Back to Journals » ImmunoTargets and Therapy » Volume 13
Advances in B Cell Targeting for Treating Muscle-Specific Tyrosine Kinase-Associated Myasthenia Gravis
Authors Hu G ![]() , Zhao X, Wang Y, Zhu X, Sun Z, Yu X, Wang J, Liu Q, Zhang J, Zhang Y, Yang J, Chang T, Ruan Z
, Zhao X, Wang Y, Zhu X, Sun Z, Yu X, Wang J, Liu Q, Zhang J, Zhang Y, Yang J, Chang T, Ruan Z ![]() , Lv J, Gao F
, Lv J, Gao F
Received 19 August 2024
Accepted for publication 30 November 2024
Published 11 December 2024 Volume 2024:13 Pages 707—720
DOI https://doi.org/10.2147/ITT.S492062
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Guanlian Hu,1,2,* Xue Zhao,1,* Yiren Wang,1 Xiaoyan Zhu,1,3 Zhan Sun,1,2 Xiaoxiao Yu,1,2 Jiahui Wang,4 Qian Liu,1 Jing Zhang,1 Yingna Zhang,1 Junhong Yang,4 Ting Chang,5 Zhe Ruan,5 Jie Lv,1 Feng Gao1
1Department of Neuroimmunology, Henan Institute of Medical and Pharmaceutical Sciences, Zhengzhou University, Zhengzhou, People’s Republic of China; 2BGI College, Zhengzhou University, Zhengzhou, People’s Republic of China; 3Department of Neurology, The Second Affiliated Hospital of Zhengzhou University, Zhengzhou, People’s Republic of China; 4Department of Encephalopathy, First Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou, People’s Republic of China; 5Department of Neurology, Second Affiliated Hospital, Air Force Medical University, Xi’an, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Feng Gao; Jie Lv, Department of Neuroimmunology, Henan Institute of Medical and Pharmaceutical Sciences, Zhengzhou University, Zhengzhou, People’s Republic of China, Email [email protected]; [email protected]
Abstract: Myasthenia gravis (MG) is a typical autoimmune disease of the nervous system. It is characterized by skeletal muscle weakness and fatigue due to impaired neuromuscular junction transmission mediated by IgG autoantibodies. Muscle-specific receptor tyrosine kinase-associated MG (MuSK-MG), a rare and severe subtype of MG, is distinguished by the presence of anti-MuSK antibodies; it responds poorly to traditional therapies. Recent research on MuSK-MG treatment has focused on specific targeted therapies. Since B cells play a critical pathogenic role in producing autoantibodies and inflammatory mediators, they are often considered the preferred target for treating MuSK-MG. Currently, various B cell-targeted drugs have been developed to treat MuSK-MG; they have shown good therapeutic effects. This review explores the evolving landscape of B cell-targeted therapies in MuSK-MG, focusing on their mechanisms, efficacy, and safety, and the current limitations associated with their use. We discuss current B cell-targeted therapies aimed at depleting or modulating B cells via both direct and indirect approaches. Furthermore, we focus on novel and promising strategies such as Chimeric Autoantibody Receptor T cell therapy, which explicitly targets MuSK-specific B cells without compromising general humoral immunity. Finally, this review provides an outlook on the potential benefits and limitations of B cell-targeted therapy in developing new therapies for MuSK-MG. We conclude by discussing future research efforts needed to optimize these therapies, expand treatment options, and improve long-term outcomes in MuSK-MG management.
Keywords: MuSK-MG, B cell-targeted therapy, direct targeting, indirect targeting, MuSK-CAR-T, MuSK-CAAR-T
Introduction
Myasthenia gravis (MG) is a disease characterized by acquired neuromuscular junction (NMJ) transmission disorders mediated by autoantibodies, giving rise to skeletal muscle weakness and fatigue.1 The disease can affect skeletal muscles throughout the body, significantly affecting the patient’s daily life. Some patients may experience rapid disease progression within a short period, leading to a myasthenic crisis or even death.2
The global incidence of MG is approximately 0.3–2.8/100,000.2 Acetylcholine receptor (AChR)-specific antibodies could be detected in most patients with MG, with approximately 5–8% having specific antibodies against muscle-specific receptor tyrosine kinases (MuSK) or Low-density lipoprotein receptor-related protein 4 (LRP4) in their serum.3,4 AChR-Ab directly causes disease by cross-linking achRs, complement binding and activation, and by inducing AChR conformational changes or blocking acetylcholine binding.5–7 MuSK-Ab carries out Fab arm exchange to bind to MuSK with functional monovalent, inhibit the dimerization and phosphorylation of MuSK, and affect the aggregation of downstream AChR.7 LRP4-Ab binds to membrane proteins in vivo and blocks Agrin-LRP4 interaction, thereby also inhibiting AChR aggregation in the membrane.8
Due to various autoantibodies that trigger MG, clinical symptoms are highly heterogeneous. MuSK-MG often develops acutely and progresses rapidly within a few weeks. It is clinically more severe than other MG subtypes, with up to 80% of patients with MuSK-MG demonstrating bulbar muscle damage, including dysarthria, dysphagia, and difficulty chewing.3,9,10 In addition, MuSK-MG patients have a higher risk of myasthenic crisis, occasional muscle atrophy, and relapse post-treatment.3
Most patients with MuSK-MG show limited or no response to anticholinesterase drug treatment, which may be harmful.11 Furthermore, patients with MuSK-MG demonstrate poor responsiveness to intravenous immunoglobulin (IVIg), thymectomy, and complement blockade therapy.12–14 The primary treatments include glucocorticoids, azathioprine, plasma exchange, rituximab (RTX) and FcRn-targeted drugs efgartigimod and rozanolixizumab, though corticosteroids and conventional immunosuppressive drugs may not adequately control long-term clinical symptoms and can induce side effects.15–17 Steroids are the standard treatment, but approximately 15% of patients exhibit refractory disease to high-dose steroids. RTX, while effective, leads to general B cell depletion, reduction in total IgG level, and immunosuppression.18–23 An ideal treatment selectively targeting MuSK-specific B cells while preserving normal B cells remains an unmet need in MuSK-MG therapy.
In recent years, immunotherapy targeting B cells has shown promising approaches for targeting MuSK-MG, with several therapies currently in development. This review discusses the latest advancements in B cell-targeted therapies for MuSK-MG, exploring their mechanisms of action, effectiveness, safety profiles, and limitations.
Role of B Cells in MuSK-MG
MuSK is an important neuromuscular junction protein crucial in forming and maintaining NMJs. It is a 100-kDa single-channel postsynaptic transmembrane receptor tyrosine kinase composed of three extracellular N-terminal immunoglobin (Ig)-like domains, a curled-like domain (Fz domain), and an intracellular kinase domain (Figure 1a).24–26 Antigen epitope mapping studies indicate that MuSK-specific antibodies mainly bind to Ig1 and Ig2, as well as to the Fz domain in some patients. However, the Ig1-like domain is the main target.27–30 MuSK interacts with some proteins through the extracellular domain to enhance signal transduction in NMJs (Figure 1b).
 |
Figure 1 The structure of MuSK and the pathogenic mechanism of MuSK-Ab. (a) The structure of the MuSK protein is made up of an extracellular domain, an intracellular kinase, and a transmembrane domain. The extracellular domain contains three Ig-like domains (Ig1-3) and a Frizzled-like domain (Fz), which is a cysteine-rich domain (CRD). (b) LRP4 simultaneously engages both MuSK and Agrin, thereby facilitating their direct interaction. Subsequently, MuSK dimerizes and activates its intracellular kinase domain upon co-stimulation of LRP4 and Agrin. Activation of MuSK further recruits and stimulates Dok7 phosphorylation, which stimulates signal transduction of the downstream AChR cluster, promoting the aggregation of AChR. (c) When MuSK-specific antibodies are present in vivo, functional monovalent MuSK-Ab binds to the extracellular domain of MuSK, thereby blocking the interaction of MuSK with LRP4 and Agrin, inhibiting the dimerization and phosphorylation of MuSK, inhibiting the aggregation of downstream AChRs, and leading to neuromuscular excitation transmission disorders. (Image created with BioRender.com). |
B cells are essential for MuSK-MG pathogenesis, mainly by producing antibodies against MuSK, which leads to the destruction and dysfunction of NMJs. The secretion of MuSK-specific antibodies is primarily attributed to short-lived plasma cells, with the majority belonging to the IgG4 subclass.31 Due to its unique hinge-region structural characteristics, IgG4 has functional monovalent and bispecific non-inflammatory properties and cannot participate in the cross-linking and internalization of target antigens.14,32,33 Furthermore, most MuSK-IgG4 antibodies undergo Fab-arm exchange, producing bispecific antibodies that bind to the MuSK extracellular domain in a functional monovalent manner, thereby blocking low-density lipoprotein receptor-related protein 4 (LRP4)-MuSK interactions, preventing MuSK dimerization and phosphorylation, inhibiting AChR aggregation, and leading to neuromuscular excitation transmission disorders (Figure 1c).11,27,34 Based on previous research, MuSK-specific antibodies contribute to the occurrence and development of MG by depleting MuSK molecules that are essential for muscle function. In recent years, most of the targeted therapies for MuSK-MG have focused on the depletion of B cells to reduce the levels of, or eliminate, MuSK-specific autoantibodies.
In addition, B cells can also play a role in antigen presentation during the autoimmune response. The B cell receptors (BCRs) directly bind to antigens and form antigen peptide-MHC molecule complexes with major histocompatibility complex (MHC) class II molecules, which are then presented to CD4+ T cells.35 Moreover, B cells can express so-called co-stimulators, such as CD40, CD80, and CD86, which further lead to the activation of pro-inflammatory T cells and thus regulate the immune response.36
Since B cells play a critical pathogenic role in producing autoantibodies and inflammatory mediators, they are the preferred target for treating MuSK-MG. Targeting B cells and the antibodies they produce may be one of the important strategies for targeted therapy of MuSK-MG. B cell-targeted therapy includes direct targeting that consumes or inhibits different B cell subpopulations, indirect targeting that inhibits B cell maturation and differentiation-related cytokines or immune cells, chimeric antigen receptor-T cell therapy (CAR-T) and reverse targeting using chimeric autoantibody receptor (CAAR) T-cell therapy (Table 1).
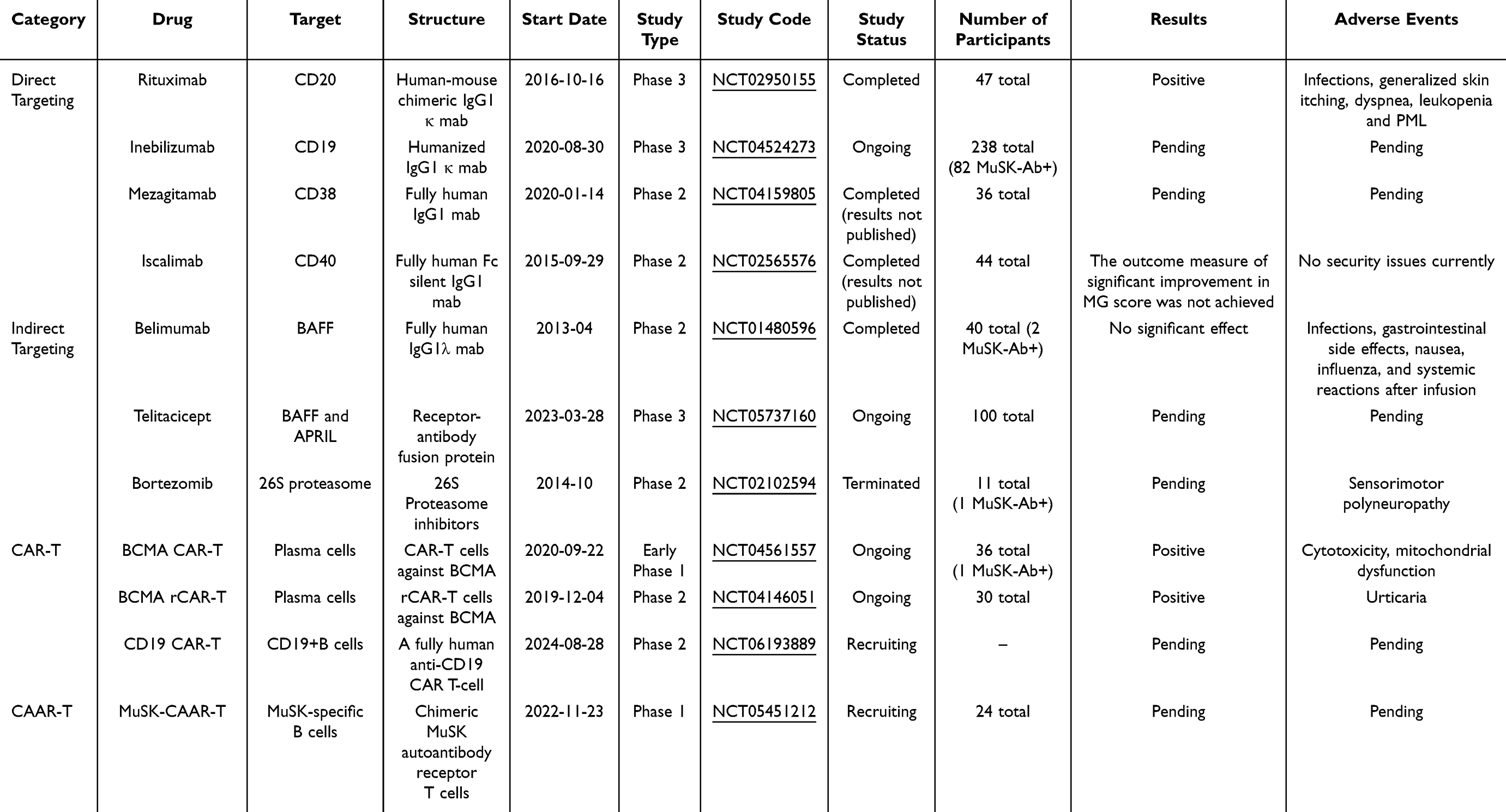 |
Table 1 Major B Cell Targeting Drugs for MuSK-MG Treatment |
Direct Targeting
Multiple CD antigens where located on the surface of B cells, which distinguish different B cell subsets and play crucial roles in their maturation, activation, differentiation, and survival. These CD molecules, whose expression can vary or become activated in pathological conditions, are valuable features on the cell surface for targeted B cell therapy. Direct targeted therapy achieves B cell depletion by specifically targeting surface-related antigens such as CD19 and CD40 (Figure 2).
 |
Figure 2 Mechanism of action of direct B cell-targeting drugs. The mechanism of action of rituximab includes antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and target cell apoptosis. Inebilizumab consumes B cells expressing CD19 through ADCC. Mezagitamab kills B cells expressing CD38 through ADCC. Iscalimab can block the interaction of CD40-CD40L, thereby inhibiting the activation of CD40-positive cells. (Image created with BioRender.com). |
CD20 (Rituximab)
RTX is a human-mouse chimeric monoclonal antibody (mAb) that can bind specifically to the transmembrane antigen CD20. RTX has gained widespread acceptance and application in the past few years.37,38 CD20 is a glycoprotein expressed on pro-B cells, naive B cells, and all mature B cells, and is critical for B cell proliferation and differentiation.39 RTX effectively depletes CD20-positive B cells, including mature B cells and memory B cells, and this reduction occurs through antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and target cell apoptosis.40,41 However, pro-B and plasma cells in the bone marrow and secondary lymphoid organs were largely unaffected.42,43 This effect lasts approximately six months until circulating B cells are replenished.38 Because AChR-Ab is mainly produced by long-lived plasma cells (which do not express CD20), RTX treatment is only effective in a subset of patients with AChR-Ab + MG.44 In contrast, MuSK-Ab is mainly produced by short-lived plasma cells. Most patients with MuSK-MG respond well to RTX, achieving a remission rate approaching 100%. A 2017 blinded, multicenter, prospective evaluation by Professor Burns et al showed that 58% of patients with MuSK-MG treated with RTX met the primary clinical endpoint, compared to 16% of the control group.18 In addition, a 2020 study by Bartoccioni et al revealed reduced titers of MuSK-IgG4 antibodies in patients and continued clinical improvement with RTX treatment.21 Experience with rituximab in the treatment of myasthenia gravis in adolescents is limited to case reports and generally works well in AChR-MG and MuSK-MG, as well as seronegative MG.45,46 In a multicenter retrospective study of 27 pediatric MG patients treated with rituximab, all patients showed improvement and no adverse events occurred during treatment.47 There is no clear consensus on the appropriate dosing regimen of rituximab in patients with MG, and although most studies have been conducted according to classical treatment guidelines (375 mg/m2 / 4 weeks or 1 g / 2 biweekly dosing), in recent years, an increasing number of authors suggest that lower doses of rituximab can achieve the same clinical results with better safety and cost-effectiveness.48–50 Despite the remarkable efficacy of RTX treatment in MuSK-MG, some patients usually relapse 1–3.5 years post-RTX treatment, and MuSK-specific B cells persist despite repeated RTX treatment. Minor patients do not respond to RTX treatment, which may be due to the existence of low expression of CD20-CD27+ B cells in peripheral blood of these patients, and the continuous production of anti-musk antibodies.19–22 The broad depletion of CD20-expressing B cells by RTX poses infection and secondary immunodeficiency risks.23 Studies have reported severe infection, generalized skin itching, dyspnea, leukopenia, or delayed progressive multifocal leukoencephalopathy (PML) after RTX treatment in some MG patients.19,50–55
CD19 (Inebilizumab)
Inebilizumab is a humanized, fucosylated IgG1 κ mAb that depletes CD19-positive B cells via the ADCC mechanism.56,57 CD19 is distributed in early pro-B cells, most plasma cells in peripheral circulation, and approximately half of the plasma cells in the central immune organs.21 This makes CD19 a potential target for depleting CD19-positive B cells, a strategy exemplified by inebilizumab. Approved in June 2020 for treating aquaporin-4 antibody-positive NMOSD, the safety and efficacy of inebilizumab are currently under evaluation for IgG4 disease.56,58 Trials involving NMOSD patients revealed that inebilizumab is well tolerated, with mild to moderate side effects consistent with other B cell-depleting drugs.56 Amgen initiated a Phase 3 study in August 2020 (NCT04524273), employing a randomized, double-masked, multicenter, placebo-controlled design to evaluate the effectiveness and safety of inebilizumab in adult patients with MG (including 42 MuSK-MG). This study is anticipated to be completed in 2027 and will clarify the effectiveness and safety of inebilizumab.
CD38 (Mezagitamab)
Mezagitamab (TAK-079) is a fully human IgG1 mAb with a high affinity for cells that express CD38, such as plasma cells and natural killer (NK) cells. It induces cell death in B cells expressing CD38 through ADCC. CD38 is a transmembrane glycoprotein with extracellular enzyme activity expressed by leukocyte subsets, with the highest density on plasma cells and plasmablasts.59 In 2020, Takeda Pharmaceuticals initiated a first-in-human Phase 1 trial (NCT02219256), which was randomized, double-masked, placebo-controlled, and single-dose, involving healthy adult subjects. It showed that TAK-079 was well tolerated, and subcutaneous injection resulted in sustained decreases in the plasmablast and NK cell counts.60 A Phase 2 randomized, placebo-controlled study (NCT04159805) by Takeda Pharmaceuticals assessed the safety, tolerability, and efficacy of TAK-079 in patients with generalized MG (including MuSK-MG). This study was completed by July 2022, but the results have not yet been published. The efficacy and safety of TAK-079 specifically still require clarification.
CD40 (Iscalimab)
Iscalimab (CFZ533) is a fully human IgG1 mAb that lacks the Fc region, blocking the CD40 signaling pathway. This mechanism prevents the activation of CD40-positive cells but does not cause exhaustion. CD40 is expressed on lymphocytes and antigen-presenting cells, while the CD40 ligand (CD40L), also known as CD154, is mainly expressed by activated CD4+ T cells.61,62 The interaction between CD40 and CD40L is essential for isotype conversion, germinal center formation, the development of memory B cells, and antibody production.63 Novartis Pharmaceuticals conducted a multicenter, randomized, double-masked, placebo-controlled phase 2 clinical trial (NCT02565576) focusing on seropositive generalized MG (including MuSK-MG), completed in 2019. Preliminary results, yet unpublished, showed no safety concerns but did not achieve a significant improvement in MG scores. Further research through large-scale and long-term clinical trials is crucial to understand the effectiveness of iscalimab fully.
Indirect Targeting
Cytokines such as B lymphocyte stimulating factor (BAFF, also known as BLyS and TNFSF13b), proliferation-inducing ligand (APRIL, TNFSF13), and their receptors play a crucial role in the growth, development, maturation, and homeostasis maintenance of B cells. Indirectly targeted therapies reduce the counts of, or eliminate, B cells and alleviate clinical symptoms in patients by blocking the functions of cytokines, such as BAFF and APRIL, or targeting plasma cell proteasome inhibitors (Figure 3).
 |
Figure 3 Mechanism of action of indirect B cell-targeting drugs. Belimumab affects B cell survival by blocking B lymphocyte stimulating factor (BAFF)-BAFF receptor (BAFFR) signaling by binding to soluble BAFF. Telitacipep can simultaneously target BAFF and April, multi-stage inhibiting the maturation and differentiation of B cells. Bortezomib binds to 26S proteasome to inhibit its enzymatic activity and leads to reducing the degradation of intracellular proteins, affecting the activation of T-cells, and promoting plasma cell apoptosis. (Image created with BioRender.com). |
B Cell Activating Factor BAFF (Belimumab)
Belimumab is a recombinant human IgG1λ mAb that neutralizes the soluble form of BAFF, a key B cell activating factor. It includes membrane-bound and secreted BAFF variants produced by non-B cells, such as monocytes, dendritic cells, and macrophages.64 BAFF binds to three different receptors: (1) BAFF receptor (BAFFR, also known as BR3 and TNFRSF13C) is mainly expressed on mature B cells; (2) B cell maturation antigen (BCMA, TNFRSF17) is only on plasma cells; and (3) transmembrane activator, calcium regulator, and cyclophilin ligand interactor (TACI, TNFRSF13B) are present on activated B cells, marginal zone B cells, switched memory B cells, and plasma cells.65–69 The dysregulated expression of BAFF affects the activation, proliferation, survival, and immunoglobulin secretion of B cells, thereby affecting the development of autoimmune diseases.70 BAFFR mediates BAFF survival signals. When BAFF binds to BAFFR, it activates the NF-κB pathway, leading to the transcription of the anti-apoptotic factor Bcl-2, whose expression is essential for the survival of B cells as they transition from immature to mature stages.71,72 Belimumab is the sole biological drug approved by the FDA to treat systemic lupus erythematosus (SLE) by targeting B cells. It blocks BAFF-BAFFR signaling by binding to soluble BAFF and promotes apoptosis of B cells.73 In a multicenter phase 3 trial, Belimumab demonstrated modest efficacy in patients with SLE.74,75 Whereas, in a phase 2 placebo-controlled, multicenter, double-masked study (NCT01480596), the patients with generalized MG who were receiving standard treatment did not reach the primary outcome, with no significant differences in Activities of Daily Living and Quantitative Myasthenia Gravis scores at week 24 compared with the placebo group. This effect was not observed significantly in patients with MuSK-MG.76
Inhibition of B Cell Proliferation, Differentiation, and Activation (Telitacicept)
Telitacicept is a TACI-Fc fusion protein, composed of the extracellular specific soluble part of TACI and the Fc part of human IgG1. It can simultaneously target BAFF and APRIL to inhibit the maturation and differentiation of B cells at multiple stages.77,78 BAFF and APRIL are two trimeric members of the tumor necrosis factor (TNF) family that are expressed in varieties of cell types.65 They are the key to stable B cells and the humoral immune proteins B cells. They combine with different receptors on B cells and plasma cells, and BAFF and APRIL both combine with TACI and BCMA. The difference is that BAFF also combines with BAFFR, but APRIL does not.65,66 BAFF is essential for the survival, differentiation, and maturation of B cells, while APRIL has a greater impact on regulating the function and survival of long-lived plasma cells, thereby affecting the production of antibodies.77 Telitacicept can recognize and bind to BAFF and APRIL, blocking their interactions with TACI, BCMA, and BAFFR, thereby inhibiting B cell proliferation and T cell maturation.77 In 2024, data from a multicenter, randomized, open-label phase 2 clinical study by Professor Fang Jianmin et al77 and Professor Li Zhijun et al79 showed that telitacicept, as a dual inhibitor of BAFF and APRIL, not only showed significant efficacy and was able to improve patients’ clinical symptoms, but also showed good tolerability and safety. Most adverse events were classified as mild or moderate, with no serious adverse reactions. The results also suggest that telitacicept may have long-term efficacy and maintain therapeutic effects even after treatment.77,79 A multicenter, randomized, double-masked, placebo-controlled phase 3 study of telitacicept (NCT05737160) is underway and is expected to be completed in 2027.
Proteasome Inhibitors (Bortezomib)
Bortezomib is a novel proteasome inhibitor that exerts its immune effects by affecting the survival of plasma cells. Its primary mechanism of action is to inhibit the normal function of proteasome responsible for protein degradation in the nucleus, thereby causing protein accumulation in the cell, leading to cell cycle arrest and apoptosis.80,81 The inhibition of proteasome function by bortezomib is particularly detrimental to the normal survival of cells with high protein turnover. Plasma cells have a high protein turnover due to the continuous release of antibodies, and they are highly sensitive to proteasome inhibitors, which can lead to the accumulation of immunoglobulin chains, which then leads to plasma cell apoptosis.82 Bortezomib is approved for the treatment of multiple myeloma and is a potential treatment option for autoimmune diseases that are resistant to various standard treatments, including generalized MG.82 A patient with MuSK-MG who had a poor response to conventional immunotherapy and RTX experienced rapid and sustained improvement after treatment with bortezomib.83 In 2014, a nonrandomized clinical trial (NCT02102594) evaluating bortezomib for antibody-mediated autoimmune diseases, including MuSK-MG, was discontinued due to recruitment difficulties; therefore, the efficacy of bortezomib in MG needs further study.84 The drug is associated with neurotoxicity and subsequent disabling peripheral neuropathy.85–87 This neurotoxicity is dose-dependent, but the risk of neuropathy can be reduced by adjusting the dose and the mode of administration.88
CAR-T Cell Therapy
Chimeric antigen receptors (chimeric antigen receptor, CAR) are synthetic proteins designed to reprogram T cells, The CAR structure consists of an extracellular antigen-recognition component (single-chain variable fragment), a transmembrane region, an intracellular costimulatory domain (typically 4–1BB or CD28), and a CD3 intracellular signaling domain (Figure 4a).89,90 CAR-T therapy modifies T cells to specifically recognize and eliminate cells expressing specific antigens (Figure 4a). In recent years, CAR-T therapy has made significant progress in the field of hematological malignancies, and has been gradually applied to the treatment of autoimmune diseases.91 An earlier phase 1 study (NCT04561557) showed that two patients (one AChR-MG and one MuSk-MG) with highly relapsed and refractory MG showed favorable safety and sustained clinical improvement over 18 months after treatment with BCMA-targeting CAR-T cells.92
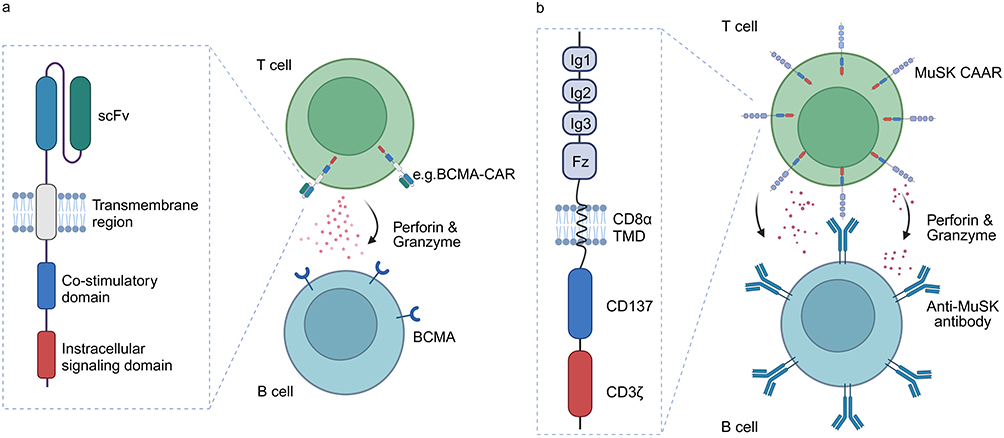 |
Figure 4 The structure and mechanism of CAR-T and MuSK-CAAR-T. (a) The CAR is composed of an extracellular antigen-recognition component (scFv), a transmembrane region, an intracellular costimulatory domain, and an intracellular signaling domain. CAR-T cells can specifically recognize and produce cytotoxic perforin/granzyme to eliminate B cells expressing specific antigens. (b) MuSK-CAAR is composed of the extracellular domain of the MuSK protein, the CD8 transmembrane domain, and the CD137-CD3ζ intracellular stimulating and signal transduction domain structure. T cells expressing MuSK-CAAR specifically recognize B cells expressing MuSK-specific antibodies or BCR and elicit cytotoxic effects against them. (Image created with BioRender.com). |
CAT-T therapy has great potential in autoimmune diseases, but the associated toxicity and the need for lymphocyte depletion limit its use in patients with autoimmune diseases. To improve the safety of CAR-T therapy, Granit et al93 used an RNA (rCAR-T) rather than a DNA approach to engineer T cells to target BCMA on the plasma cell surface and attempted to use rCAR-T to treat patients with MG. Unlike conventional DNA-engineered CAR T cells, their RNA-engineered counterparts do not persist for long and do not require demanding lymphocyte depletion protocols. In a prospective, multicenter, open-label, nonrandomized phase 1b/2a study of rCAR-T in MG (NCT04146051), Descartes-08 was reported to be safe and well tolerated, with clinically meaningful improvements observed at up to 9 months of follow-up. There were no adverse events (eg, cytokine release syndrome) that were similar to those seen with DNA CAR T cells.93
CAAR-T Cell Therapy
Patients with acquired autoimmune diseases harbor antigen-specific autoreactive B cells in their bodies. These B cells express BCRs on their surfaces that are capable of binding specifically to their target antigens, a feature absent in normal B cells. Studies have shown that B cells can rapidly internalize their cognate antigens upon contact. This specificity of BCR antigens forms the basis of reverse BCR-targeted therapy, known as BAR (BCR antigen for reverse targeting). This approach enables targeted recognition and elimination of B cells expressing specific BCRs. Recently, BAR has been successfully applied to the precise elimination of antigen-specific B lymphocytes associated with several autoimmune diseases.
CAAR-T cell therapy is a further refinement of CAR-T therapy, which is specifically designed to target B cells that produce pathogenic antibodies. The recombinant autoantibody receptor of CAAR-T cells replaces the single-chain variable fragment (scFv) antigen recognition domain of CAR-T cells. This modification allows CAAR-T cells to precisely target the specific BCR on the surface of B cells, thereby selectively eliminating antigen-specific B cells without affecting normal B cells (Figure 4b).94,95 In model animals, reinfusion of CAAR-T cells can significantly reduce the titer of autoantibodies without affecting other B cells. This approach aims to avoid the chronic immunosuppression associated with current treatments and potentially offer therapeutic efficacy comparable to or superior to CAR-T therapy. In 2023, Sangwook Oh et al designed MuSK chimeric autoantibody receptor (MuSK-CAAR)-T cells incorporating a CD137-CD3ζ signaling domain to precisely target B cells that express anti-MuSK autoantibodies (Figure 4b). Their results showed that MuSK-CAAR-T cell treatment in the mouse model of MuSK-MG reduced the anti-MuSK antibody levels but did not affect the overall B cell counts or IgG levels, reflecting the depletion of MuSK-specific B cells.95 Furthermore, recruitment is currently underway for a phase 1, open-label study (NCT05451212) initiated by Cabaletta Bio in November 2022 to evaluate the safety of various dosing regimens for the treatment of MuSK-MG. This study is expected to be completed by 2028.
Outlook: Advantages, Limitations, and Challenges Associated with B Cell-Targeted Therapy for MuSK-MG
MuSK-MG is a serious and intractable NMJ disease. Establishing strategies for the precise and effective treatment of MuSK-MG, especially for patients with refractory MG is urgently needed. Compared with traditional immunotherapy methods, B cell-targeted therapy is expected to improve patients’ clinical symptoms while minimizing side effects and enhancing patient compliance more precisely and rapidly.
Direct B cell-targeted therapy offers several advantages. For example, monoclonal antibodies exhibit high specificity and provide lasting and potent therapeutic effects by directly eliminating B cells without affecting other immune cells. In addition, they are associated with fewer off-target effects. However, one of the limitations associated with their use is their inability to distinguish between normal and autoreactive B cells, which potentially compromises normal humoral immunity. Furthermore, their effectiveness may vary as they cannot comprehensively eliminate autoreactive B cells across different developmental stages. Indirect B cell-targeted therapy functions mainly through immune regulation, lacking direct B cell elimination capabilities. In a phase 2 clinical study, telitacicept has shown significant efficacy and good safety by inhibiting B cell maturation and differentiation at multiple stages. However, it does not completely eliminate autoreactive B and plasma cells.
CAR-T therapy has shown the light of cure in the treatment of MG. However, the neurotoxicity, cytokine release syndrome (CRS), hypogammaglobulinemia and other related toxicities of DNA CAR-T therapy, and the need for lymphocyte depletion have limited its application in autoimmune diseases. To improve the safety of CAR-T therapy, Granit et al93 designed rCAR-T, which is the expression of CAR through RNA engineering. Because the CAR-encoding mRNA does not replicate with activated and proliferating rCAR-T cells, amplification of the CAR signal is avoided, and the CAR+ load is dose-dependent and decreases over time, enabling more precise pharmacokinetic control and reducing the potential hematologic toxicity and tumor risk of genomic integration. In addition, since this method uses ex vivo T cell proliferation, there is also no need for depletion of lymphocytes to induce a specific cytokine milieu prior to administration. At present, CAR-T cell therapy provides a potential revolutionary treatment for immune-mediated nervous system diseases, bringing new hope for patients who fail to respond to traditional treatment. Although more clinical trials are needed to verify its safety and efficacy, and further research is needed to optimize the treatment regimen and reduce the cost of treatment. However, the long-term remission potential of CAR-T cell therapy and the advantages of individualized treatment indicate that it may become a powerful tool for the treatment of such diseases.
CAAR-T therapy represents a cutting-edge precision medicine approach. It selectively targets immune cells expressing specific autoantibodies, such as those seen in MuSK-MG, without inducing broad immunosuppression. Preclinical studies in mouse models have shown that CAAR-T cells selectively eliminate MuSK-specific B cells, demonstrating potential for use as targeted therapies for treating MuSK-MG. While both MUSK-CAART and CAR-T target the BCR complex on plasma cells and kill plasma cells, MUSK-CAART chimeric autoantigen MuSK receptors that bind specifically to the variable region of the BCR and kill only plasma cells that produce antibodies against MuSK. In contrast to CAR-T (which kills all plasma cells), MUSK-CAART chimeric autoantigen MuSK receptors bind specifically to the variable region of the BCR and kill only plasma cells that produce antibodies against MuSK. It has obvious advantages. However, the circulating autoantibodies could significantly affect the treatment of CAAR-T, and further studies are still needed to optimize the treatment plan. Although their use comes with a few challenges, including the requirement for demanding and expensive procedures for the in vitro engineering and expansion of patient-specific autologous T cells; this limits the widespread clinical application of CAAR-T cells. However, it has great potential research value in the treatment of autoimmune diseases.
In summary, many promising B cell-targeted therapies for MuSK-MG have encountered setbacks during preclinical and clinical development stages. These failures are primarily due to weak immunosuppressive reactions, nonspecific immunogenicity, or security issues. Although currently used targeted therapies in clinical settings are effective, they are also associated with limitations. Thus, exploring and identifying more precise approaches to improve the efficacy and safety of these critical therapies is needed. Strategies such as targeting only antigen-specific B cells, combination therapies addressing multiple targets, antibody-drug conjugates, and dual-targeted drugs that circumvent the need for autologous T cell modification hold promise for developing disease-specific targeted therapies.
Acknowledgments
We are grateful for the support provided by the Henan Engineering Technology Research Center for Accurate Diagnosis Neuroimmunity and BGI College, Zhengzhou University; and the Key Laboratory of Pharmacology for Liver Diseases of Henan Province for the smooth implementation of this study. We want to thank Editage (www.editage.cn) for English language polish.
Disclosure
The author states that this article was published in the research and /or financial support. This study was supported by projects of the Basic Research Fund of Henan Institute of Medical and Pharmacological Sciences (grant numbers: 2022BP0116 and 2023BP0201); Henan Province scientific and technological research grant (grant numbers: 232102310408 and 232102311196); the Special Project of Scientific Research for advancing “Double First-class” Traditional Chinese Medicine (HSRP-DFCTCM-2023-1-27); Key R&D Plan of Shanxi Province (grant number: 2021ZDLSF02-01) and the Key Scientific and Technological Project of Henan Province (No. 242102310369). The authors declare there are no other conflicts of interest in this work.
References
1. Gilhus NE, Longo DL. Myasthenia gravis. N Engl J Med. 2016;375(26):2570–2581. doi:10.1056/NEJMra1602678
2. Chen J, Tian D-C, Zhang C, et al. Incidence, mortality, and economic burden of myasthenia gravis in China: a nationwide population-based study. Lancet Reg Health West Pac. 2020;5:100063. doi:10.1016/j.lanwpc.2020.100063
3. Rodolico C, Bonanno C, Toscano A, Vita G. MuSK-associated myasthenia gravis: clinical features and management. Front Neurol. 2020;11. doi:10.3389/fneur.2020.00660
4. Koneczny I, Herbst R. Myasthenia gravis: pathogenic effects of autoantibodies on neuromuscular architecture. Cells. 2019;8(7):671. doi:10.3390/cells8070671
5. Gilhus NE. Myasthenia and the neuromuscular junction. Curr Opin Neurol. 2012;25(5):523–529. doi:10.1097/WCO.0b013e3283572588
6. Querol L, Illa I. Myasthenia gravis and the neuromuscular junction. Curr Opin Neurol. 2013;26(5):459–465. doi:10.1097/WCO.0b013e328364c079
7. Verschuuren JJ, Huijbers MG, Plomp JJ, et al. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun Rev. 2013;12(9):918–923. doi:10.1016/j.autrev.2013.03.001
8. Shen C, Lu Y, Zhang B, et al. Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J Clin Invest. 2013;123(12):5190–5202. doi:10.1172/jci66039
9. Evoli A, Alboini PE, Iorio R, Damato V, Bartoccioni E. Pattern of ocular involvement in myasthenia gravis with MuSK antibodies. J Neurol Neurosurg. 2017;88(9):761–763. doi:10.1136/jnnp-2017-315782
10. Evoli A, Alboini PE, Damato V, et al. Myasthenia gravis with antibodies to MuSK: an update. Ann N Y Acad Sci. 2018;1412(1):82–89. doi:10.1111/nyas.13518
11. Koneczny I, Stevens JAA, De Rosa A, et al. IgG4 autoantibodies against muscle-specific kinase undergo Fab-arm exchange in myasthenia gravis patients. J Autoimmun. 2017;77:104–115. doi:10.1016/j.jaut.2016.11.005
12. Borges LS, Richman DP. Muscle-specific kinase myasthenia gravis. Front Immunol. 2020;11:707. doi:10.3389/fimmu.2020.00707
13. Clifford KM, Hobson-Webb LD, Benatar M, et al. Thymectomy may not be associated with clinical improvement in MuSK myasthenia gravis. Muscle Nerve. 2019;59(4):404–410. doi:10.1002/mus.26404
14. Vakrakou AG, Karachaliou E, Chroni E, et al. Immunotherapies in MuSK-positive myasthenia gravis; an IgG4 antibody-mediated disease. Front Immunol. 2023;14:1212757. doi:10.3389/fimmu.2023.1212757
15. Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14(10):1023–1036. doi:10.1016/S1474-4422(15)00145-3
16. Howard JF, Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2021;20(7):526–536. doi:10.1016/s1474-4422(21)00159-9
17. Bril V, Drużdż A, Grosskreutz J, et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): a randomised, double-blind, placebo-controlled, adaptive phase 3 study. Lancet Neurol. 2023;22(5):383–394. doi:10.1016/s1474-4422(23)00077-7
18. Hehir MK, Hobson-Webb LD, Benatar M, et al. Rituximab as treatment for anti-MuSK myasthenia gravis. Neurology. 2017;89(10):1069–1077. doi:10.1212/wnl.0000000000004341
19. Díaz-Manera J, Martínez-Hernández E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. 2012;78:189–193. doi:10.1212/WNL.0b013e3182407982
20. Cortes-Vicente E, Rojas-Garcia R, Diaz-Manera J, et al. The impact of rituximab infusion protocol on the long-term outcome in anti-MuSK myasthenia gravis. Ann Clin Transl Neurol. 2018;5(6):710–716. doi:10.1002/acn3.564
21. Marino M, Basile U, Spagni G, et al. Long-lasting rituximab-induced reduction of specific—but not total—IgG4 in MuSK-positive myasthenia gravis. Front Immunol. 2020;11:613. doi:10.3389/fimmu.2020.00613
22. Fichtner ML, Hoehn KB, Ford EE, et al. Reemergence of pathogenic, autoantibody-producing B cell clones in myasthenia gravis following B cell depletion therapy. Acta Neuropathol Commun. 2022;10(1):154. doi:10.1186/s40478-022-01454-0
23. Szepanowski F, Warnke C, Meyer Zu Hörste G, et al. Secondary immunodeficiency and risk of infection following immune therapies in neurology. CNS Drugs. 2021;35(11):1173–1188. doi:10.1007/s40263-021-00863-4
24. Till JH, Becerra M, Watty A, et al. Crystal structure of the MuSK tyrosine kinase: insights into receptor autoregulation. Structure. 2002;10(9):1187–1196. doi:10.1016/S0969-2126(02)00814-6
25. Stiegler AL, Burden SJ, Hubbard SR. Crystal structure of the frizzled-like cysteine-rich domain of the receptor tyrosine kinase MuSK. J Mol Biol. 2009;393(1):1–9. doi:10.1016/j.jmb.2009.07.091
26. Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S. Myasthenia gravis — autoantibody characteristics and their implications for therapy. Nat Rev Neurol. 2016;12(5):259–268. doi:10.1038/nrneurol.2016.44
27. Huijbers MG, Vergoossen DL, Fillié-Grijpma YE, et al. MuSK myasthenia gravis monoclonal antibodies. Neurol Neuroimmunol Neuroinflamm. 2019;6(3):e547. doi:10.1212/nxi.0000000000000547
28. Takata K, Stathopoulos P, Cao M, et al. Characterization of pathogenic monoclonal autoantibodies derived from muscle-specific kinase myasthenia gravis patients. JCI Insight. 2019;4(12):e127167. doi:10.1172/jci.insight.127167
29. Huijbers MG, Vink AFD, Niks EH, et al. Longitudinal epitope mapping in MuSK myasthenia gravis: implications for disease severity. J Neuroimmunol. 2016;291:82–88. doi:10.1016/j.jneuroim.2015.12.016
30. Lim JL, Augustinus R, Plomp JJ, et al. Development and characterization of agonistic antibodies targeting the Ig-like 1 domain of MuSK. Sci Rep. 2023;13(1):7478. doi:10.1038/s41598-023-32641-1
31. McConville J, Farrugia ME, Beeson D, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55(4):580–584. doi:10.1002/ana.20061
32. Lighaam LC, Rispens T. The Immunobiology of Immunoglobulin G4. Semin Liver Dis. 2016;36(3):200–215. doi:10.1055/s-0036-1584322
33. van der Neut Kolfschoten M, Schuurman J, Losen M, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic fab arm exchange. Science. 2007;317(5844):1554–1557. doi:10.1126/science.1144603
34. Vergoossen DLE, Augustinus R, Huijbers MG. MuSK antibodies, lessons learned from poly- and monoclonality. J Autoimmun. 2020;112:102488. doi:10.1016/j.jaut.2020.102488
35. Kohler S, Keil TOP, Swierzy M, et al. Disturbed B cell subpopulations and increased plasma cells in myasthenia gravis patients. J Neuroimmunol. 2013;264(1–2):114–119. doi:10.1016/j.jneuroim.2013.09.006
36. Rivera A, Chen -C-C, Ron N, Dougherty JP, Ron Y. Role of B cells as antigen-presenting cells in vivo revisited: antigen-specific B cells are essential for T cell expansion in lymph nodes and for systemic T cell responses to low antigen concentrations. Int Immunol. 2001;13(12):1583–1593. doi:10.1093/intimm/13.12.1583
37. Kaegi C, Wuest B, Schreiner J, et al. Systematic review of safety and efficacy of rituximab in treating immune-mediated disorders. Front Immunol. 2019;10:1990. doi:10.3389/fimmu.2019.01990
38. Stathopoulos P, Dalakas MC. Evolution of anti-B cell therapeutics in autoimmune neurological diseases. Neurotherapeutics. 2022;19(3):691–710. doi:10.1007/s13311-022-01196-w
39. Pavlasova G, Mraz M. The regulation and function of CD20: an “enigma” of B-cell biology and targeted therapy. Haematologica. 2020;105(6):1494–1506. doi:10.3324/haematol.2019.243543
40. Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22(47):7359–7368. doi:10.1038/sj.onc.1206939
41. Cerny T, Borisch B, Introna M, Johnson P, Rose AL. Mechanism of action of rituximab. Anticancer Drugs. 2002;13(Suppl 2):S3–10. doi:10.1097/00001813-200211002-00002
42. Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nat Clin Pract Neuro. 2008;4(10):557–567. doi:10.1038/ncpneuro0901
43. Leandro MJ. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res Ther. 2013;15(Suppl 1):S3. doi:10.1186/ar3908
44. Di Stefano V, Lupica A, Rispoli MG, Di Muzio A, Brighina F, Rodolico C. Rituximab in AChR subtype of myasthenia gravis: systematic review. J Neurol Neurosurg Psychiatry. 2020;91(4):392–395. doi:10.1136/jnnp-2019-322606
45. Govindarajan R, Iyadurai SJ, Connolly A, Zaidman C. Selective response to rituximab in a young child with MuSK-associated myasthenia gravis. Neuromuscul Disord. 2015;25(8):651–652. doi:10.1016/j.nmd.2015.03.014
46. Koul R, Al Futaisi A, Abdwani R. Rituximab in severe seronegative juvenile myasthenia gravis: review of the literature. Pediatr Neurol. 2012;47(3):209–212. doi:10.1016/j.pediatrneurol.2012.05.017
47. Molimard A, Gitiaux C, Barnerias C, et al. Rituximab therapy in the treatment of juvenile myasthenia gravis: the French experience. Neurology. 2022;98(23):e2368–e2376. doi:10.1212/wnl.0000000000200288
48. Tandan R, Hehir MK 2nd, Waheed W, Howard DB. Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve. 2017;56(2):185–196. doi:10.1002/mus.25597
49. Li T, Zhang GQ, Li Y, et al. Efficacy and safety of different dosages of rituximab for refractory generalized AChR myasthenia gravis: a meta-analysis. J Clin Neurosci. 2021;85:6–12. doi:10.1016/j.jocn.2020.11.043
50. Topakian R, Zimprich F, Iglseder S, et al. High efficacy of rituximab for myasthenia gravis: a comprehensive nationwide study in Austria. J Neurol. 2019;266(3):699–706. doi:10.1007/s00415-019-09191-6
51. Keung B, Robeson KR, DiCapua DB, et al. Long-term benefit of rituximab in MuSK autoantibody myasthenia gravis patients. J Neurol Neurosurg Psychiatry. 2013;84(12):1407–1409. doi:10.1136/jnnp-2012-303664
52. Nowak RJ, Dicapua DB, Zebardast N, Goldstein JM. Response of patients with refractory myasthenia gravis to rituximab: a retrospective study. Ther Adv Neurol Disord. 2011;4(5):259–266. doi:10.1177/1756285611411503
53. Dos Santos A, Noury JB, Genestet S, et al. Efficacy and safety of rituximab in myasthenia gravis: a French multicentre real-life study. Eur J Neurol. 2020;27(11):2277–2285. doi:10.1111/ene.14391
54. Caballero-ávila M, Álvarez-Velasco R, Moga E, et al. Rituximab in myasthenia gravis: efficacy, associated infections and risk of induced hypogammaglobulinemia. Neuromuscul Disord. 2022;32(8):664–671. doi:10.1016/j.nmd.2022.06.006
55. Afanasiev V, Demeret S, Bolgert F, Eymard B, Laforêt P, Benveniste O. Resistant myasthenia gravis and rituximab: a monocentric retrospective study of 28 patients. Neuromuscul Disord. 2017;27(3):251–258. doi:10.1016/j.nmd.2016.12.004
56. Frampton JE. Inebilizumab: first approval. Drugs. 2020;80(12):1259–1264. doi:10.1007/s40265-020-01370-4
57. Ali F, Sharma K, Anjum V, Ali A. Inebilizumab-cdon: USFDA approved for the treatment of NMOSD (Neuromyelitis Optica Spectrum Disorder). Curr Drug Discov Technol. 2022;19(1):e140122193419. doi:10.2174/1570163818666210519103001
58. Levy M, Fujihara K, Palace J. New therapies for neuromyelitis optica spectrum disorder. Lancet Neurol. 2021;20(1):60–67. doi:10.1016/S1474-4422(20)30392-6
59. Piedra-Quintero ZL, Wilson Z, Nava P, Guerau-de-Arellano M. CD38: an immunomodulatory molecule in inflammation and autoimmunity. Front Immunol. 2020;11:597959. doi:10.3389/fimmu.2020.597959
60. Fedyk ER, Zhao L, Koch A, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of the anti‐CD38 cytolytic antibody TAK‐079 in healthy subjects. Br J Clin Pharmacol. 2020;86(7):1314–1325. doi:10.1111/bcp.14241
61. Karnell JL, Rieder SA, Ettinger R, Kolbeck R. Targeting the CD40-CD40L pathway in autoimmune diseases: humoral immunity and beyond. Adv Drug Deliv Rev. 2019;141:92–103. doi:10.1016/j.addr.2018.12.005
62. Alabbad S, AlGaeed M, Sikorski P, Kaminski HJ. Monoclonal antibody-based therapies for myasthenia gravis. BioDrugs. 2020;34(5):557–566. doi:10.1007/s40259-020-00443-w
63. Ristov J, Espie P, Ulrich P, et al. Characterization of the in vitro and in vivo properties of CFZ533, a blocking and non-depleting anti-CD40 monoclonal antibody. Am J Transplant. 2018;18(12):2895–2904. doi:10.1111/ajt.14872
64. Eslami M, Schneider P. Function, occurrence and inhibition of different forms of BAFF. Curr Opin Immunol. 2021;71:75–80. doi:10.1016/j.coi.2021.06.009
65. Zhang Y, Tian J, Xiao F, et al. B cell-activating factor and its targeted therapy in autoimmune diseases. Cytokine Growth Factor Rev. 2022;64:57–70. doi:10.1016/j.cytogfr.2021.11.004
66. Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F. The BAFF/April system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013;24(3):203–215. doi:10.1016/j.cytogfr.2013.04.003
67. Gross JA, Johnston J, Mudri S, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404(6781):995–999. doi:10.1038/35010115
68. Mantchev GT, Cortesão CS, Rebrovich M, Cascalho M, Bram RJ. TACI is required for efficient plasma cell differentiation in response to T-independent type 2 antigens. J Immunol. 2007;179(4):2282–2288. doi:10.4049/jimmunol.179.4.2282
69. Ozcan E, Garibyan L, Lee JJ, Bram RJ, Lam KP, Geha RS. Transmembrane activator, calcium modulator, and cyclophilin ligand interactor drives plasma cell differentiation in LPS-activated B cells. J Allergy Clin Immunol. 2009;123(6):1277–86.e5. doi:10.1016/j.jaci.2009.03.019
70. Berrih-Aknin S, Ragheb S, Le Panse R, Lisak RP. Ectopic germinal centers, BAFF and anti-B-cell therapy in myasthenia gravis. Autoimmun Rev. 2013;12(9):885–893. doi:10.1016/j.autrev.2013.03.011
71. Rauch M, Tussiwand R, Bosco N, Rolink AG. Crucial role for BAFF-BAFF-R signaling in the survival and maintenance of mature B cells. PLoS One. 2009;4(5):e5456. doi:10.1371/journal.pone.0005456
72. Smulski CR, Eibel H. BAFF and BAFF-receptor in B cell selection and survival. Front Immunol. 2018;9:2285. doi:10.3389/fimmu.2018.02285
73. Möckel T, Basta F, Weinmann-Menke J, Schwarting A. B cell activating factor (BAFF): structure, functions, autoimmunity and clinical implications in Systemic Lupus Erythematosus (SLE). Autoimmun Rev. 2021;20(2):102736. doi:10.1016/j.autrev.2020.102736
74. Furie R, Petri M, Zamani O, et al. A Phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63(12):3918–3930. doi:10.1002/art.30613
75. Navarra SV, Guzmán RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9767):721–731. doi:10.1016/s0140-6736(10)61354-2
76. Hewett K, Sanders DB, Grove RA, et al. Randomized study of adjunctive belimumab in participants with generalized myasthenia gravis. Neurology. 2018;90(16):e1425–e1434. doi:10.1212/wnl.0000000000005323
77. Yin J, Zhao M, Xu X, et al. A multicenter, randomized, open‐label, phase 2 clinical study of telitacicept in adult patients with generalized myasthenia gravis. Eur J Neurol. 2024;31:e16322. doi:10.1111/ene.16322
78. Shi F, Xue R, Zhou X, Shen P, Wang S, Yang Y. Telitacicept as a BLyS/April dual inhibitor for autoimmune disease. Immunopharmacol Immunotoxicol. 2021;43(6):666–673. doi:10.1080/08923973.2021.1973493
79. Lin J, Li Y, Gui M, Bu B, Li Z. Effectiveness and safety of telitacicept for refractory generalized myasthenia gravis: a retrospective study. Ther Adv Neurol Disord. 2024;17:17562864241251476. doi:10.1177/17562864241251476
80. Tan CRC, Abdul-Majeed S, Cael B, Barta SK. Clinical pharmacokinetics and pharmacodynamics of bortezomib. Clin Pharmacokinet. 2019;58(2):157–168. doi:10.1007/s40262-018-0679-9
81. Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017;36(4):561–584. doi:10.1007/s10555-017-9707-8
82. Kohler S, Märschenz S, Grittner U, Alexander T, Hiepe F, Meisel A. Bortezomib in antibody-mediated autoimmune diseases (TAVAB): study protocol for a unicentric, non-randomised, non-placebo controlled trial. BMJ Open. 2019;9(1):e024523. doi:10.1136/bmjopen-2018-024523
83. Schneider-Gold C, Reinacher-Schick A, Ellrichmann G, Gold R. Bortezomib in severe MuSK-antibody positive myasthenia gravis: first clinical experience. Ther Adv Neurol Disord. 2017;10(10):339–341. doi:10.1177/1756285617721093
84. Alé A, Bruna J, Navarro X, Udina E. Neurotoxicity induced by antineoplastic proteasome inhibitors. Neurotoxicology. 2014;43:28–35. doi:10.1016/j.neuro.2014.02.001
85. Yamamoto S, Egashira N. Pathological mechanisms of bortezomib-induced peripheral neuropathy. Int J Mol Sci. 2021;22(2):888. doi:10.3390/ijms22020888
86. Yang Y, Zhao B, Lan H, Sun J, Wei G. Bortezomib-induced peripheral neuropathy: clinical features, molecular basis, and therapeutic approach. Crit Rev Oncol Hematol. 2024;197:104353. doi:10.1016/j.critrevonc.2024.104353
87. Yan W, Wu Z, Zhang Y, et al. The molecular and cellular insight into the toxicology of bortezomib-induced peripheral neuropathy. Biomed Pharmacother. 2021;142:112068. doi:10.1016/j.biopha.2021.112068
88. Klimas R, Sgodzai M, Motte J, et al. Dose-dependent immunomodulatory effects of bortezomib in experimental autoimmune neuritis. Brain Commun. 2021;3(4):fcab238. doi:10.1093/braincomms/fcab238
89. Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545(7655):423–431. doi:10.1038/nature22395
90. Singh AK, McGuirk JP. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 2020;21(3):e168–e178. doi:10.1016/s1470-2045(19)30823-x
91. Haghikia A, Schett G, Mougiakakos D. B cell-targeting chimeric antigen receptor T cells as an emerging therapy in neuroimmunological diseases. Lancet Neurol. 2024;23(6):615–624. doi:10.1016/s1474-4422(24)00140-6
92. Tian DS, Qin C, Dong MH, et al. B cell lineage reconstitution underlies CAR-T cell therapeutic efficacy in patients with refractory myasthenia gravis. EMBO Mol Med. 2024;16(4):966–987. doi:10.1038/s44321-024-00043-z
93. Granit V, Benatar M, Kurtoglu M, et al. Safety and clinical activity of autologous RNA chimeric antigen receptor T-cell therapy in myasthenia gravis (MG-001): a prospective, multicentre, open-label, non-randomised phase 1b/2a study. Lancet Neurol. 2023;22(7):578–590. doi:10.1016/s1474-4422(23)00194-1
94. Ellebrecht CT, Bhoj VG, Nace A, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353(6295):179–184. doi:10.1126/science.aaf6756
95. Oh S, Mao X, Manfredo-Vieira S, et al. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat Biotechnol. 2023;41(9):1229–1238. doi:10.1038/s41587-022-01637-z
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.