, El-Rayes B. Markers of resistance to anti-EGFR therapy in colorectal cancer. J Gastrointest Oncol. 2013;4(3):308–318. | ||
Dienstmann R, Tabernero J, Van Cutsem E, et al. Proof-of-concept study of Sym004, an anti-EGFR monoclonal antibody (mAb) mixture, in patients (pts) with anti-EGFR mab-refractory KRAS wild-type (wt) metastatic colorectal cancer (mCRC). J Clin Oncol. 2013;31 Suppl:3551. | ||
Schaefer G, Haber L, Crocker LM, et al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell. 2011;20(4):472–486. | ||
Huang S, Li C, Armstrong EA, et al. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013;73(2):824–833. | ||
Cervantes-Ruiperez A, Juric D, Hidalgo M, et al. A phase I study of MEHD7945A (MEHD), a first-in-class HER3/EGFR dual-action antibody, in patients (pts) with refractory/recurrent epithelial tumors: expansion cohorts. J Clin Oncol. 2012;30 Suppl:2568. | ||
Gerdes CA, Nicolini VG, Herter S, et al. GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res. 2013;19(5):1126–1138. | ||
Cervantes-Ruiperez A, Markman B, Siena S, et al. The GAIN-C study (BP25438): randomized phase II trial of RG7160 (GA201) plus FOLFIRI, compared to cetuximab plus FOLFIRI or FOLFIRI alone in second-line KRAS wild type (WT) or mutant metastatic colorectal cancer (mCRC). J Clin Oncol. 2012;30 Suppl:TPS3637. | ||
Delord JP, Tabernero J, Garcia-Carbonero R, et al. Long-term efficacy and pharmacodynamic parameter analysis in pretreated KRAS-mutant metastatic colorectal carcinoma (mCRC) patients treated with RG7160 (GA201), an antibody-dependent cellular cytotoxicity (ADCC)-enhanced monoclonal anti-EGFR antibody. J Clin Oncol. 2013;31 Suppl 4:379. | ||
Bertotti A, Migliardi G, Galimi F, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1(6):508–523. | ||
Kopetz S, Desai J, Chan E, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol. 2010;28(15 Suppl):3534. | ||
Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. | ||
Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2(3):227–235. | ||
Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. | ||
Yang H, Higgins B, Kolinsky K, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012;72(3):779–789. | ||
Yaeger R, Cercek A, O’Reilly EM, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21(6):1313–1320. | ||
Van Geel R, Elez E, Bendell JC, et al. Phase I study of the selective BRAFV600 inhibitor encorafenib (LGX818) combined with cetuximab and with or without the α-specific PI3K inhibitor BYL719 in patients with advanced BRAF-mutant colorectal cancer. J Clin Oncol. 2014;32(5 Suppl):3514. | ||
Bendell JC, Atreya CE, André T, et al. Efficacy and tolerability in an open-label phase I/II study of MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in combination in patients (pts) with BRAF V600E mutated colorectal cancer (CRC). J Clin Oncol. 2014;32(5 Suppl):3515. | ||
Fujita S, Sugano K. Expression of c-met proto-oncogene in primary colorectal cancer and liver metastases. Jpn J Clin Oncol. 1997;27(6):378–383. | ||
Zeng ZS, Weiser MR, Kuntz E, et al. c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett. 2008;265(2):258–269. | ||
Otte JM, Schmitz F, Kiehne K, et al. Functional expression of HGF and its receptor in human colorectal cancer. Digestion. 2000;61(4):237–246. | ||
Di Renzo MF, Olivero M, Giacomini A, et al. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res. 1995;1(2):147–154. | ||
Kataoka H, Hamasuna R, Itoh H, Kitamura N, Koono M. Activation of hepatocyte growth factor/scatter factor in colorectal carcinoma. Cancer Res. 2000;60(21):6148–6159. | ||
Song EK, Tai WM, Messersmith WA, et al. Potent antitumor activity of cabozantinib, a c-MET and VEGFR2 inhibitor, in a colorectal cancer patient-derived tumor explant model. Int J Cancer. 2015;136(8):1967–1975. | ||
Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–928. | ||
Flanigan SA, Pitts TM, Newton TP, et al. Overcoming IGF1R/IR resistance through inhibition of MEK signaling in colorectal cancer models. Clin Cancer Res. 2013;19(22):6219–6229. | ||
Wilky BA, Rudek MA, Ahmed S, et al. A phase I trial of vertical inhibition of IGF signalling using cixutumumab, an anti-IGF-1R antibody, and selumetinib, an MEK 1/2 inhibitor, in advanced solid tumours. Br J Cancer. 2015;112(1):24–31. | ||
McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–1284. | ||
Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. | ||
Sarker D, Ang JE, Baird R, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21(1):77–86. | ||
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. | ||
Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. | ||
Sartore-Bianchi A, Martini M, Molinari F, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69(5):1851–1857. | ||
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. | ||
Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28(6):1075–1083. | ||
Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103(2):253–262. | ||
O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508. | ||
Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–3074. | ||
Bauer TM, Patel MR, Infante JR. Targeting PI3 kinase in cancer. Pharmacol Ther. 2015;146:53–60. | ||
Sturgill TW, Hall MN. Activating mutations in TOR are in similar structures as oncogenic mutations in PI3KCα. ACS Chem Biol. 2009;4(12):999–1015. | ||
Zitzmann K, Ruden J, Brand S, et al. Compensatory activation of Akt in response to mTOR and Raf inhibitors – a rationale for dual-targeted therapy approaches in neuroendocrine tumor disease. Cancer Lett. 2010;295(1):100–109. | ||
She QB, Halilovic E, Ye Q, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18(1):39–51. | ||
Holt SV, Logie A, Odedra R, et al. The MEK1/2 inhibitor, selumetinib (AZD6244;ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer. 2012;106(5):858–866. | ||
Khan KH, Yan L, Mezynski J, et al. A phase I dose escalation study of oral MK-2206 (allosteric Akt inhibitor) with oral selumetinib (AZD6244;ARRY-142866) (MEK 1/2 inhibitor) in patients with advanced or metastatic solid tumors. J Clin Oncol. 2012;30 Suppl:e13599. | ||
Neuzillet C, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E. MEK in cancer and cancer therapy. Pharmacol Ther. 2014;141(2):160–171. | ||
Heist RS, Gandhi L, Shapiro G, et al. Combination of a MEK inhibitor, pimasertib (MSC1936369B), and a PI3K/mTOR inhibitor, SAR245409, in patients with advanced solid tumors: Results of a phase IB dose-escalation trial. J Clin Oncol. 2013;31 Suppl:2530. | ||
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. | ||
de Reynies A, Boige V, Milano G, Faivre J, Laurent-Puig P. KRAS mutation signature in colorectal tumors significantly overlaps with the cetuximab response signature. J Clin Oncol. 2008;26(13):2228–2230; author reply 2230–2231. | ||
Lievre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26(3):374–379. | ||
Wee S, Jagani Z, Xiang KX, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69(10):4286–4293. | ||
Rad R, Cadinanos J, Rad L, et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell. 2013;24(1):15–29. | ||
Mao M, Tian F, Mariadason JM, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19(3):657–667. | ||
Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17(1):45–51. | ||
Mologni L, Dekhil H, Ceccon M, et al. Colorectal tumors are effectively eradicated by combined inhibition of β-catenin, KRAS, and the oncogenic transcription factor ITF2. Cancer Res. 2010;70(18):7253–7263. | ||
Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16(12):3153–3162. | ||
Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13(7):513–532. | ||
Lepourcelet M, Chen YN, France DS, et al. Small-molecule antagonists of the oncogenic Tcf/β-catenin protein complex. Cancer Cell. 2004;5(1):91–102. | ||
Emami KH, Nguyen C, Ma H, et al. A small molecule inhibitor of β-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci U S A. 2004;101(34):12682–12687. | ||
Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-β-catenin signaling axis. Science. 2005;310(5753):1504–1510. | ||
Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 stimulates the β-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280(28):26565–26572. | ||
El-Khoueiry AB, Ning Y, Yang D, et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J Clin Oncol. 2013;31 Suppl:2501. | ||
Chen B, Dodge ME, Tang W, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5(2):100–107. | ||
Yeh JR, Peterson RT. Novel Wnt antagonists target porcupine and Axin. Nat Chem Biol. 2009;5(2):74–75. | ||
Wang Y, Palmer M, Jaeger S, et al. Dual Wnt and EGFR-MAPK dependency of BRAFV600E-mutant colorectal cancer. Poster presented at: 106th Annual Meeting of the American Association for Cancer Research; April 18–22, 2015; Philadelphia, PA. | ||
Waaler J, Machon O, Tumova L, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012;72(11):2822–2832. | ||
Gurney A, Axelrod F, Bond CJ, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109(29):11717–11722. | ||
Jimeno A, Gordon MS, Chugh R, et al. A first-in-human phase 1 study of anticancer stem cell agent OMP-54F28 (FZD8-Fc), decoy receptor for WNT ligands, in patients with advanced solid tumors. J Clin Oncol. 2014;32(5 Suppl):2505. | ||
Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol. 2000;228(2):151–165. | ||
Kovall RA. More complicated than it looks: assembly of Notch pathway transcription complexes. Oncogene. 2008;27(38):5099–5109. | ||
Strosberg JR, Yeatman T, Weber J, et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer. 2012;48(7):997–1003. | ||
Meng RD, Shelton CC, Li YM, et al. γ-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009;69(2):573–582. | ||
Richter S, Bedard PL, Chen EX, et al. A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL-078/CTEP 8575). Invest New Drugs. 2014;32(2):243–249. | ||
Arcaroli JJ, Powell RW, Varella-Garcia M, et al. ALDH+ tumor-initiating cells exhibiting gain in NOTCH1 gene copy number have enhanced regrowth sensitivity to a γ-secretase inhibitor and irinotecan in colorectal cancer. Mol Oncol. 2012;6(3):370–381. | ||
Messersmith WA, Shapiro GI, Cleary JM, et al. A phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res. 2015;21(1):60–67. | ||
Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059–3087. | ||
McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of Hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114. | ||
Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432(7015):324–331. | ||
Kinzler KW, Bigner SH, Bigner DD, et al. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236(4797):70–73. | ||
Kubo M, Nakamura M, Tasaki A, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004;64(17):6071–6074. | ||
Ma X, Chen K, Huang S, et al. Frequent activation of the hedgehog pathway in advanced gastric adenocarcinomas. Carcinogenesis. 2005;26(10):1698–1705. | ||
Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–856. | ||
Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgård R, Undén AB. Deregulation of the Hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. 2006;208(1):17–25. | ||
Zurawel RH, Allen C, Chiappa S, et al. Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosomes Cancer. 2000;27(1):44–51. | ||
di Magliano MP, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer. 2003;3(12):903–911. | ||
Ruel L, Rodriguez R, Gallet A, Lavenant-Staccini L, Therond PP. Stability and association of Smoothened, Costal2 and Fused with Cubitus interruptus are regulated by Hedgehog. Nat Cell Biol. 2003;5(10):907–913. | ||
Douard R, Moutereau S, Pernet P, et al. Sonic Hedgehog-dependent proliferation in a series of patients with colorectal cancer. Surgery. 2006;139(5):665–670. | ||
Varnat F, Siegl-Cachedenier I, Malerba M, Gervaz P, Altaba AR. Loss of WNT-TCF addiction and enhancement of HH-GLI1 signalling define the metastatic transition of human colon carcinomas. EMBO Mol Med. 2010;2(11):440–457. | ||
Gulino A, Ferretti E, De Smaele E. Hedgehog signalling in colon cancer and stem cells. EMBO Mol Med. 2009;1(6–7):300–302. | ||
Iwasaki H, Nakano K, Shinkai K, et al. Hedgehog Gli3 activator signal augments tumorigenicity of colorectal cancer via upregulation of adherence-related genes. Cancer Sci. 2013;104(3):328–336. | ||
Bassilana F, Carlson A, DaSilva JA, et al. Target identification for a Hedgehog pathway inhibitor reveals the receptor GPR39. Nat Chem Biol. 2014;10(5):343–349. | ||
Banerjee U, Hadden MK. Recent advances in the design of Hedgehog pathway inhibitors for the treatment of malignancies. Expert Opin Drug Discov. 2014;9(7):751–771. | ||
Berlin J, Bendell J, Hart LL, et al. A phase 2, randomized, double-blind, placebo-controlled study of Hedgehog pathway inhibitor (HPI) GDC-0449 in patients with previously untreated metastatic colorectal cancer (mCRC). Ann Oncol. 2010;21 Suppl 8:viii10. | ||
Sahebjam S, Siu LL, Razak AA. The utility of Hedgehog signaling pathway inhibition for cancer. Oncologist. 2012;17(8):1090–1099. | ||
Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011;71(3):1092–1102. | ||
Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326(5952):572–574. | ||
Dijkgraaf GJ, Alicke B, Weinmann L, et al. Small molecule inhibition of GDC-0449 refractory Smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71(2):435–444. | ||
Buonamici S, Williams J, Morrissey M, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2(51):51ra70. | ||
Neuzillet C, Tijeras-Raballand A, Cohen R, et al. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31. | ||
Aza-Blanc P, Cooper CL, Wagner K, Batalov S, Deveraux QL, Cooke MP. Identification of modulators of TRAIL-induced apoptosis via RNAi-based phenotypic screening. Mol Cell. 2003;12(3):627–637. | ||
Soria JC, Mark Z, Zatloukal P, et al. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(33):4442–4451. | ||
Thorburn A, Behbakht K, Ford H. TRAIL receptor-targeted therapeutics: resistance mechanisms and strategies to avoid them. Drug Resist Updat. 2008;11(1–2):17–24. | ||
Rosevear HM, Lightfoot AJ, Griffith TS. Conatumumab, a fully human mAb against death receptor 5 for the treatment of cancer. Curr Opin Investig Drugs. 2010;11(6):688–698. | ||
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. | ||
Chung KY, Gore I, Fong L, et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol. 2010;28(21):3485–3490. | ||
Lipson EJ, Sharfman WH, Drake CG, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19(2):462–468. | ||
Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. | ||
Lieu C, Bendell J, Powderly JD, et al. Safety and efficacy of MPDL3280A (anti-PDL1) in combination with bevacizumab (bev) and/or chemotherapy (chemo) in patients (pts) with locally advanced or metastatic solid tumours. Ann Oncol. 2014;25 Suppl 4:iv361–iv372. | ||
Segal NH, Senzer N, Gada P, et al. Imprime PGG plus cetuximab therapy for advanced KRAS mutant colorectal cancer. Annals of Oncology. 2011;22 Suppl 5:v22. | ||
Vetvicka V, Thornton BP, Ross GD. Soluble β-glucan polysaccharide binding to the lectin site of neutrophil or natural killer cell complement receptor type 3 (CD11b/CD18) generates a primed state of the receptor capable of mediating cytotoxicity of iC3b-opsonized target cells. J Clin Invest. 1996;98(1):50–61. | ||
Kohrt HE, Colevas AD, Houot R, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest. 2014;124(6):2668–2682. | ||
Willingham SB, Volkmer JP, Gentles AJ, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662–6667. | ||
Tseng D, Volkmer JP, Willingham SB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110(27):11103–11108. | ||
Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–237. | ||
Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66(2):605–612. | ||
De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–286. | ||
Jinushi M, Komohara Y. Tumor-associated macrophages as an emerging target against tumors: creating a new path from bench to bedside. Biochim Biophys Acta. 2015;1855(2):123–130. | ||
Welford AF, Biziato D, Coffelt SB, et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest. 2011;121(5):1969–1973. | ||
Wang-Gillam A, Nywening TM, Sanford DE, et al. Phase IB study of FOLFIRINOX plus PF-04136309 in patients with borderline resectable and locally advanced pancreatic adenocarcinoma (PC). J Clin Oncol. 2015;33 Suppl 3:338. | ||
Demaria S, Ng B, Devitt ML, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58(3):862–870. | ||
Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805(1):53–71. | ||
Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–265. | ||
Ferrara TA, Hodge JW, Gulley JL. Combining radiation and immunotherapy for synergistic antitumor therapy. Curr Opin Mol Ther. 2009;11(1):37–42. | ||
Dewan MZ, Galloway AE, Kawashima N, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15(17):5379–5388. | ||
Wong SH, Sung JJ, Chan FK, et al. Genome-wide association and sequencing studies on colorectal cancer. Semin Cancer Biol. 2013;23(6 Pt B):502–511. | ||
Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19(5):619–625. | ||
Schlicker A, Beran G, Chresta CM, et al. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med Genomics. 2012;5:66. | ||
Pihl E, Hughes ES, McDermott FT, Johnson WR, Katrivessis H. Lung recurrence after curative surgery for colorectal cancer. Dis Colon Rectum. 1987;30(6):417–419. | ||
Young J, Simms LA, Biden KG, et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. Am J Pathol. 2001;159(6):2107–2116. | ||
Bertario L, Russo A, Sala P, et al. Survival of patients with hereditary colorectal cancer: comparison of HNPCC and colorectal cancer in FAP patients with sporadic colorectal cancer. Int J Cancer. 1999;80(2):183–187. | ||
Tamas K, Walenkamp AM, de Vries EG, et al. Rectal and colon cancer: not just a different anatomic site. Cancer Treat Rev. 2015;41(8):671–679. | ||
Loupakis F, Yang D, Yau L, et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J Natl Cancer Inst. 2015;107(3).pii: dju427. | ||
Nagtegaal ID, Marijnen CA, Kranenbarg EK, et al. Local and distant recurrences in rectal cancer patients are predicted by the nonspecific immune response; specific immune response has only a systemic effect – a histopathological and immunohistochemical study. BMC Cancer. 2001;1:7. | ||
Algarra I, Collado A, Garrido F. Protein bound polysaccharide PSK abrogates more efficiently experimental metastases derived from H-2 negative than from H-2 positive fibrosarcoma tumor clones. J Exp Clin Cancer Res. 1997;16(4):373–380. | ||
Sadahiro S, Suzuki T, Maeda Y, et al. Effects of preoperative immunochemoradiotherapy and chemoradiotherapy on immune responses in patients with rectal adenocarcinoma. Anticancer Res. 2010;30(3):993–999. | ||
Kanazawa M, Mori Y, Yoshihara K, et al. Effect of PSK on the maturation of dendritic cells derived from human peripheral blood monocytes. Immunol Lett. 2004;91(2–3):229–238. | ||
Tamas K, Domanska UM, van Dijk TH, et al. CXCR4 and CXCL12 expression in rectal tumors of stage IV patients before and after local radiotherapy and systemic neoadjuvant treatment. Curr Pharm Des. 2015;21(17):2276–2283. | ||
Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–699. | ||
Kreso A, van Galen P, Pedley NM, et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014;20(1):29–36. | ||
Kahn M. Symmetric division versus asymmetric division: a tale of two coactivators. Future Med Chem. 2011;3(14):1745–1763. | ||
Justilien V, Fields AP. Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin Cancer Res. 2015;21(3):505–513. | ||
Bass AJ, Lawrence MS, Brace LE, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 2011;43(10):964–968. | ||
Seshagiri S, Stawiski EW, Durinck S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488(7413):660–664. | ||
Yu J, Wu WK, Li X, et al. Novel recurrently mutated genes and a prognostic mutation signature in colorectal cancer. Gut. 2015;64(4):636–645. | ||
Rubio-Perez C, Tamborero D, Schroeder MP, et al. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 2015;27(3):382–396. | ||
Cheng WC, Chung IF, Chen CY, et al. DriverDB: an exome sequencing database for cancer driver gene identification. Nucleic Acids Res. 2014;42(D1):D1048–D1054. | ||
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. | ||
Tabernero J, Shapiro GI, LoRusso PM, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3(4):406–417. | ||
Baumer S, Baumer N, Appel N, et al. Antibody-mediated delivery of anti-KRAS-siRNA in vivo overcomes therapy resistance in colon cancer. Clin Cancer Res. 2015;21(6):1383–1394. | ||
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. | ||
Hiraki M, Nishimura J, Takahashi H, et al. Concurrent targeting of KRAS and AKT by MiR-4689 is a novel treatment against mutant KRAS colorectal cancer. Mol Ther Nucleic Acids. 2015;4:e231. | ||
Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21(2):121–131. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.