Back to Journals » Cancer Management and Research » Volume 10
Advances in antitumor effects of NSAIDs
Authors Zhang Z, Chen FL, Shang L
Received 24 May 2018
Accepted for publication 13 July 2018
Published 15 October 2018 Volume 2018:10 Pages 4631—4640
DOI https://doi.org/10.2147/CMAR.S175212
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Harikrishna Nakshatri
Zhen Zhang,1,2 Fulin Chen,1,2 Lijun Shang1–3
1Laboratory of Tissue Engineering, Faculty of Life Science, Northwest University, Xi’an, Shaanxi Province 710069, People’s Republic of China; 2Provincial Key Laboratory of Biotechnology of Shaanxi, Northwest University, Xi’an, Shaanxi Province 710069, People’s Republic of China; 3School of Chemistry and Biosciences, Faculty of Life Sciences, University of Bradford, Bradford, UK
Abstract: In recent years, the reports on using nonsteroidal anti-inflammatory drugs (NSAIDs) for cancer prevention and treatment have been on the rise. In 2017, the US Preventive Services Working Group issued primary prevention guidelines on the use of NSAIDs, especially aspirin, for cardiovascular disease and colorectal cancer, and formally established the role and status of aspirin in cancer prevention. However, the mechanism of NSAIDs on preventing cancer is still not clear. In this paper, the progress of the application of NSAIDs, especially aspirin, in the prevention and treatment of tumors in recent years is summarized, and new ideas and directions for the follow-up study are also discussed.
Keywords: NSAIDs, aspirin, platelets, cancer
Introduction
In 1897, German chemist Felix Hoffmann successfully applied salicylic acid and acetic anhydride to synthesize acetyl salicylic acid (acetylated salicylic acid) and obtained excellent effects in the treatment of rheumatoid arthritis for his father. This drug was named aspirin. Since then, this anti-inflammatory drug has been officially accepted in clinical practice, making it the most widely used antipyretic, analgesic and anti-inflammatory drug in the world. In addition to treatment, aspirin has been utilized as a standard preparation for the evaluation of other drugs. The molecular structure of aspirin is simple but used in vivo aspirin has shown a variety of effects over the years. It has been used as analgesic, antipyretic, anti-inflammatory and antirheumatic treatment in cardiovascular and cerebrovascular thrombosis. While its side effects cannot be ignored, especially long-term use of aspirin can lead to increased risk of gastrointestinal bleeding, increased uric acid, allergic asthma, coagulation inhibition amongst others.1 In recent years, different types of NSAIDs have been developed and applied to clinical treatment, but among all NSAIDs, aspirin still seems to be the best and first choice.2
Some large epidemiological studies have displayed that aspirin shows great effect in the primary and secondary prevention of cancer and in a variety of malignant tumor treatments (especially in colon cancer, stomach cancer etc.).3–6 In May 2009, a meta-analysis of aspirin in the prevention of colorectal cancer (CRC) and other cancers in recent years7 reported that regular administration of aspirin is associated with the reduction of the incidence of cancer. This attracted scientific interest and posed the question: could anti-inflammatory drugs be used for the prevention and treatment of the tumor? There are some problems raised in this hypothesis, such as how to avoid the gastrointestinal bleeding caused by long-term use of aspirin and the optimal dosage of aspirin. Therefore using aspirin in cancer prevention and treatment is still very controversial. In 2012, Rothwell8 indicated that regular administration of aspirin (≥75 mg/daily) could reduce the incidence of adenocarcinoma, CRC, and lung cancer, inhibit tumor metastases, and improve the survival rate of cancer patients. Additionally, they hypothesized that aspirin could inhibit tumor metastasis by eliminating platelet aggregation.
Early theories indicated that anti-inflammatory drugs could remove inflammatory factors in the surrounding inflammatory microenvironment of the tumor, so that the tumor cells can be directly exposed to immune cells, and then immune cells can function through immune recognition to achieve the role of cleaning up tumor cells. It is known that the body can self-recover after short-term inflammatory response, and this is a normal body self-protection mechanism, but long-term chronic inflammation may lead to cancer. Colotta et al9 proposed that tumors are caused by inflammation factors (IL-1, IL-6, IL-12, IL-17, TNF-α, TGF-β, etc.) assembling and the reactive oxygen species (ROS) produced by inflammatory infiltrating cells in the inflammatory microenvironment causing DNA damage (such as P53 gene mutation) and further induce normal cell carcinogenesis. Based on this, Okada et al10 created gp91phox gene knockout mouse implanted with or without degenerated fibro sarcoma cells (QR cells) and wild-type mouse macrophages. Compared to the gene knockout mouse, mouse implanted with wild-type mouse macrophages increased frequency of tumor development, and showed macrophage-driven ROS participated in the development of tumors. Amino guanidine is a broad inhibitor of inducible nitric oxide synthase, and its regulation can partially and significantly inhibit the transformation of degenerating QR cells into fibrosarcoma cells, therefore Futoshi Okada et al10 speculated that in addition to ROS there was nitrous oxide (NO) involved in the process of tumor deterioration. In addition, the inflammatory cytokines, chemokines (IL family) and NK cells can accumulate in the surrounding microenvironment of the tumor, and mediate the growth and metastasis of tumor cells (related to the upregulation of the expression of vascular growth factor in the surrounding tumor microenvironment).11,12 Additionally, the inflammatory factor and histamine can increase the permeability of the vessel wall and the lymphatic wall, and assist the distal metastasis of the tumor.13 This article was therefore regarded as the cornerstone of the researches.
Aspirin used in primary tumor prevention
A great number of large epidemiological studies found that long-term use of low-dose aspirin (75–300 mg/daily) can effectively inhibit a variety of cancer incidence, malignant cancer metastasis rate, and provide patients with a high survival rate. In 2012, Rothwell et al14 conducted a meta-analysis of the six trials of daily low-dose aspirin in primary prevention (35,535 participants) and pointed out that the regular administration of low-dose aspirin (75–300 mg/daily) for more than 3 years can reduce cancer risk by 24% (324 vs 421 cases; OR = 0.76, 95% CI 0.66–0.88, P=0.0003). In addition, the aspirin group reduced the risk of CRC within 20 years in six randomized trials and 26 case–control studies. At the same time, the risk of esophageal cancer, gastric cancer, cholangiocarcinoma and breast cancer in the aspirin group was significantly decreased. Rothwell et al15 analyzed five large randomized clinical trials which examined the effects of aspirin (75 mg/daily or more) on tumors. The results of the analysis showed that aspirin could reduce the incidence of distant metastases by 36% and reduce the risk of cancer-specific mortality by 50% compared with the control group. For patients with adenocarcinoma, the risk of morbidity can be reduced by 46%. In patients with no metastatic adenocarcinoma at the time of initial diagnosis, the use of aspirin in the subsequent long-term observation can reduce the associated cancer metastasis rate by 70%. In 2014, Cuzick et al16 also concluded that taking low doses of aspirin (30–100 mg/daily) for 10 years can reduce the risks of gastric cancer, esophageal cancer, and CRC by 30%. Liao et al17 found regular use of aspirin (twice a week or more, standard dose of 325 mg aspirin tablets) after diagnosis, compared with wild-type PIK3CA cancer patients, mutant PIK3CA CRC patients can obtain more benefits, and this once again determined the value of aspirin in primary prevention of CRC. A prospective study showed that among the registered 130,251 healthy subjects, after a 32-year follow-up survey, 27,985 people had cancer (20,414 women).18 Those who regularly take aspirin (0.5–1.5 standard aspirin tablets/week, 163 mg to 488 mg) for at least 6 years would reduce the overall risk of cancer by 3%, gastrointestinal cancer incidences reduce, including CRC (19% lower risk) and gastroesophageal cancer, but the risks of non-gastrointestinal cancer (such as breast cancer, advanced prostate cancer, lung cancer, etc.) were not significantly reduced. Importantly, retrospective epidemiological studies have shown that low-dose aspirin (75 mg/day) is more effective in reducing morbidity and mortality associated with CRC than high-dose aspirin (325–1200 mg/day).8
In April 2016, the USPSTF19 reviewed a number of large randomized controlled trials (RCTs) in the use of aspirin to prevent cancer, and found that taking aspirin for 3.6–10.1 years showed no significant difference in the overall cancer mortality and incidence between the aspirin group and the normal control group. The 20-year mortality rate of CRC was significantly lower in the aspirin group (four RCTs, RR=0.67), and the incidence of CRC was reduced for those taking aspirin for 10–19 years (three RCTs, total number of cases 47464, relative risk [RR]=0.60). In view of this, the USPSTF eventually issued aspirin as a guide for cardiovascular disease (CVD) and primary prevention of CRC, and ultimately recommended that: adults aged 50–69 years, 10% or greater 10-year CVD risk, are not at increased risk of bleeding, have a life expectancy of at least 10 years, and are willing to take low-dose aspirin (75 mg/daily) for at least 10 years, should consider taking low-dose aspirin to prevent CVD and CRC. The working group here considered the benefits of the various protocols that have been demonstrated in primary prevention trials, including doses of 75 and 100 mg per day and doses of 100 and 325 mg aspirin every day. The daily dose of 75 mg seems to be as effective as the higher dose (325 mg). However, the risk of gastrointestinal bleeding may increase with the increased doses. Comprehensive consideration is given that aspirin should be taken at 50–325 mg/day. Meanwhile, the guidelines also emphasize the need for trade-offs and individualization of aspirin-induced bleeding and other risks and benefits. It is worth mentioning that in the USA, the most commonly prescribed dose is 81 mg per day. In addition, we have summarized the main recommendations of the USPSTF in Table 1.
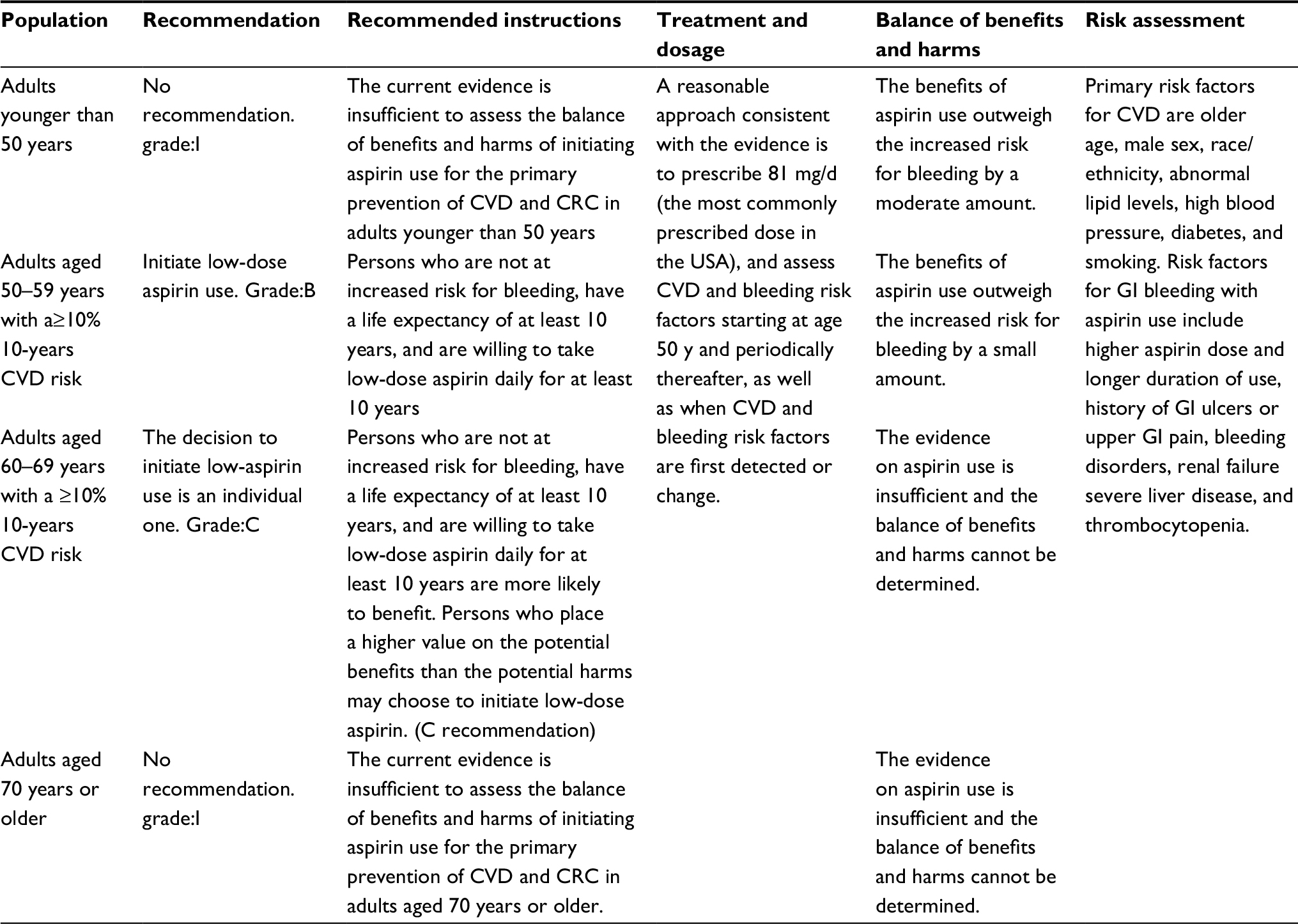 | Table 1 Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer in the USA: clinical summary19 Abbreviations: CVD, cardiovascular disease; CRC, colorectal cancer; GI, gastrointestinal. |
Antitumor mechanism of non-steroidal anti-inflammatory drugs (NSAIDs)
Inhibition of COX-2
Most of the NSAIDs are thought to act as tumor suppressors by inhibiting cyclooxygenase-COX.20 COX is known to convert arachidonic acid to prostaglandins (PGs), and the PGs overexpression are thought to be early events in a variety of oncogenic events. COX has three structural subtypes, structural COX-1 and inducible COX-2 (selectivity), COX-3 (found in the nervous system appears late in the inflammatory response).21,22 COX-1 is mainly present in normal tissue cells and catalyzes the production of PGs that maintain normal physiological function. COX-2 is a membrane-bound protein. In normal physiological status, the majority of tissue cells do not express COX-2; but in inflammation, tumor and other pathological conditions, due to inflammatory substances and carcinogenic substances such as the inflammatory mediators induced, COX-2 shows an increasing expression, and participates in a variety of pathophysiological processes, causing canceration (large number of COX-2-dependent genes are involved in this tumorigenesis process).23,24 Krysan et al25 found that COX-2 is usually upregulated in carcinoma in situ and non-small-cell lung cancer. Chen et al26 showed that the high expression of COX-2 in tumor tissue upregulated the expression of bcl-2 and thus played the role of an antiapoptotic effect of tumor cells. In addition, COX-2 and PG overexpression in chronic inflammation suggest that PGs produced by COX-2 catalysis can induce neovascularization to a certain extent and provide nutrition for tumor survival and proliferation.27 Finally, the COX-2 can affect upregulation of matrix metalloproteinase-2 expression which then promotes tumor invasion and metastasis.28 Recent studies have shown that the specific COX-2 enzyme inhibitor celecoxib (a NSAID) can effectively inhibit a variety of tumor cell proliferation and promote apoptosis on Raji cells, SK-N-SH cells and K562 cells in vitro.29–31 In addition, Harris et al32 conducted a case–control study of NSAID in 489 lung cancer patients and 978 control subjects. The results showed that a daily intake of aspirin or ibuprofen for at least 2 years reduces the relative risk of lung cancer by 68% (P<0.01). The detection of COX-2 in patients also suggested that patients taking NSAID significantly reduced the content. These results, with molecular evidence suggested that NSAID intake may prevent cancer by blocking COX-2 expression.
Induced tumor cell apoptosis
Bcl-2 gene inhibits apoptosis predominantly in the following three ways: 1) Bcl-2 can change the mitochondrial thiol reduction state to control the membrane potential and regulate apoptosis; 2) Bcl-2 can regulate the mitochondrial membrane permeability of some apoptotic protein precursors; 3) Bcl-2 can locate the apoptotic protein precursor Apaf-1 to the mitochondrial membrane so that it cannot play a role in apoptosis.
Yang et al and Xu et al33,34 used aspirin to treat human small cell lung cancer A549 cells, ovarian cancer SK-OV-3 cells and showed the expression of bcl-2 gene decreased and the expression of bax gene increased. Kim35 found that strong NSAIDs such as naproxen used on culture bladder cancer cells caused significant cell apoptosis, which was mainly due to the downregulation of bcl-2 gene and the upregulation of bax gene. In addition, Bank et al36 found that NSAIDs sulindac acid can degrade antiapoptotic protein bcl-XL, block bcl-XL antagonism of bax, make pro-apoptotic protein bax activation caspase cascade reaction, then activate the cell apoptosis program. Raza et al37 found that amino salicylic acid can increase the production of intracellular ROS, reduce intracellular glutathione storage, and inhibit mitochondrial respiratory chain enzyme complex, electron transport chain complex Body I, electron transport chain complex IV, mitochondrial matrix enzyme, cis-aconitase and other enzyme activity. The mitochondrial permeability changes can then lead to the synthesis of ATP being blocked, bcl-2 expression downregulated, and the release of cytochrome C will eventually trigger apoptosis. Furthermore, celecoxib can increase the wild-type p53 gene cancer overexpressing the p53 protein.38 The upregulation of p53 protein expression could further upregulate the expression of p53up-regulated modulator of apoptosis (PUMA) protein, which is the p53 gene downstream positive regulator. PUMA encodes the BH3-only protein of the bcl-2 family, whereas the BH3-only protein is a key factor in the initiation of apoptosis by the p53 pathway.
Downregulation of EGFR expression
Recently, Dong et al39 found that in 80% of patients with colorectal cancer EGFR overexpression occurred, but it can be downregulated by regular use of aspirin. Similarly, they noted that EGFR and COX-2 were co-located in patients with familial multiple adenomas and that COX-2 overexpression triggers proto-oncogene c-Jun-dependent transcription factors which bind to the EGFR promoter activation of protein-1 and can further promote the accumulation of EGFR cells in cells and promote their transformation to malignant.
Protect and repair DNA damage
Oxidative stress plays an important role in the development of various diseases such as cancer, cardiovascular disease, etc40,41 and can damage nucleic acid, protein, membrane and other cell elements. Banti et al42 mentioned that aspirin counteracts exogenous bacterial infections by inhibiting the production of peroxides, and oxygen radicals produced by bacteria, and can therefore cause great damage to cellular DNA. Chen et al43 confirmed that aspirin can inhibit the formation of nucleic acid cleavage and hydroxyl radicals caused by peroxynitrite, which play a role in protecting DNA. Goel et al44 found that aspirin could induce high expression of DNA mismatch repair proteins hMLH1, hMSH2, hMSH6 and hPMS2 in colon cancer cells, and the high expression of hMLH1 and hMSH2 could induce tumor cell apoptosis. Dibra et al45 found that aspirin can induce X-ray repair of cross-complementary gene 3 (XRCC3) and its protein expression, thus promoting DNA self-healing.
The general antitumor mechanism of NSAIDs is summarized in Table 2.
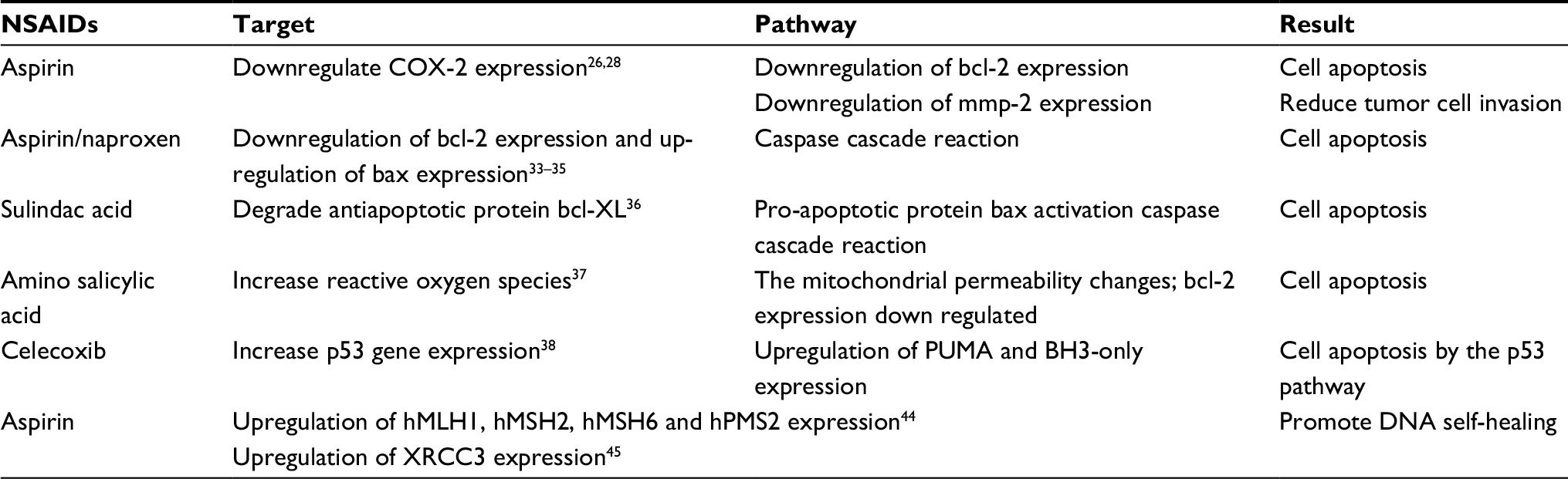 | Table 2 Traditional pathway of NSAIDs inhibiting tumors Abbreviations: COX-2, cyclooxygenase-2; NSAIDs, nonsteroidal anti-inflammatory drugs; XRCC3, X-ray repair of cross-complementary gene 3. |
The new thinking on the apparent phenomenon
Individual differences bring non-universality
A recent study in 2012 set out aspirin as a research boom in CRC secondary prevention. Liao et al17 retrospectively analyzed 964 patients with CRC and found that CRC-related mortality was significantly reduced (RR=0.18) and the overall survival extended (RR =0.54) in patients with CRC who had been diagnosed with aspirin-treated PIK3CA mutations. In contrast, in PIK3CA wild-type patients, regular aspirin administration (twice a week or more, standard dose of 325 mg aspirin tablets) after diagnosis did not improve CRC patients specific survival (RR=0.96) or overall survival (RR=0.94). In addition, some studies46,47 have shown that patients with 15-PGDH (15-hydroxyprogesterone dehydrogenase) and antigen-specific HLA-1 high expression in colon cancer can achieve more benefits in aspirin therapy (twice a week or more, standard dose of 325 mg aspirin tablets). Based on this, whether PIK3CA, 15-PGDH and HLA-1 can be used as key molecular biomarkers in the treatment of CRC is worthy of consideration.
NSAIDs prevents cancer from escaping immune surveillance
In 2015, Zelenay et al48 demonstrated that skin cancer, breast cancer and CRC cells often produce large amounts of PG E2 (PGE2), which can attenuate the immune system’s normal response to diseased cells and help cancer cells to hide. This process allows the tumor to grow rapidly, and therefore explains why some immunotherapy has difficulty in achieving the desired effect. Aspirin is a kind of COX inhibitor, which can inhibit the synthesis of PGE2 to achieve the effect of re-awakening the immune system. Therefore, compared with the use of immunotherapy alone, the combination of immunotherapy with aspirin or other COX inhibitors can significantly reduce the growth of CRC or melanoma in mice. In addition, a study showed that platelets can also help tumor cells escape the immune system.49 In vivo platelet-specific Glycoprotein A Repetitions Predominant (GARP) can activate platelet-derived or non-platelet-derived TGFβ, and GARP-TGFβ complex can effectively reduce the activity of T-cell activity at the physiological level. Thus most tumor cells can escape the clearance of the immune system, and the use of platelet antagonists will significantly improve the killing of tumor cells by the immune system. Therefore, the administration of COX inhibitor or platelet antagonists simultaneously with the patient’s immunotherapy can make cancer immunotherapy more effective and may become a new treatment with great potential.
Aspirin, platelet, tumor
Recent reports show that patients with a high blood platelet count are significantly more likely to develop cancer than those with a low blood platelet count, suggesting that tumor development may be associated with platelets.50 It is known that platelets, in addition to coagulation, play a role in the formation of new blood vessels.51 This effect is usually beneficial, such as the formation of a new clot after injury, and the need for new blood vessels to regulate blood flow. However, the same effect will also help the tumor growth. Platelets can enhance the invasiveness of cancer cells,49 promote epithelial-mesenchymal transition (EMT) transformation,52 and contribute to the deterioration of cancer to a large extent. In addition, cancer as a long-term chronic injury process will inevitably produce various mechanical damage etc. and promote the self-aggregation of platelets in the cancer site. There is already evidence that platelets can be spontaneously recruited to colon cancer cells in patients with colon cancer,53 which will undoubtedly greatly increase the progression of cancer. So we speculate that use of platelet antagonists can effectively slow down the progression of cancer. Lichtenberger et al54 indicated that aspirin can inhibit the activity of COX-1 enzyme, control the number of circulating platelets and their activity levels, and therefore block the interaction between platelets and cancer cells. They used platelet co-culture of human and murine colon cancer cell layers with two different aspirin preparations (normal aspirin and combination of phosphatidylcholine-specific aspirin) and found that platelets can significantly promote tumor cell proliferation and enhance its ability to migrate, while the use of aspirin can significantly decline the effect, and the effect of special aspirin (aspirin and phosphatidylcholine mixture) had increased efficacy. Other previous studies55 used platelets as a carrier to deliver PD-L1 antibody to the tumor site to achieve targeted killing of tumor cells and suggested that the role of platelets in the development of tumors is not absolute. Therefore, changing the traditional thoughts on cancer treatment would make us gain more benefits in the fight against tumors.
Aspirin against cancer by slowing up the mutation accumulates
It is known that tumorigenesis is caused by long-term accumulation of genetic mutations. Kostadinov et al56 suggested that aspirin can prevent cancer by slowing the accumulation of DNA mutations in abnormal cells in precancerous lesions. They analyzed 13 biopsy samples from patients with Barrett’s esophagus and conducted a 6–19 years follow-up studies. In the “observational crossover” studied, some patients began taking aspirin daily initially for several years and then stopped taking it, while others took aspirin until the end of the observation. The biopsy results showed that patients who did not use aspirin showed more than 10 times faster accumulations of gene mutations than those who used aspirin long term.
Different forms of NSAIDs may have different effects
In addition to traditional formulations of NSAIDs for the prevention and treatment of tumor, various forms of NSAIDs become available for tumor prevention or treatment either in vivo or in vitro. One example is the use of aspirin-phosphatidylcholine in vitro achieved better results in human or murine colon cancer cell lines at the cellular level compared to traditional aspirin.54 In addition, another study57 showed that the use of nano-forms of aspirin and ibuprofen to treat lung cancer patient’s whole blood in vitro could effectively reduce the DNA damage of white blood cells, which suggested that the nano-form of NSAIDs is more likely to enter the cell membrane and bind to DNA. It is worth mentioning that the use of metal complexes could also enhance the affinity of the complex for DNA binding.42 Therefore, NSAIDs and metal complexes should be taken into consideration in the treatment of cancer. Compared with traditional NSAIDs, NSAIDs with bio-metals show better pharmacological activity,58 and lower gastrointestinal toxicity.59 In addition, different metal and NSAIDs complexes are selective for different cell lines, for example, the platinum complexes of the NSAIDs tomeidine and indomethacin have the higher selectivity on non-cancerous cells BHK21 and L929.42 This suggests that we can choose different metal complexes as ligands to modify NSAIDs and to achieve selective killing of tumor cells.
In addition, as early as 2013, Liu et al60 conducted a meta-analysis of 10 studies between 2003 and 2013 and concluded that the use of aspirin and other NSAIDs did not significantly affect the incidence of brain tumors and was not associated with special subtypes of brain tumor. However, Simms et al61 showed that in vivo the liquid containing aspirin may be able to extend the life of thousands of patients with brain cancer. This is a breakthrough result in which they found that liquid aspirin can pass through the blood–brain barrier (known in the treatment of brain tumor, the blood–brain barrier can prevent chemotherapy drugs passing into the brain to kill brain tumor cells), and early tests showed that compared to existing chemotherapy drugs, liquid aspirin can kill brain cancer cells to improve the efficiency ten-fold. Therefore, we speculate that the role of aspirin in tumor treatment may be associated with the use of drug forms.
Enhance the effect of antitumor therapy
Some NSAIDs can enhance the sensitivity of the tumor to chemotherapy drugs, thus antitumor drugs and NSAIDs combination can produce a better prognosis in tumor therapy. Huang et al62 found that for in vitro culture of glioma cells, the use of indomethacin and adriamycin can significantly inhibit tumor cell growth than indomethacin use only. The result of QT-PCR and Western blot showed that indomethacin could downregulate the expression of ABCG2, MDR1 and MRP1, which were colloidal tumor-associated resistance genes, so that the antitumor effect of chemotherapeutic drugs was significantly improved. In addition, indomethacin does not only downregulate the resistance genes, but also enhances the sensitivity of colon cancer cells to chemotherapy drugs. 5-fluorouracil (5Fu) is known as a common cancer chemotherapy drug. Réti et al63 found that in patient body 80% 5Fu could metabolize into 5, 6-dihydro-5-fluorouracil by dihydropyrimidine dehydrogenase and lost the activity. And taking indomethacin could increase the concentration of 5Fu in vivo by inhibiting the activity of dihydropyrimidine dehydrogenase; thereby the increasing concentration of 5Fu can enhance the toxicity to tumor cells. Cheng et al64 demonstrated a new technique by using nucleophilic substitution to combine tumor chemotherapeutic drug cisplatin and aspirin which can significantly reduce the drug resistance of cisplatin in tumor therapy. Finally, in a recent study,49 the researchers found that when using combination of aspirin and clopidogrel (antiplatelet drugs) to treat melanoma-bearing mice, the mice obtained longer survival and fewer relapses when receiving adoptive T-cell therapy, suggesting that antiplatelet drugs could attenuate the ability of platelets to help tumor cells escape immune clearance. It is fair to say that the combination of NSAIDs and traditional cancer therapies or adoptive T cell therapy therefore could lead to a further breakthrough in cancer therapy with “1+1>2.”
All the above new thoughts are summarized in Table 3.
 | Table 3 The new thinking about NSAIDs inhibiting tumors Abbrevations: GARP, Glycoprotein A Repetitions Predominant; NSAIDs, nonsteroidal anti-inflammatory drugs; PGE2, prostaglandin E2. |
Conclusion
Based on a large amount of clinical data, NSAIDs, especially aspirin chemical prophylaxis has been gradually extended to clinical cancer treatment. The National Comprehensive Cancer Network colorectal cancer guide (2017 version) established the role of aspirin in the secondary prevention of cancer. However, taking into account the side effects of long-term use of aspirin, the large-scale use of aspirin as a cancer chemical prophylaxis still needs to weigh up the pros and cons for individual patients. The US “Guidelines for the Use of Preventive Drugs” clearly state that daily intake of “low-dose” aspirin has anticancer effects; here “low-dose” means 75–100 mg per day. But it is worth noting that the mechanism of anti-inflammatory drugs in tumor prevention and control is still not clear, future studies need to focus on the precise molecular classification and reasonable choice of population and dose, to achieve the largest anticancer benefits. As the body’s own platelets are linked to tumor growth and metastasis during tumorigenesis,54 our in vitro study found that platelets promote the proliferation of tumor cells and this effect can be inhibited by aspirin (unpublished data). In addition, a prospective cohort study50 showed that thrombocytosis is positively associated with tumor incidence. Of all the 9,435 male thrombocythemia patients, 1,098 were diagnosed with cancer (11.6%; 95% CI=11.0–12.3), however, only 106 out of 2,599 men without thrombocytosis had cancer (4.1%; 95% CI=3.4–4.9). Similarly, a total of 1,355 out of 21,826 females with thrombocytosis developed cancer (6.2%; 95% CI=5.9–6.5), compared with 119 of 5,370 females without (2.2%; 95% CI=1.8–2.6). Our previous in vitro studies also showed that the effect of platelets on adenoma cells is two-sided, but this effect can be attenuated by the platelet antagonist aspirin (unpublished data). Furthermore, our in vivo experiments have shown that aspirin has a certain effect on inhibiting tumor metastasis and both the duration and varying time periods of drug use could affect the survival of tumor-bearing mice (unpublished data). Further research work should focus on exploring the relationship between platelet, cancer and aspirin, clarifying the role of platelets in tumorigenesis and development, evaluating which stage of aspirin intervention could gain more benefits, proposing potential molecular mechanisms and new ideas for aspirin in clinical cancer prevention and treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
Derry S, Loke YK, Li Z. Risk of gastrointestinal haemorrhage with long term use of aspirin: meta-analysis. Chinese Edition of BMJ. 2001;04:186–190. | ||
Yujie G. Clinical application and safety evaluation of non-steroidal anti-inflammatory drugs. Pract Pharm Clin Remedies. 2009;02:122–124. | ||
Kang SU, Lee BS, Lee SH, Baek SJ, Shin YS, Kim CH. Expression of NSAID-activated gene-1 by EGCG in head and neck cancer: involvement of ATM-dependent p53 expression. J Nutr Biochem. 2013;24(6):986–999. | ||
Kawahara T, Ishiguro H, Hoshino K, et al. Analysis of NSAID-activated gene 1 expression in prostate cancer. Urol Int. 2010;84(2):198–202. | ||
Retsky M, Rogers R, Demicheli R, et al. NSAID analgesic ketorolac used perioperatively may suppress early breast cancer relapse: particular relevance to triple negative subgroup. Breast Cancer Res Treat. 2012;134(2):881–888. | ||
Seufert BL, Poole EM, Whitton J, et al. IκBKβ and NFκB1, NSAID use and risk of colorectal cancer in the Colon Cancer Family Registry. Carcinogenesis. 2013;34(1):79–85. | ||
Cuzick J, Otto F, Baron JA, et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10(5):501–507. | ||
Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379(9826):1591–1601. | ||
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–1081. | ||
Okada F. Inflammation-related carcinogenesis: current findings in epidemiological trends, causes and mechanisms. Yonago Acta Med. 2014;57(2):65–72. | ||
Dengbo J, Jingrong C. Relationship between the initiation, development and therapy of tumor and inflammation. Chinese J New Drugs. 2010;17:1551–1555. | ||
Shoubao M, Dandan L, Haiyan L. Inflammatory cytokines as therapeutic targets in tumor immunotherapy. Chinese Bull Life Sci. 2016;02:182–191. | ||
Wang Y, Li Yan BFS, Chen GJ. Inflammation: a driver for tumor development. J Int Pharm Res. 2014;01:63–67. | ||
Rothwell PM, Price JF, Fowkes FG, et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet. 2012;379(9826):1602–1612. | ||
Rothwell PM, Wilson M, Elwin CE, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376(9754):1741–1750. | ||
Cuzick J, Thorat MA, Bosetti C, et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann Oncol. 2015;26(1):47–57. | ||
Liao X, Lochhead P, Nishihara R, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367(17):1596–1606. | ||
Zolot J, Rosenberg K. Long-term aspirin use may reduce cancer risk. Am J Nurs. 2016;116(6):69. | ||
Bibbins-Domingo K, U.S. Preventive Services Task Force. Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2016;164(12):836–845. | ||
Benamouzig R, Uzzan B, Martin A, et al. Cyclooxygenase-2 expression and recurrence of colorectal adenomas: effect of aspirin chemoprevention. Gut. 2010;59(5):622–629. | ||
Willoughby DA, Moore AR, Colville-Nash PR. COX-1, COX-2, and COX-3 and the future treatment of chronic inflammatory disease. Lancet. 2000;19;355(9204):646–8. | ||
Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med. 2007;356(21):2131–2142. | ||
Kim ES, Hong WK, Lee JJ, et al. Biological activity of celecoxib in the bronchial epithelium of current and former smokers. Cancer Prev Res. 2010;3(2):148–159. | ||
Xu J, Yin Z, Gao W, et al. Meta-analysis on the association between nonsteroidal anti-inflammatory drug use and lung cancer risk. Clin Lung Cancer. 2012;13(1):44–51. | ||
Krysan K, Reckamp KL, Sharma S, Dubinett SM. The potential and rationale for COX-2 inhibitors in lung cancer. Anticancer Agents Med Chem. 2006;6(3):209–220. | ||
Chen XL, Su BS, Sun RQ, Zhang J, Wang YL. Relationship between expression and distribution of cyclooxygenase-2 and bcl-2 in human gastric adenocarcinoma. World J Gastroenterol. 2005;11(8):1228–1231. | ||
Kim BH, Kim CI, Chang HS, et al. Cyclooxygenase-2 overexpression in chronic inflammation associated with benign prostatic hyperplasia: is it related to apoptosis and angiogenesis of prostate cancer? Korean J Urol. 2011;52(4):253–259. | ||
Leung E, Mcarthur D, Morris A, Williams N. Cyclooxygenase-2 inhibition prevents migration of colorectal cancer cells to extracellular matrix by down-regulation of matrix metalloproteinase-2 expression. Dis Colon Rectum. 2008;51(3):342–347. | ||
Feng WY, Zhou Y, Zhong YG, Fu JP. Effect of SN-38 on prolifera- tion inhibition and apoptosis induction in K562 cells and synergistic effect of SN-38 and celecoxib. Acta Medicinae Universitatis Scientiae et Technologiae Huazhong. 2012;02:156–160. | ||
Lu Y, Wang W, Wang HY. Study on effect of celecoxib on growth of SK- N-SH cells and its mechanism. Chinese J Cancer Prev Treat. 2008;16:1218–1221. | ||
Huang XC, Li XL, Huang RX, et al. Effect of celecoxib and adriamycin on proliferation and apoptosis of Raji cells and its mechanism. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2010;35(10):1085–1090. | ||
Harris RE, Beebe-Donk J, Schuller HM. Chemoprevention of lung cancer by non-steroidal anti-inflammatory drugs among cigarette smokers. Oncol Rep. 2002;9(4):693–695. | ||
Yang JL, Han Y, Zhou YQ. The role of Bcl-2 family in the apoptosis of human small cell lung cancer A549 cells induced by aspirin. Guangdong Med J. 2010;11:1389–1391. | ||
Xu H, An CP, Chen RM. Effect of aspirin on apoptosis of ovar- ian carcinoma cells and expression of Bcl-2 bax mRNA. Hebei Med J. 2009;04:404–406. | ||
Kim MS, Kim JE, Lim DY, et al. Naproxen induces cell-cycle arrest and apoptosis in human urinary bladder cancer cell lines and chemically induced cancers by targeting PI3K. Cancer Prev Res. 20142014;7(2):236–245. | ||
Bank A, Yu J, Zhang L. NSAIDs downregulate Bcl-X(L) and dissociate BAX and Bcl-X(L) to induce apoptosis in colon cancer cells. Nutr Cancer. 2008;60(Suppl 1):98–103. | ||
Raza H, John A, Benedict S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. Eur J Pharmacol. 2011;668(1−2):15–24. | ||
Liu HF, Hsiao PW, Chao JI. Celecoxib induces p53-PUMA pathway for apoptosis in human colorectal cancer cells. Chem Biol Interact. 2008;176(1):48–57. | ||
Li H, Zhu F, Boardman LA, et al. Aspirin prevents colorectal cancer by normalizing EGFR expression. EBioMedicine. 2015;2(5):447–455. | ||
Fang C, Bourdette D, Banker G. Oxidative stress inhibits axonal transport: implications for neurodegenerative diseases. Mol Neurodegener. 2012;7:29. | ||
Wang Y, Yu Q, Fan D, Cao F. Coronary heart disease in type 2 diabetes: mechanisms and comprehensive prevention strategies. Expert Rev Cardiovasc Ther. 2012;10(8):1051–1060. | ||
Banti CN, Hadjikakou SK. Non-steroidal anti-inflammatory drugs (NSAIDs) in metal complexes and their effect at the cellular level. Eur J Inorg Chem. 2016;19:3048–3071. | ||
Chen W, Zhu H, Jia Z, et al. Inhibition of peroxynitrite-mediated DNA strand cleavage and hydroxyl radical formation by aspirin at pharmacologically relevant concentrations: implications for cancer intervention. Biochem Biophys Res Commun. 2009;390(1):142–147. | ||
Goel A, Chang DK, Ricciardiello L, Gasche C, Boland CR. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clin Cancer Res. 2003;9(1):383–390. | ||
Dibra HK, Brown JE, Hooley P, Nicholl ID. Aspirin and alterations in DNA repair proteins in the SW480 colorectal cancer cell line. Oncol Rep. 2010;24(1):37–46. | ||
Fink SP, Yamauchi M, Nishihara R, et al. Aspirin and the risk of colorectal cancer in relation to the expression of 15-hydroxyprostaglandin dehydrogenase (HPGD). Sci Transl Med. 2014;6(233):233re2. | ||
Reimers MS, Bastiaannet E, Langley RE, et al. Expression of HLA class I antigen, aspirin use, and survival after a diagnosis of colon cancer. JAMA Intern Med. 2014;174(5):732–739. | ||
Zelenay S, vanderVeen AG, Böttcher JP, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162(6):1257–1270. | ||
Rachidi S, Metelli A, Riesenberg B, et al. Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci Immunol. 2017;2(11):eaai7911. | ||
Bailey SER, Ukoumunne OC, Shephard EA, Hamilton W. Clinical relevance of thrombocytosis in primary care: a prospective cohort study of cancer incidence using English electronic medical records and cancer registry data. Br J Gen Pract. 2017;67(659):e405–e413. | ||
Mohle R, Green D, Moore MAS, Nachman RL, Rafii S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci. 1997;94(2):663–668. | ||
Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–590. | ||
Baker-Groberg SM, Itakura A, Gruber A, McCarty OJT. Role of coagulation in the recruitment of colon adenocarcinoma cells to thrombus under shear. Am J Physiol Cell Physiol. 2013;305(9):C951–C959. | ||
Lichtenberger LM, Fang D, Bick RJ, et al. Unlocking aspirin’s chemopreventive activity: role of irreversibly inhibiting platelet cyclooxygenase-1. Cancer Prev Res. 2017;10(2):142–152. | ||
Wang C, Sun W, Ye Y, et al. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat Biomed Eng. 2017;1(2):0011. | ||
Kostadinov RL, Kuhner MK, Li X, et al. NSAIDs modulate clonal evolution in Barrett’s esophagus. PLoS Genet. 2013;9(6): e1003553. | ||
Najafzadeh M, Normington C, Jacob BK, Isreb M, Gopalan RC, Anderson D. DNA Damage in healthy individuals and respiratory patients after treating whole blood in vitro with the bulk and nano forms of NSAIDs. Front Mol Biosci. 2016;3(288). | ||
Krstic NS, Nikolic RS, Stankovic MN, Nikolic NG, Dordevic DM. Coordination compounds of M(II) biometal ions with acid-type anti-inflammatory drugs as ligands – a review. Trop J Pharm Res. 2015;14(2):337–349. | ||
Weder JE, Dillon CT, Hambley TW, et al. Copper complexes of non-steroidal anti-inflammatory drugs: an opportunity yet to be realized. Coord Chem Rev. 2002;232(1-2):95–126. | ||
Liu Y, Lu Y, Wang J, et al. Association between nonsteroidal anti-inflammatory drug use and brain tumour risk: a meta-analysis. Br J Clin Pharmacol. 2014;78(1):58–68. | ||
Simms C, Howarth A, Pilkington G, Hill R. HG-124 autophagy modulation using repurposed drugs in the treatment of glioblastoma multiforme. Neuro Oncol. 2016;18(suppl 3):iii76.4–iii76. | ||
Huang N, Cheng Y. Enhanced cell toxicity of adriamycin by indomethacin on U251 glioma cell. Chinese J Cell Biol. 2013;05:649–654. | ||
Réti A, Pap Éva, Adleff V, et al. Enhanced 5-fluorouracil cytotoxicity in high cyclooxygenase-2 expressing colorectal cancer cells and xenografts induced by non-steroidal anti-inflammatory drugs via downregulation of dihydropyrimidine dehydrogenase. Cancer Chemother Pharmacol. 2010;66(2):219–227. | ||
Cheng Q, Shi H, Wang H, Min Y, Wang J, Liu Y. The ligation of aspirin to cisplatin demonstrates significant synergistic effects on tumor cells. Chem Commun. 2014;50(56):7427–7430. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.