")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
Advanced Glycation End Products Downregulate Connexin 43 and Connexin 40 in Diabetic Atrial Myocytes via the AMPK Pathway
Authors Yang F, Liu HH, Zhang L, Zhang XL, Zhang J, Li F, Zhao N, Zhang ZY, Kong Q, Liu XY, Wu Y, Yu ZM, Qian LL, Wang RX
Received 28 April 2023
Accepted for publication 26 September 2023
Published 2 October 2023 Volume 2023:16 Pages 3045—3056
DOI https://doi.org/10.2147/DMSO.S419189
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gian Paolo Fadini
Fan Yang,1,* Huan-Huan Liu,2,* Lei Zhang,1,* Xiao-Lu Zhang,1 Jie Zhang,1 Feng Li,1 Ning Zhao,2 Zhi-Yuan Zhang,1 Qi Kong,1 Xiao-Yu Liu,1 Ying Wu,1 Zhi-Ming Yu,1 Ling-Ling Qian,1 Ru-Xing Wang1,2
1Department of Cardiology, the Affiliated Wuxi People’s Hospital of Nanjing Medical University, Wuxi, 214023, People’s Republic of China; 2Wuxi School of Medicine, Jiangnan University, Wuxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ling-Ling Qian; Ru-Xing Wang, Department of Cardiology, the Affiliated Wuxi People’s Hospital of Nanjing Medical University, Wuxi, 214023, People’s Republic of China, Tel +0086-510-85351593, Email [email protected]; [email protected]
Purpose: Diabetes mellitus is an independent risk factor for atrial fibrillation (AF), which may be related to accumulation of advanced glycation end products (AGEs). However, the mechanisms involved are not completely clear. Abnormality of gap junction proteins, especially connexin 43 (Cx43) and connexin 40 (Cx40) in atrial myocytes, is an important cause of increased susceptibility of AF. The aim of our work is to investigate the mechanism of dysregulated Cx43 and Cx40 in atrial myocytes of diabetic rats.
Methods: We established a type 1 diabetic rat model by intraperitoneal injection of streptozotocin. HL-1 cells and primary rat atrial myocytes were treated with AGEs in vitro. Using Western blotting, immunofluorescence staining, immunohistochemistry, and lucifer yellow diffusion measurements, we investigated dysregulation of Cx43 and Cx40 and its mechanism in atrial myocytes of diabetic rats.
Results: Accumulation of AGEs was found in diabetic rats. The expression of Cx43 and Cx40 was reduced in the atrium of diabetic rats, accompanied by the decrease of phosphorylated Adenosine 5’-monophosphate-activated protein kinase (p-AMPK). Similar results were found in cultured HL-1 cells and primary rat atrial myocytes, suggesting a role of AGEs on gap junction proteins. An AMPK agonist, 5-Aminoimidazole-4-carboxamide ribonucleoside (AICAR), reversed the down-regulated Cx43 expression induced by AGEs stimulation. More importantly, lucifer yellow diffusion assay showed that AGEs significantly affected gap junctional function, and these changes were reversed by AICAR.
Conclusion: Thus, we conclude that AGEs cause dysregulation of Cx43 and Cx40 in diabetic atria via the AMPK pathway, thereby leading to gap junction dysfunction, which may contribute to the increased AF susceptibility in diabetes.
Keywords: diabetes, atrial fibrillation, advanced glycosylation end products, adenosine 5’-monophosphate-activated protein kinase, connexin
Introduction
Diabetes mellitus (DM) is a chronic progressive metabolic disease with a serious threat to human health. The International Diabetes Federation announced that the amount of diabetic patients has increased to 537 million worldwide in 2021. The number is still growing at an alarming rate globally and it seems to affect 693 million adults by 2045.1 DM disrupts vital organs in the body and leads to various long-term complications, of which cardiovascular complications are major causes of increased morbidity and mortality, mainly including cardiac arrhythmias, heart failure, myocardial infarction and even sudden cardiac death.2–4 It has been found that atrial fibrillation (AF) is an independently associated risk factor for individuals with DM.5 DM plays an important role in structural remodeling, electrical remodeling, autonomic remodeling, connexin remodeling, and oxidative stress, thus promoting the occurrence of AF.5 However, the specific pathophysiology of AF complicated by DM has not been fully elucidated.
Advanced glycation end products (AGEs) are the biochemical end products of non-enzymatic glycosylation reactions (Maillard reaction).6 It is a stable covalent addition produced from the spontaneous reaction of macromolecules such as proteins, lipids or nucleic acids with glucose or other reducing monosaccharides.6 AGEs have two sources in the body: the synthesis of AGEs in the body from excess sugar and protein, and the ingestion of AGEs present in food through diets. The production of AGEs is moderate under normal physiological conditions. However, it is significantly accelerated due to increased glucose utilization in the case of diabetes.7 Thus, AGEs are considered to be a key factor in the progression of diabetic complications.7–9 Recently, studies have reported that AGEs are related to arrhythmias both in patients10 and animal models.11 AGEs induced senescence of atrial myocytes and increase susceptibility of AF in diabetic mice via reversing electrical remodeling.12 However, knowledge of the role of AGEs in DM-related AF remains limited.
Connexin is a membrane protein that is an important component for gap junction channels in the myocardium and responsible for the communication between neighboring cardiomyocytes, of which connexin 43 (Cx43) is the most abundant one in cardiomyocytes. Besides, Cx40 is selectively expressed in atrial myocytes and is mainly involved in coordinating the electrical activation of atria.13 It has been shown that connexin plays a key role in propagating action potentials between cardiomyocytes. The pathogenesis of AF is significantly associated with slower conduction and reentry formation resulting from downregulation of Cx43, disruption of atrial gap junctions and enhanced fibrosis.14 Cx40 has also been reported to be engaged in the onset and progression of AF to varying extents.15 However, whether AGEs play a role in regulating gap junctional proteins is still unknown. Therefore, this study examined the role of AGEs in regulating gap junction proteins, which may underlie DM-related AF.
Materials and Methods
Experimental Animal Models
Type 1 diabetic animal models were established as previously reported.16 Briefly, 8-week-old male Sprague-Dawley (SD) rats, weighing approximately 180 g (n = 28, purchased from Cavins Laboratory Animals, Changzhou, China) were injected intraperitoneally with streptozotocin (STZ, 60mg/kg, S0130, Sigma-Aldrich, St. Louis, MO, USA). One week after injection, blood was drawn from the tail veins and random blood glucose was measured using an Accu-Chek glucose meter (Roche Diagnostics GmbH Company, Germany). Rats with blood glucose concentration >16.7 mmol/L were enrolled in this study as the diabetic group, and vehicle-injected, age-matched rats served as controls. All rats were raised at a standard of care-specific pathogen-free (SPF) conditions (temperature 22–24°C and humidity 55–65%) in a light/dark cycle adapted to 12h. After 8 weeks, body weight and random blood glucose were measured. The rats were then sacrificed, and their hearts were harvested and stored at −80°C. All experiments were conducted according to the internationally accepted principles by the ethical guidelines by the International Council for Laboratory Animal Science (ICLAS) and all animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Nanjing Medical University.
Isolation of Neonatal Rat Atrial Myocytes
Primary rat atrial myocytes were extracted from the atrial tissue of neonatal rats using the cardiomyocyte isolation kit (88281Y, Pierce Thermo Fisher Inc., Rockford, IL, USA) according to manufacturer’s instructions as previously reported.12,17 The rat bodies were disinfected with 75% ethanol, and then the hearts were rapidly isolated and placed in 1.5 mL sterile tubes containing 500 μL of phosphate buffered saline. Atrium were cut into 1 mm3 pieces and washed. An appropriate amount of Cardiomyocyte isolation enzyme 1 (88288E, Thermo Fisher) and cardiomyocyte isolation enzyme 2 (88289E, Thermo Fisher) was added into the shredded tissue. Then, we incubated tubes at 37°C. After 30 minutes, the tissues were washed twice with PBS. DMEM containing 10% fetal bovine serum (FBS) was added to each tube, mixed by pipetting, and then seeded and incubated in 1×106/100-mm Petri dishes. The unattached cells were seeded at 2×105/well in 6-well plates and initially incubated at 37°C and 5% CO2 after 2 hours. Primary atrial myocytes were supplemented with fresh culture medium 2–3 days after isolation and treated in 5–7 days after isolation. To investigate the effect AGEs on Cx43 and Cx40 expression, primary atrial myocytes were exposed to 100 μg/mL AGEs-BSA (ab51995, Abcam) for 24 hours. Cells cultured with 100 μg/mL non-glycated BSA (A3803, Sigma) were used as a control.
HL-1 Cell Culture
HL-1 cells (m077, iCell, Shanghai, China) were cultured in DMEM containing 10% FBS. When cell fusion reached approximately 70%, HL-1 cells were treated with AGEs-BSA or BSA similar as primary rat atrial myocytes. To observe whether Adenosine 5’-monophosphate-activated protein kinase (AMPK) agonist AICAR (2840, TOCRIS, Bristol, UK) could reverse the effect of AGEs, AGEs+AICAR group and BSA+AICAR group were set. Cells were first treated with AGEs for 24 hours then with 10 μmol/L AICAR for another 24 hours,18,19 to test whether post-AGE AMPK activation leads to normalization of Cx43 and Cx40 expression.
siRNA Transfection
AMPK α1 siRNA (S: 5′-CUGUGGCUCACCCAAUUAUTT-3′, AS: 5′-AUAAUUGGGUGAGCCACAGTT-3′), AMPK α2 siRNA (S: 5′-GAGCGACUAUCAAAGACAUTT-3′, AS: 5′-AUGUCUUUGAUAGUCGCUCTT-3′) and negative control (NC) siRNA (S: 5′-UUCUCCGAACGUGUCACGUTT-3′, AS: 5′-ACGUGACACGUUCGGAGAATT-3′) were purchased from Gene Pharma (Shanghai, China). HL-1 cells were cultured into 6-well plates and transfected with siRNA (100 nmol/L final) for 8 hours using Lipofectamine 2000 liposome (11668019, Thermo Fisher, USA). The ratio of Lipofectamine 2000 over siRNA in a well was 10 μL: 10 μL. The siRNA-transfected cells were used for Western blotting analysis at 48–72h post infection.
Western Blot
Atrial tissues, HL-1, primary rat atrial myocytes were homogenized and lysed with ice-cold RIPA buffer containing protease and phosphatase inhibitors. Equal amounts of protein (30 μg) were separated on a 10% SDS-polyacrylamide gel and transferred to PVDF membranes. After blocking with 5% non-fat milk in tris-buffered saline containing Triton-X100, membranes were hybridized overnight with the specific primary antibodies AGE (1:1000, ab23722, Abcam), rabbit polyclonal to RAGE (1:1000, ab65965, Abcam), p-AMPK (1:1000, 2535S, Cell Signaling Technology), AMPK (1:1000, 2532S, Cell Signaling Technology), Cx43 (1:500, sc-365107, Santa Cruz Biotechnology, Dallas, Texas, USA) and Cx40 (1:500, sc-271837, Santa Cruz Biotechnology). Then, the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG or anti-mouse IgG for 1.5 hours. Expression of β-tubulin (1:10000, ab21058, Abcam) was determined as a housekeeping control. After washing, membranes were developed with electrochemiluminescence (ECL) reagents (wbkls0500, Millipore) and images were taken by the ECL imager (Tanon-5200 Multi, Tanon Technology Co., Ltd). Quantification was performed with ImageJ.
Immunohistochemistry
Hearts were harvested and fixed in 4% paraformaldehyde and cut into 5 µm slices after embedding in paraffin after 8 weeks. Then, the sections were incubated with anti-Cx40 and anti-Cx43 antibodies overnight at 4°C. The next day, the primary antibody was eluted and the secondary antibody was incubated at 37° for 30 min, followed by incubation with SABC at 37° for 30 min, DAB development, and then counterstaining with hematoxylin. Following dehydration, coverslips were mounted with neutral balsam). Finally, the images were captured with a light microscope. The positive signals for Cx40 and Cx43 proteins are brown.
Lucifer Yellow Diffusion
Neonatal rat atrial myocytes cultured in 6-well plates were washed with PBS. After aspiration of the liquid, 0.05% fluorescent dye fluorescein yellow (L0259, Sigma) was added to the wells and immediately scratched with a scalpel blade.20 Cells were rinsed with PBS three times and fixed with 4% paraformaldehyde. Images were observed under a fluorescence microscope (EVOS, Thermo Fisher).
Statistical Analysis
Data were presented as mean ± SEM. The normality of the distributions was analyzed by Shapiro Wilk test. Student’s t-test was used to compare data between two groups if data normally distributed; otherwise, the Mann–Whitney U-test was used. One-way ANOVA was used to compare data from multiple groups if data normally distributed, and LSD or Tamhane’s were adopt as post hoc tests; otherwise, the Kruskal–Wallis test followed by Dunn’s was used. Statistical significance was defined as P < 0.05. Graphing and statistical analysis were performed with GraphPad Prism 8 and SPSS 27.0.
Results
Establishment of Type 1 Diabetes Model Induced by Streptozotocin
Eight weeks after STZ injection, DM rats showed significant increase in blood glucose and decrease in body weight, while control rats showed no significant changes in blood glucose but increase in body weight (Figure 1A and B). The protein levels of AGE and RAGE in the atrial tissue of two groups of rats were measured. As is shown in Figure 1C and D, the expressions of AGE and RAGE were significantly higher in the atria of diabetic rats compared to the normal group. This result indicates that diabetes increases the accumulation of AGEs.
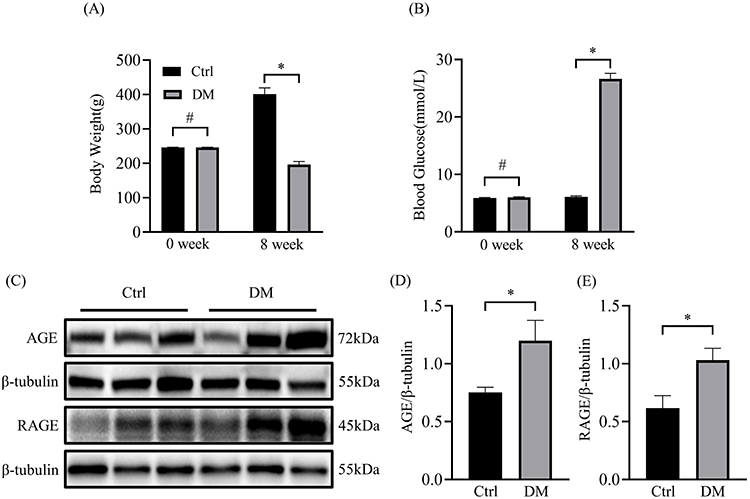 |
Figure 1 Body weight and blood glucose of diabetic rats and the expression of AGE and RAGE in the atrium. (A) Changes in body weight of two groups of rats in 0 week and 8 week (n=12). (B) Blood glucose changes in two groups of rats before and after modeling (n=12). (C) Representative blots of AGE and RAGE proteins in atrium of diabetic rat (n=6). (D) Densitometry analysis of AGE and RAGE proteins in atrium of diabetic rat (n=6). *p<0.05, #p>0.05. |
Expression of Cx43 and Cx40 in Myocardial Tissue of Diabetic Rats
The protein levels of Cx43 and Cx40 in the atrial tissue of two groups were analyzed by Western Blot. As is shown in Figure 2A-C, both Cx43 and Cx40 were significantly downregulated in the atria of diabetic rats compared to the control rats. Consistently, as assessed by immunohistochemistry, the expression of Cx43 and Cx40 was downregulated in DM group, and their distribution in the atria became disturbed (Figure 2D). These results suggest remodeling of connexins was detected in the atria of diabetic rats.
 |
Figure 2 Expression of Cx43 and Cx40 proteins in atrial tissues from Control and Diabetic groups. (A) Representative blots of Cx43 and Cx40 proteins in atrium of diabetic rat (n=6). (B and C) Densitometry analysis of Cx43 and Cx40 proteins in atrium of diabetic rat (n=6). (D) Representative histological fixed sections of atrial tissues stained with Immunohistochemistry. *p<0.05. |
Effects of AGEs on Cx43 and Cx40 in Cultured HL-1 and Primary Neonatal Rat Atrial Myocytes
To determine whether AGEs play a role on the regulation of Cx43 and Cx40 protein expression, in vitro experiments were performed on cultured HL-1 cells and primary rat atrial myocytes. Our results showed that AGEs significantly decreased the expression of Cx43 and Cx40 in both HL-1 cells (Figure 3A-C) and primary rat atrial myocytes (Figure 3D-F), suggesting that AGEs have a negative effect on atrial gap junction proteins.
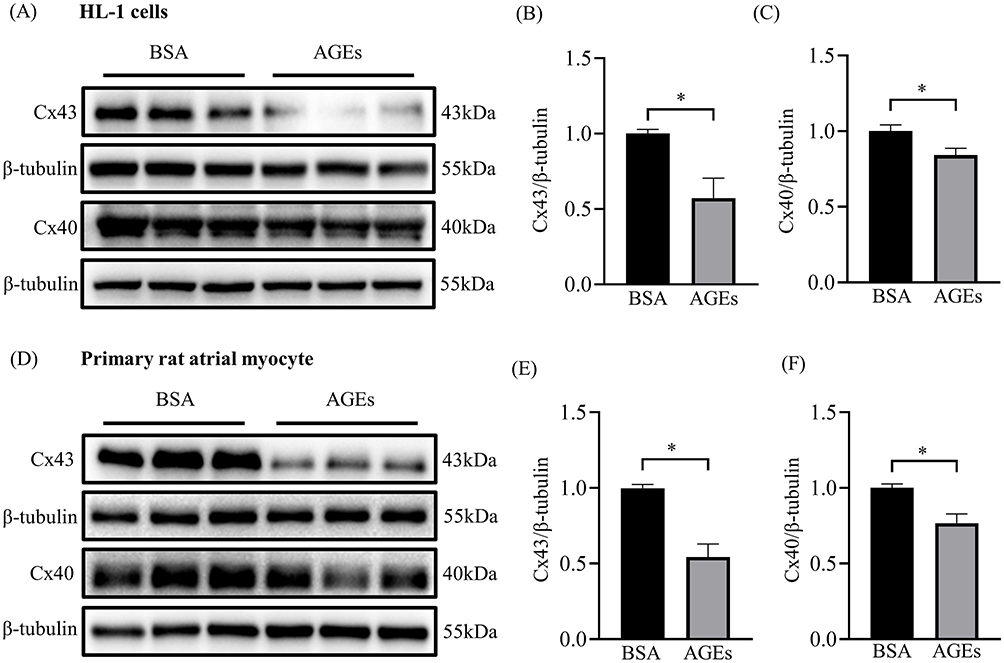 |
Figure 3 Altered protein expression levels of Cx43 and Cx40 in HL-1 cells and primary atrial myocytes treated with AGEs. (A) Representative blots of Cx43 and Cx40 proteins in each HL-1 cell group (n=6). (B and C) Densitometry analysis of Cx43 and Cx40 proteins in each HL-1 cell group (n=6). (D) Representative blots of Cx43 and Cx40 proteins in each primary rat atrial myocyte group (n=6). (E and F) Densitometry analysis of Cx43 and Cx40 proteins in each primary rat atrial myocyte group. *p<0.05. |
Role of AMPK Pathway in AGEs Regulating of Connexin Expression in Atrial Myocytes
As a key energy metabolism molecule in cell signaling, AMPK plays a critical role in the pathophysiological development of diseases. To explore the mechanism of AGEs regulating the expression of Cx43 and Cx40, we further determined the expression of p-AMPK and AMPK in the atrial tissue of two groups of rats. p-AMPK was significantly inhibited in diabetic atrial tissues while the total protein level of AMPK had no significant change (Figure 4A-C). In vitro studies show p-AMPK levels was significantly lower in the AGEs group compared to the BSA control group both in HL-1 cells and primary rat atrial myocytes, while the level of AMPK did not change significantly (Figure 4D-I), indicating AGEs is involved in AMPK inactivation.
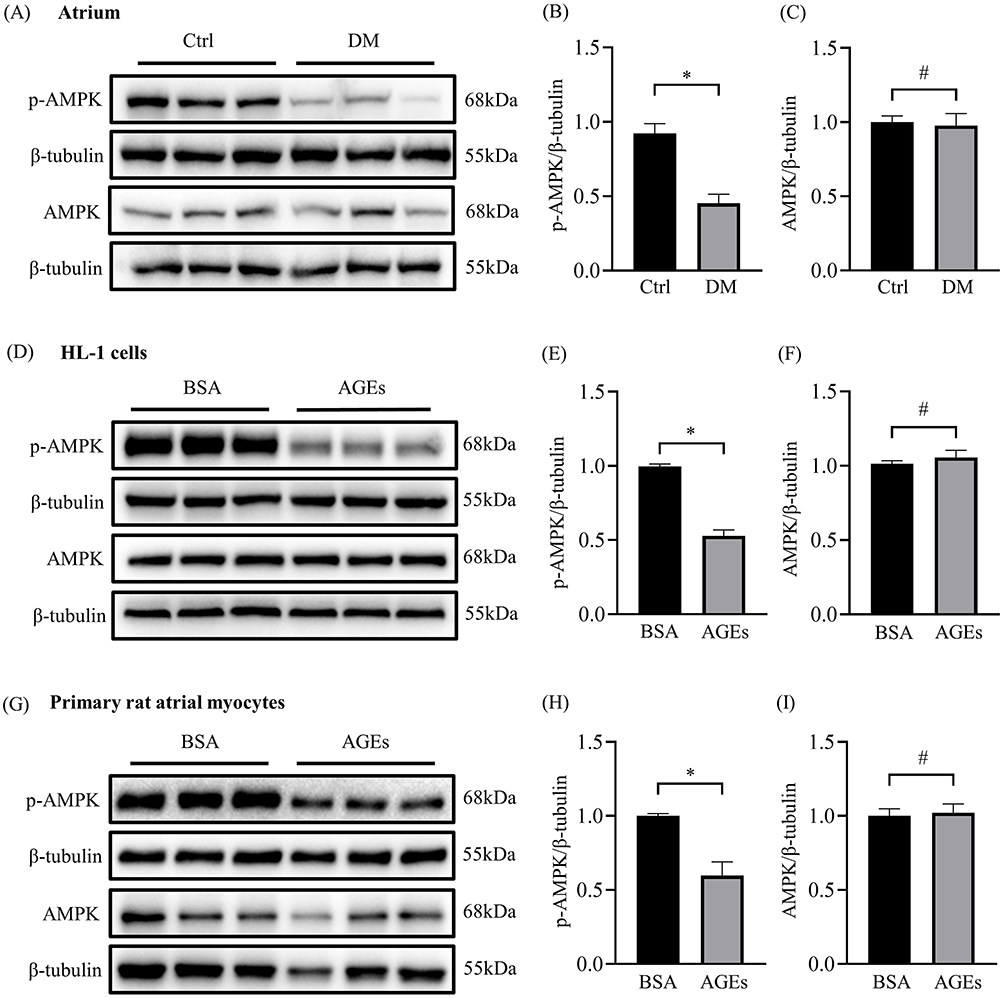 |
Figure 4 Inactivation of AMPK in atrial of diabetic rats and AGEs treated atrial cells. (A) Representative blots of p-AMPK (n=6) and AMPK proteins (n=12) in atrium of diabetic rat. (B and C) Densitometry analysis of p-AMPK (n=6) and AMPK (n=12) proteins in atrium of diabetic rat. (D) Representative blots of p-AMPK and AMPK proteins in each HL-1 cell group (n=6). (E and F) Densitometry analysis of p-AMPK and AMPK proteins in each HL-1 cell group (n=6). (G) Representative blots of p-AMPK and AMPK proteins in each primary rat atrial myocyte group (n=6). (H and I) Densitometry analysis of p-AMPK and AMPK proteins in each primary rat atrial myocyte group (n=6). *p<0.05, #p>0.05. |
Effect of AICAR on AGEs‑induced Dysregulation of Cx43 and Cx40
AGEs downregulate the expression of Cx43 and Cx40 in HL-1 and primary rat atrial myocytes, causing gap junction dysfunction and remodeling. Therefore, to investigate whether AGEs induce dysregulation of atrial junctional proteins by downregulating the expression of p-AMPK and thus the effect on this process, we analyzed the levels of Cx43 and Cx40 by AICAR treatment after 24 hours of AGEs incubation. AICAR could restore the expression of p-AMPK (Figure 5A-C). The results showed that AGEs significantly reduced the expression of Cx43 and Cx40 in HL-1 cells, whereas AICAR restored the expression of Cx43 and Cx40 (Figure 5A, D and E). These results suggest that AICAR prevents AGEs-induced Connexin dysregulation.
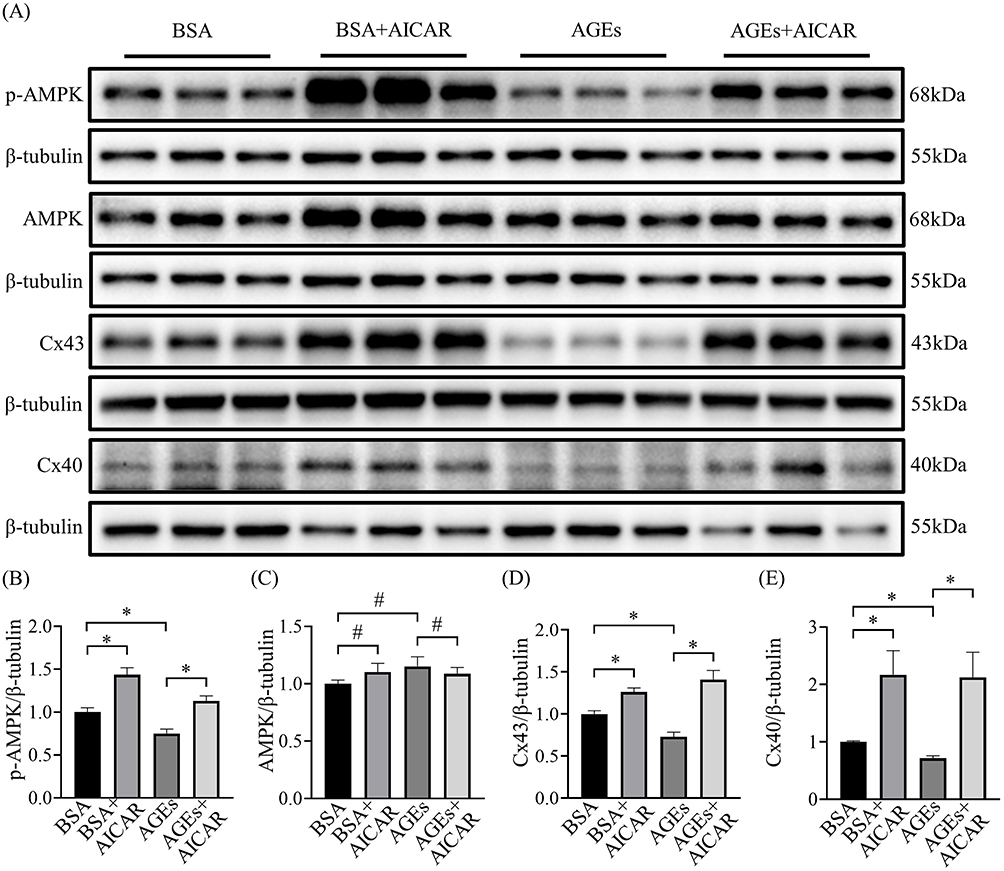 |
Figure 5 Effects of AMPK activation on AGEs-induced Cx43 and Cx40 alteration in atrial cells. (A) Representative blots of p-AMPK, AMPK, Cx43 and Cx40 proteins in each cell group (n=6-9). (B-E) Densitometry analysis of p-AMPK, AMPK, Cx43 and Cx40 proteins in each cell group (n=6-9). *p<0.05, #p>0.05. |
Knockdown of AMPK Decreased Expression of Cx43 and Cx40
To further investigate whether AMPK plays a regulatory role on connexin, we detected the protein levels of Cx43 and Cx40 after knocking down of AMPK α1 and α2 subunit respectively in HL-1 cells. The results showed that knockdown of AMPK α1 or α2 subunit significantly reduced the expression of Cx43 and Cx40 in HL-1 cells (Figure 6A-H).
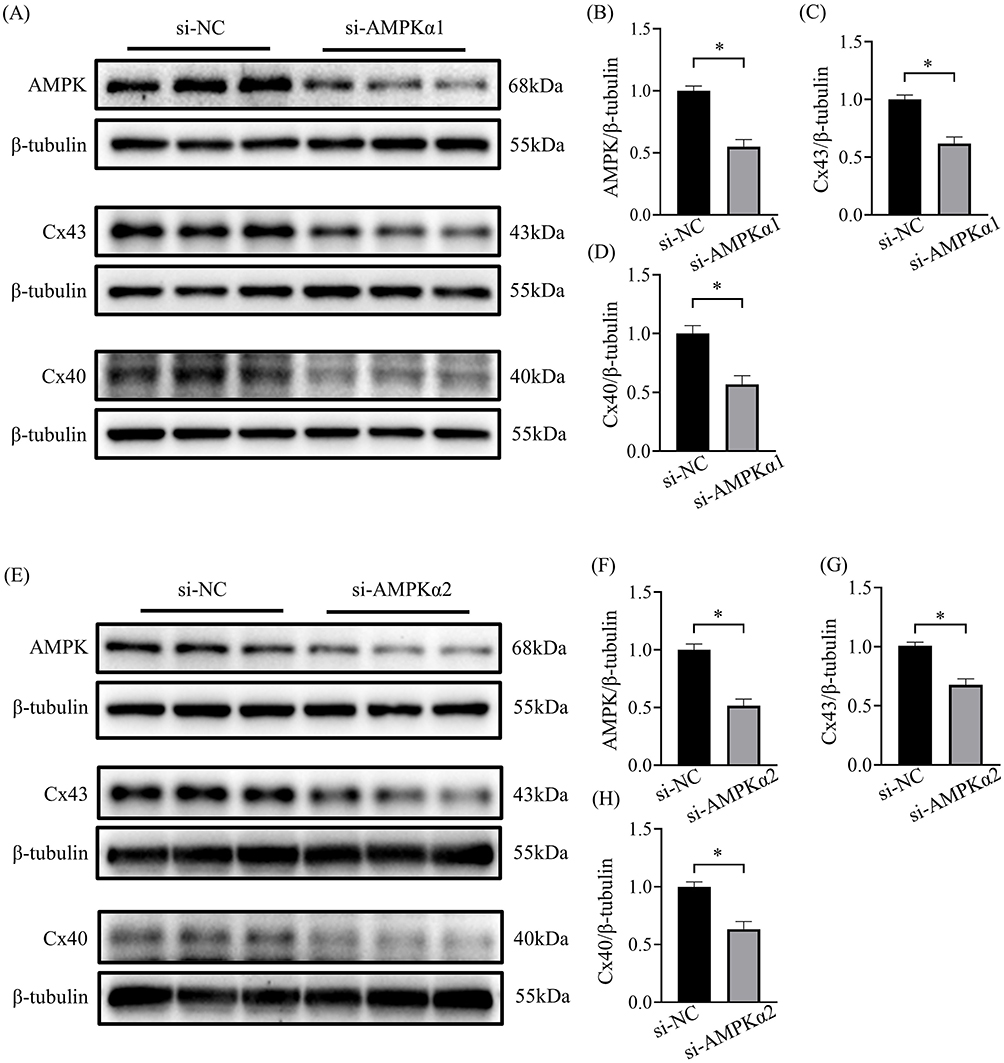 |
Figure 6 Decreased expression of Cx43 and Cx40 by downregulation of AMPK. (A) Representative blots of AMPK, Cx43 and Cx40 proteins in HL-1 cells transfected with AMPK α1 siRNA (n=6-9). (B-D) Representative densitometry analysis of AMPK, Cx43 and Cx40 proteins in HL-1 cells transfected with AMPK α1 siRNA (n=6-9). (E) Representative blots of AMPK, Cx43 and Cx40 proteins in HL-1 cells transfected with AMPK α2 siRNA (n=6-9). (F-H) Representative densitometry analysis of AMPK, Cx43 and Cx40 proteins in HL-1 cells transfected with AMPK α2 siRNA (n=6-9). *p<0.05. |
AMPK Activation Increases Gap Junction Permeability
Altered gap junction channel proteins in cardiomyocytes are associated with the development of diabetic atrial fibrillation. Since activation of AMPK by AICAR upregulated the expression of Cx43 and Cx40, we further explored whether AICAR affects intercellular conduction in cardiac myocytes. We performed fluorescein yellow dye diffusion assays to detect gap junction function. AGEs treatment resulted in slower intercellular conduction as shown by a reduction in dye diffusion distance and AICAR significantly reversed this change (Figure 7). The result suggests that AMPK activation increases gap junction permeability in cardiomyocytes.
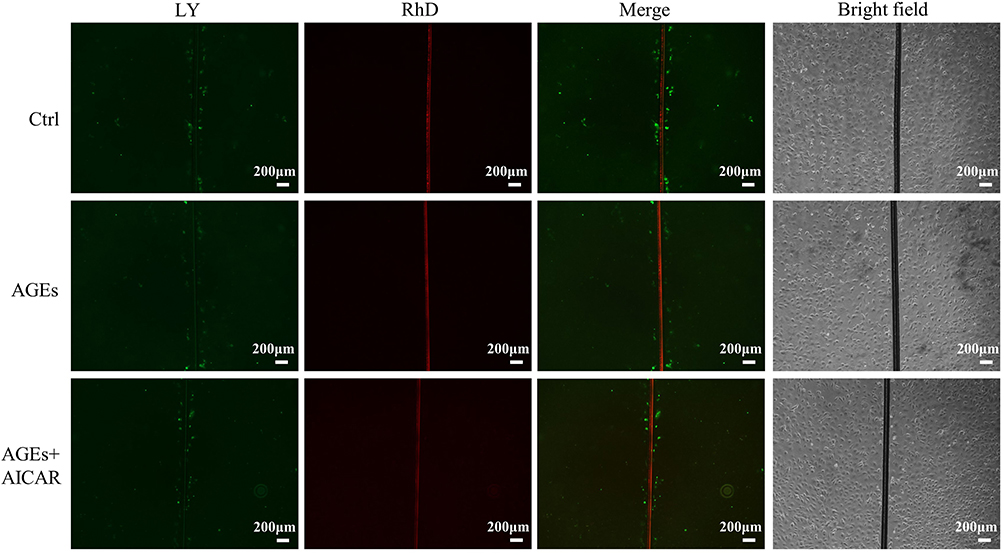 |
Figure 7 AMPK activation diminished AGEs-induced gap junctional dysfunction in primary atrial myocytes. Representative images of fluorescein yellow (LY) and rhodamine (RD) diffusion in each group of cells indicate lower diffusion through gap junctions in the AGEs group and increased diffusion in the AICAR group compared to the AGEs group. |
Discussion
The increased AGEs are closely associated with the development of diabetic-related AF.12,21 However, the exact mechanism remains unknown. Our work focused on the study of the regulatory relationship between AGEs and gap junction proteins. The main findings of our study are: (1) Expression of AGEs and RAGE are increased in atrium of diabetic rats. (2) AGEs cause a decrease in the expression of Cx43 and Cx40 as well as abnormal distribution, which subsequently leads to conduction dysfunction in atrial myocytes. (3) Knockdown of AMPK in vitro downregulates the expression of Cx43 and Cx40. (4) Phosphorylated AMPK was reduced by AGEs stimulation and activation of AMPK restored the expression of connexin and gap junction function. DM, a serious long-term metabolic disorder, promotes an excessive generation of AGEs, which is caused by the acceleration of glycation reaction in chronic hyperglycemia condition.22 Consistent with previous studies, AGEs are substantially increased in combination with RAGE in DM and are involved in the pathogenesis of diabetic cardiovascular diseases.12,23 Our results showed that the accumulation of AGEs in the atrial tissues of diabetic rats was significantly higher than the control group rats, along with a significant increase in RAGE. Several studies have proved that increased concentrations of AGEs in the circulation are closely linked with inflammation, microvascular dysfunction, and diabetic adverse cardiac remodeling.24,25 Besides, it was revealed that AGEs could increase lipotoxicity by inhibiting sterol regulating element-binding protein (SREBP) signaling which played an important role in keeping balance between lipid and glucose, thus aggravating myocardial injury in diabetic cardiomyopathy.26 Furthermore, a recent study demonstrated that AGEs treatment could prolong the action potential duration, decrease current density of ICa,L, IKur in HL‐1 cells and reduce protein expression of Cav1.2, Kv1.5 both in atrium of diabetic mice and HL-1 cells, consequently increasing susceptibility to AF in diabetic mice.12 Nonetheless, the concrete mechanism by which AGEs promote AF susceptibility remains to be explored.
Gap junctions (GJ) composed of connexins are essential for electrical coupling and rapid action potential conduction synchronously between cardiomyocytes. Growing evidence has shown that connexin remodeling induce altered atrial conduction heterogeneity in DM and reduced GJ function may play a critical role in promoting AF.27–29 In patients with DM, the expressions of Cx43 and Cx40 were downregulated in the myocardium.30 Yan.et al31 discovered that long-term JNK activation downregulates the protein level of Cx43 via c-jun suppressed transcriptional activity of the Cx43 gene promoter. Also, some other transcription factors were reported to induce AF through regulating the expression of atrial connexins.29,32 However, the modulation of atrial gap junction in atrial though AGEs has not been reported. Our results revealed that Cx43 and Cx40 were significantly downregulated in diabetic rats and an increase in AGEs could cause a decrease in expression and function of Cx43 and Cx40 in atrial myocytes.
It has been demonstrated that the differences of protein expression between the left atrial appendage tissues of patients with AF and healthy individuals were closely related to energy metabolism.33 AMPK is known as an “energy detector” in cells and closely associated with maintaining the balance of cellular energy supply and demand by influencing multiple aspects of cellular substance metabolism. A recent study proved that the deletion of AMPK in the atria promotes both the triggers and the substrate that predispose to the development of atrial fibrillation.15 The p-AMPK, as active form of AMPK, plays a major role in vivo. The expression of p-AMPK was significantly decreased in diabetic mice and in cardiomyocytes cultured with high glucose.34 Our work also proved that the expression of p-AMPK decreased in diabetic atrial tissue and AGEs cultured cells.
Dufeys’s study explored that Cx43 in myofibroblast was significantly reduced in a mouse model of conditionally knocked out for myofibroblast-restricted AMPK α1.13 Similarly, we knocked down AMPK in HL-1 cells and the expression of Cx40 and Cx43 was significantly downregulated. Hence, we hypothesize that AGEs impaired AMPK activity, resulting in a decrease in Cx43 and Cx40 and our experiments proved that both Cx43 and Cx40 were significantly decreased after intervention with AGEs in HL-1 cells and primary atrial myocytes.
AICAR has been one of the most commonly used pharmacological modulators of AMPK activity.35 The protein level of Cx43 and Cx40 which was inhibited by AGEs was both restored by AICAR treatment for 24 hours after AGEs incubation for 24 hours and functional assays showed that AICAR also partially restored gap channel function. Different from post-AGEs treatment, AICAR pretreatment for 6 hours did not affect the Cx40 expression. This suggests different activation time or duration may affect the role of AMPK in AGEs induced – Cx40 regulation (data is shown in Supplement Figure 1). It has been reported that AMPK deficiency in mice atria promotes triggers and substrates that induce AF by disrupting the expression of Gja5 (the gene encoding Cx40).15 Consistently, we found Cx40 and Cx43 were decreased by AMPK downregulation with siRNA. However, the exact mechanism remains to be further explored.
There are several limitations in this study. First, the in vivo experiments suggest that AGEs reduce the expression of connexins, but no in vivo experiments were performed to show the role of AGEs in connexins and electrical conduction regulation. It would be best to test the conduction in vivo models by inhibiting AGEs formation or in ex vivo models of whole hearts or dissected atria. Second, immunofluorescence of Cx40 and Cx43 was not performed. Immunofluorescence can visually observe the expression and distribution of connexin. Third, the mechanisms underlying the inhibitory effects of AGEs on AMPK phosphorylation in atrium need to be further explored.
Conclusion
The present study demonstrates that AGEs decreased myocardial Cx43 and Cx40 expression through inhibition of the AMPK signaling pathway, which increase the susceptibility to DM-related atrial fibrillation. Reducing the production of AGEs and improving diabetic myocardial energy metabolism may influence AF by modulating gap junction function. Therefore, AGEs may be an intervention target for the prevention and treatment of diabetic atrial fibrillation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sun H, Saeedi P, Karuranga S, et al. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119. doi:10.1016/j.diabres.2021.109119
2. Svane J, Pedersen-Bjergaard U, Tfelt-Hansen J. Diabetes and the Risk of Sudden Cardiac Death. Curr Cardiol Rep. 2020;22(10):112. doi:10.1007/s11886-020-01366-2
3. Cosentino F, Grant PJ, Aboyans V, et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur Heart J. 2020;41(2):255–323. doi:10.1093/eurheartj/ehz486
4. Yu M, Zhan X, Yang Z, Huang Y. Measuring the global, regional, and national burden of type 2 diabetes and the attributable risk factors in all 194 countries. J Diabetes. 2021;13(8):613–639. doi:10.1111/1753-0407.13159
5. Wang A, Green JB, Halperin JL, Piccini JP. Atrial Fibrillation and Diabetes Mellitus: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;74(8):1107–1115. doi:10.1016/j.jacc.2019.07.020
6. Twarda-Clapa A, Olczak A, Białkowska AM, Koziołkiewicz M. Advanced Glycation End-Products (AGEs): formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells. 2022;11(8):1312. doi:10.3390/cells11081312
7. Khalid M, Petroianu G, Adem A. Advanced Glycation End Products and Diabetes Mellitus: mechanisms and Perspectives. Biomolecules. 2022;12(4):542. doi:10.3390/biom12040542
8. Peppa M, Uribarri J, Vlassara H. Glucose, Advanced Glycation End Products, and Diabetes Complications: what Is New and What Works. Clin Diabetes. 2003;21(4):186–187. doi:10.2337/diaclin.21.4.186
9. Fishman SL, Sonmez H, Basman C, et al. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: a review. Mol Med. 2018;24(1):59. doi:10.1186/s10020-018-0060-3
10. Bohm A, Urban L, Tothova L, et al. Advanced glycation end products predict long-term outcome of catheter ablation in paroxysmal atrial fibrillation. J Interv Card Electrophysiol. 2022;64(1):17–25. doi:10.1007/s10840-021-00972-6
11. Chen YC, Lu YY, Wu WS, et al. Advanced glycation end products modulate electrophysiological remodeling of right ventricular outflow tract cardiomyocytes: a novel target for diabetes-related ventricular arrhythmogenesis. Physiol Rep. 2022;10(21):e15499. doi:10.14814/phy2.15499
12. Zheng DL, Wu QR, Zeng P, et al. Advanced glycation end products induce senescence of atrial myocytes and increase susceptibility of atrial fibrillation in diabetic mice. Aging Cell. 2022;21(12):e13734. doi:10.1111/acel.13734
13. Gollob MH, Jones DL, Krahn AD, et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354(25):2677–2688. doi:10.1056/NEJMoa052800
14. Dufeys C, Daskalopoulos EP, Castanares-Zapatero D, et al. AMPKα1 deletion in myofibroblasts exacerbates post-myocardial infarction fibrosis by a connexin 43 mechanism. Basic Res Cardiol. 2021;116(1):10. doi:10.1007/s00395-021-00846-y
15. Su KN, Ma Y, Cacheux M, et al. Atrial AMP-activated protein kinase is critical for prevention of dysregulation of electrical excitability and atrial fibrillation. JCI Insight. 2022;7(8):e141213. doi:10.1172/jci.insight.141213
16. Zhang ZY, Dang SP, Li SS, et al. Glucose Fluctuations Aggravate Myocardial Fibrosis via the Nuclear Factor-κB-Mediated Nucleotide-Binding Oligomerization Domain-Like Receptor Protein 3 Inflammasome Activation. Front Cardiovasc Med. 2022;9:748183. doi:10.3389/fcvm.2022.748183
17. Wu LD, Liu Y, Li F, et al. Glucose fluctuation promotes cardiomyocyte apoptosis by triggering endoplasmic reticulum (ER) stress signaling pathway in vivo and in vitro. Bioengineered. 2022;13(5):13739–13751. doi:10.1080/21655979.2022.2080413
18. Gong H, Chen H, Xiao P, et al. MiR-146a impedes the anti-aging effect of AMPK via NAMPT suppression and NAD+/SIRT inactivation. Signal Transduct Target Ther. 2022;7(1):66. doi:10.1038/s41392-022-00886-3
19. Yang Q, Shi Y, Jin T, et al. Advanced Glycation End Products Induced Mitochondrial Dysfunction of Chondrocytes through Repression of AMPKα-SIRT1-PGC-1α Pathway. Pharmacology. 2022;107(5–6):298–307. doi:10.1159/000521720
20. Qin W, Zhang L, Li Z, et al. Metoprolol protects against myocardial infarction by inhibiting miR-1 expression in rats. J Pharm Pharmacol. 2020;72(1):76–83. doi:10.1111/jphp.13192
21. Yamagishi SI, Sotokawauchi A, Matsui T. Pathological Role of Advanced Glycation End Products (AGEs) and their Receptor Axis in Atrial Fibrillation. Mini Rev Med Chem. 2019;19(13):1040–1048. doi:10.2174/1389557519666190311140737
22. Al-Saoudi E, Christensen MMB, Nawroth P, et al. Advanced glycation end-products are associated with diabetic neuropathy in young adults with type 1 diabetes. Front Endocrinol. 2022;13:891442. doi:10.3389/fendo.2022.891442
23. Méndez JD, Xie J, Aguilar-Hernández M, Méndez-Valenzuela V. Trends in advanced glycation end products research in diabetes mellitus and its complications. Mol Cell Biochem. 2010;341(1–2):33–41. doi:10.1007/s11010-010-0434-5
24. Schalkwijk CG, Stehouwer CDA. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol Rev. 2020;100(1):407–461. doi:10.1152/physrev.00001.2019
25. Maasen K, Eussen SJPM, Dagnelie PC, et al. Habitual intake of dietary methylglyoxal is associated with less low-grade inflammation: the Maastricht Study. Am J Clin Nutr. 2022;116(6):1715–1728. doi:10.1093/ajcn/nqac195
26. Thakur M, Tupe RS. Lipoxin and glycation in SREBP signaling: insight into diabetic cardiomyopathy and associated lipotoxicity. Prostaglandins Other Lipid Mediat. 2023;164:106698. doi:10.1016/j.prostaglandins.2022.106698
27. Patti G, Lucerna M, Cavallari I, et al. Insulin-Requiring Versus Noninsulin-Requiring Diabetes and Thromboembolic Risk in Patients With Atrial Fibrillation: PREFER in AF. J Am Coll Cardiol. 2017;69(4):409–419. doi:10.1016/j.jacc.2016.10.069
28. Shah MS, Brownlee M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ Res. 2016;118(11):1808–1829. doi:10.1161/CIRCRESAHA.116.306923
29. Noureldin M, Chen H, Bai D. Functional Characterization of Novel Atrial Fibrillation-Linked GJA5 (Cx40). Mutants Int J Mol Sci. 2018;19(4):977. doi:10.3390/ijms19040977
30. Chen Y, Liu H, Zheng Q, et al. Promotion of tumor progression induced by continuous low-dose administration of antineoplastic agent gemcitabine or gemcitabine combined with cisplatin. Life Sci. 2022;306:120826. doi:10.1016/j.lfs.2022.120826
31. Yan J, Thomson JK, Zhao W, et al. The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J Mol Cell Cardiol. 2018;114:105–115. doi:10.1016/j.yjmcc.2017.11.006
32. Ma JF, Yang F, Mahida SN, et al. TBX5 mutations contribute to early-onset atrial fibrillation in Chinese and Caucasians. Cardiovasc Res. 2016;109(3):442–450. doi:10.1093/cvr/cvw003
33. Tu T, Zhou S, Liu Z, Li X, Liu Q. Quantitative proteomics of changes in energy metabolism-related proteins in atrial tissue from valvular disease patients with permanent atrial fibrillation. Circ J. 2014;78(4):993–1001. doi:10.1253/circj.CJ-13-1365
34. Zhang Z, Ni L, Zhang L, et al. Empagliflozin Regulates the AdipoR1/p-AMPK/p-ACC Pathway to Alleviate Lipid Deposition in Diabetic Nephropathy. Diabetes Metab Syndr Obes. 2021;14:227–240. doi:10.2147/DMSO.S289712
35. Višnjić D, Lalić H, Dembitz V, Tomić B, Smoljo T. AICAr, a Widely Used AMPK Activator with Important AMPK-Independent Effects: a Systematic Review. Cells. 2021;10(5):1095. doi:10.3390/cells10051095
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.