Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 8
Adult-onset Still's disease: current challenges and future prospects
Authors Siddiqui M, Putman M, Dua A
Received 3 November 2015
Accepted for publication 31 December 2015
Published 15 March 2016 Volume 2016:8 Pages 17—22
DOI https://doi.org/10.2147/OARRR.S83948
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Chuan-Ju Liu
Mariam Siddiqui,1 Michael S Putman,2 Anisha B Dua,1
1Department of Rheumatology, 2Department of Internal Medicine, The University of Chicago Medical Center, Chicago, IL, USA
Abstract: Adult-onset Still's disease (AOSD) – a multi-systemic inflammatory condition characterized by high fevers, polyarthritis, an evanescent rash, and pharyngitis – has been a challenging condition to diagnose expediently and treat effectively. Questions remain regarding the underlying pathophysiology and etiology of AOSD. Pathognomonic diagnostic tests and reliable biomarkers remain undiscovered. Over the past decade, important progress has been made. Diagnostic criteria employing glycosylated ferritin have improved specificity. More important, novel biologic therapies have offered important clues to AOSD's underlying pathophysiology. Cytokine-specific biologic therapies have been instrumental in providing more effective treatment for disease refractory to conventional treatment. While IL-1 therapy has demonstrated efficacy in refractory disease, novel therapies targeting IL-6 and IL-18 show great promise and are currently under investigation.
Keywords: adult-onset Still's disease, biomarkers, therapeutics
Introduction
Adult-onset Still’s disease (AOSD) is a rare multisystemic inflammatory condition characterized by high fevers, polyarthritis or arthralgias, an evanescent rash, and pharyngitis. Though first noted in children as early as 1897 by Dr George Still, AOSD remained largely unrecognized until its description by Bywaters in 1971.1 Since then, AOSD and systemic juvenile idiopathic arthritis (sJIA), which share many clinical features, have been more clearly defined from a clinical perspective and have been affected by the revolution of biologic therapies.2,3 Despite this progress, many challenges remain. The etiologic factors and pathogenesis of AOSD are largely obscure. Diagnostic delays and disease complications are exacerbated by the lack of pathognomonic serologic or clinical disease markers. Clinical research has essentially been limited to case reports and small retrospective series. Going forward, research on the mechanisms that underlie AOSD may provide new avenues for identifying at-risk groups, diagnosing AOSD more expediently, identifying prognostic markers, and providing more targeted therapy.
Etiology and pathogenesis
Despite decades of research, disease triggers, genetic markers, and pathogenesis remain unclear. In a large case–control study of age-matched siblings, AOSD patients had no increased rates of smoking, substance abuse, surgery, pregnancy, transfusions, or dietary changes.4 Further, AOSD does not show increased incidence in families, ethnic groups, or certain geographic areas, suggesting against a single-gene association.5 Several other case studies have looked at distinct HLA alleles in their association with AOSD. In a series of 29 cases, HLA-DR4 was found to be prevalent.6 A subsequent survey in Canada described strong correlations between AOSD and HLA-B17, B18, B35, and DR2.7 Others have shown links between HLA-DR7 and HLA-DRB1 genotypes and AOSD.8,9 Thus, no consistent results have been obtained from association studies and HLA gene loci. In general, strong relationships have been observed between the inflammatory cytokines IL-6, IL-1, and IL-18 in both AOSD and sJIA.2,10
Infections and malignancies have been suspected as etiologies of AOSD, largely due to their similar clinical presentations and temporal relationships with AOSD diagnoses. Many microorganisms, including parvovirus B19, rubella, echovirus 7, Yersinia spp., Epstein–Barr virus, cytomegalovirus, coxsackie B4, and chlamydia, have been observed concurrently with AOSD.2,11–14 Similarly, individual instances of AOSD have been seen in association with esophageal cancer, breast cancer, renal cell carcinoma, ovarian cancer, melanoma, papillary thyroid cancer, non-Hodgkin’s lymphoma, and multiple other cancers.15–21 However, a causal relationship between infection or malignancy and AOSD has never been clearly established. In short, various genetic, infectious, and malignant etiologies have been suggested but none have been reproducible in larger series.
Diagnostic challenges
Diagnosing AOSD or sJIA can be difficult given the lack of sensitivity of their principal features – high fevers, arthritis, and rash. This presentation can easily be confused with infections, other autoimmune diseases, and malignancies. In one series of patients presenting with fever of unknown origin, 90% of those eventually diagnosed with AOSD also received antibiotics.22 Moreover, AOSD and sJIA occur rarely, with incidences estimated at 0.16 cases per 100,000 people per year23 and 0.6 per 100,000 people per year, respectively.10 Due to the infrequency of the condition and the nonspecific clinical presentation, there are often delays in diagnosis. A recent retrospective series of 57 patients found a mean time to diagnosis of 4 months.5 In addition, patients presenting with arthritis and a rash may receive empiric nonsteroidal anti-inflammatory medications or even a course of steroids, which may inadvertently treat the episode of AOSD.
Complicating expedient diagnosis of AOSD, a definitive diagnostic test has yet to be discovered. Depending on which criteria are employed, rheumatoid factor and antinuclear antibody tests are negative by definition. Nonspecific elevations of inflammatory markers, such as C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR), are present in almost 100% of cases.5,7 Given a differential diagnosis of infections, neoplasms, and other autoimmune diseases, these are not generally helpful. Bone marrow biopsies typically reveal nonspecific granulocyte hyperplasia consistent with reactive bone marrow,5,7 and biopsies of other tissues are primarily helpful in ruling out malignancy.
The most promising index to date – the glycosylated ferritin – has been useful but not definitive. While 50%–80% of serum ferritin is glycosylated in healthy controls, studies have shown a significant reduction among those with AOSD.24 Using serum ferritin levels five times the normal value and glycosylated ferritin ≤20% yields a specificity of 92.9%, which is higher than the majority of published diagnostic criteria.25,26 Yet, this measure is only 43.2% sensitive, and is not routinely checked as part of a diagnostic workup. Moreover, although markedly elevated ferritin levels are seen in 70% of those with AOSD, high ferritin levels are also a common feature of malignancy and infection.27 Consequently, measures of ferritin may only be useful when they are extremely elevated, clinical suspicion is already high, or as a part of fulfilling diagnostic criteria.
Given these limitations, multiple clinical criteria have been proposed.28–33 The Yamaguchi criteria have the highest sensitivity (93.5%) and the Fautrel criteria have the highest specificity (98.5%).32,33 The Yamaguchi criteria have become the most widely accepted, but they do not include glycosylated ferritin and require the exclusion of neoplasms, infections, and autoimmune diseases that mimic AOSD (Table 1). Using a 2002 retrospective analysis of 72 patients with AOSD and 130 controls, Fautrel et al developed updated criteria with a sensitivity of 80.6% and a specificity of 98.5% (Table 1).33 Because of the higher specificity and lack of exclusions, these criteria may form the basis for more precise research going forward. However, the relatively low sensitivity means that the Yamaguchi criteria should still be used to rule out AOSD.
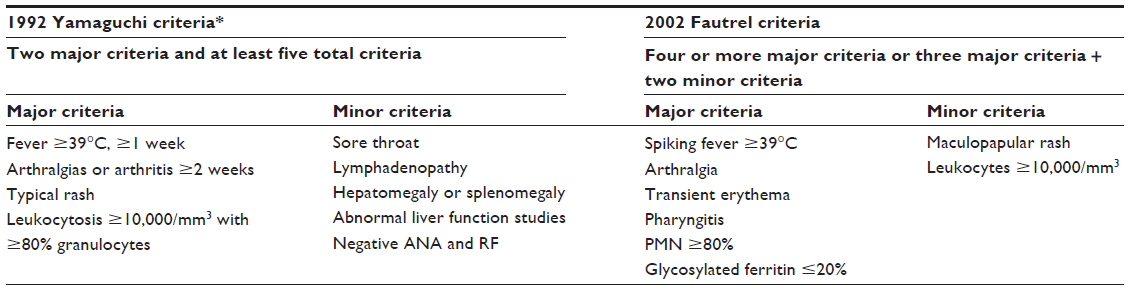 | Table 1 Two proposed sets of diagnostic criteria for AOSD |
Future prospects in diagnostic biomarkers
While glycosylated ferritin shows promise, other novel biomarkers have also been identified. A recent study demonstrated higher levels of germinal center kinase-like kinase (GLK)-expressing T-cells in sera of AOSD patients (31.85% vs 8.93% for healthy volunteers, P<0.001). These findings also correlated with disease activity and GLK expression levels that decreased after therapy for AOSD.34 In a study of the calcium-binding proteins S100A8, S100A9, and IL-18 on 36 patients with AOSD, S100A8/A9 levels were higher than in patients with rheumatoid arthritis or healthy controls, correlated with known inflammatory markers and disease activity scores, and significantly decreased after treatment of AOSD. IL-18 levels performed even better for diagnosis, with a sensitivity of 91.7% and a specificity of 99.1% at a cutoff of 366.1 pg/mL, but did not decrease after treatment of AOSD.35 Finally, in a study of 30 patients with AOSD and age-matched controls, mean calprotectin levels were significantly higher (61 ng/ml vs 35 ng/ml, P<0.05) and levels decreased after treatment of AOSD.36 None of these markers have been tested in a large cohort of patients with infections, neoplasms, or a variety of autoimmune diseases. These need to be evaluated in larger studies in order to find more sensitive, specific, and reliable markers in diagnosing AOSD.
Prognostic factors
Traditionally, three patterns of AOSD manifest after initial diagnosis – systemic monocyclic, intermittent/polycyclic, and chronic articular.29 Polyarthritis and joint erosions at disease onset are predictive of chronic articular disease.5–7,28 Still’s rash has also been reported as a predictor of poor functional outcomes.7 Several investigators have attempted to identify those at increased risks of morbidity and mortality, but data regarding disease course, ultimate outcomes, and therapeutic responses of AOSD are limited. Macrophage activation syndrome (MAS), disseminated intravascular coagulopathy, thrombotic thrombocytopenic purpura, diffuse alveolar hemorrhage, pulmonary arterial hypertension, and rarely respiratory and hepatic failure have all been described as secondary complications of AOSD.37 The estimated incidence of MAS in AOSD is 12%–14%; not only is this higher than in other rheumatologic diseases,38,39 but in sJIA 53% of patients had bone marrow biopsies suggestive of MAS.40 To date, the majority of prognostic factors have been identified in retrospective studies; ideally, future prospective studies will more accurately define prognostic factors.
Research limitations
As with many rare medical conditions, progress on AOSD has been hampered by the difficulty of enrolling a sufficient number of patients for large randomized trials. A recent review in Rheumatology of all biologic therapies for AOSD covering the years 2000–2012 identified only one randomized controlled trial out of 78 published articles.3 Most of the data came from retrospective case series, and over half of published papers were case reports. Though compiling dozens of case reports may be instructive, without randomized controlled trials, this research is vulnerable to both selection bias and publication bias and should be questioned regularly. The paucity of large randomized trials in AOSD has likely been secondary to the rarity of the disease. Rare diseases, defined as affecting <200,000 people in the US, present unique challenges to the medical community. Specifically, affected populations encounter difficulty obtaining timely and accurate diagnoses, struggle to find reliable resources or experienced providers, and often must pay exorbitant fees for what drugs do become available.41
Current treatments
The treatment of AOSD has been largely derived from case reports and retrospective data. While mild cases can be treated with nonsteroidal anti-inflammatory drugs (NSAIDs), more than 80% of AOSD patients do not achieve remission and require the use of corticosteroids.5 Both of these pharmaceutical classes cause significant side effects. In one recent series, almost half of the cohort developed corticosteroid-dependent disease and 75% of the total cohort suffered corticosteroid-related side effects, such as Cushing syndrome, osteoporosis, and aseptic osteonecrosis.5 In the event of corticosteroid treatment failure or dependence, disease-modifying antirheumatic drugs (DMARDs) have been used. Modest success has been achieved with several DMARDs, including methotrexate, sulfasalazine, cyclosporin A, leflunomide, azathioprine, hydroxychloroquine, and tacrolimus.5,7,42,43 Of these DMARDs, methotrexate remains the first-line steroid sparing agent in the management of AOSD.5 In a retrospective series of 26 patients with AOSD refractory to prednisone who were treated with methotrexate, 18 patients achieved remission and eleven patients were able to discontinue prednisone entirely.44 Yet one-third of methotrexate-treated patients experience elevated liver enzymes, drug-induced cytopenias, or cough.5 In an era of biologic medicines, several promising targets have emerged in the treatment of AOSD. These therapies have also brought to light potential information regarding disease pathogenesis and are an essential aspect of the future prospects of AOSD.
IL-1 inhibition
Of these novel therapies, the recombinant IL-1 receptor antagonist anakinra has been most widely used and remains the only biologic therapy yet subjected to a randomized controlled trial. A retrospective study of 28 patients who had received anakinra for refractory AOSD found a rapid improvement in all patients, and 12 patients experienced persistent complete remission at a mean of 23 months.45 In a similarly encouraging retrospective study, eleven out of 15 patients with AOSD achieved 50% improvement in all disease markers.46 Most important, anakinra demonstrated efficacy in a randomized multicenter trial of anakinra vs DMARDs (methotrexate, azathioprine, leflunomide). When compared to conventional DMARDs, a greater number of patients receiving anakinra achieved remission over 24 weeks (P=0.22) and scores on a Medical Outcomes Study Short-Form 36 (SF-36) physical health summary significantly improved.47
Given these encouraging results for IL-1 blockade with short-acting anakinra, canakinumab, a monoclonal IL-1β antibody with a half-life of 26 days, is being further investigated. A randomized controlled trial in sJIA found more children achieving JIA American College of Rheumatology 30 response at 15 days (84% canakinumab vs 10% placebo, P<0.001) and more without a disease flare in a withdrawal phase of the trial (74% canakinumab vs 25% placebo, P=0.003).48 Data remain limited in AOSD, but a retrospective case series of two patients treated with canakinumab, who had previously failed to respond to anakinra therapy, demonstrated sustained improvement in clinical and laboratory parameters.49 With these encouraging results, a randomized controlled trial of canakinumab vs placebo is currently recruiting patients.50 Though there has been concern that agents such as anakinra increase the risk of malignancy, a recent Cochrane review did not find this, instead indicating that injection site issues, serious infections, and tuberculosis were more common among those taking biologic response modifiers and other DMARD therapies.51
IL-6 inhibition
The monoclonal antibody to IL-6, tocilizumab, has also shown promise in refractory AOSD. In a retrospective study on 16 patients with AOSD, half of the patients initially receiving either infliximab or etanercept were changed to another agent, while ten of eleven eventually treated with tocilizumab responded well and two discontinued corticosteroids entirely.52 In another retrospective study of 14 patients who had failed methotrexate and anakinra, treatment with tocilizumab led to a reduction in mean corticosteroid dosing by 56% and European League Against Rheumatism remission in 57% at 6 months.53 In sJIA, serum IL-6 levels are elevated, as compared with healthy controls, and correlate with joint symptoms and platelet counts.54 Data from recent randomized controlled trials of tocilizumab in sJIA have been encouraging. In a Phase II trial, investigators assigned 112 children with active sJIA to tocilizumab or placebo; at week 12, 85% of those receiving tocilizumab had improved compared with 24% in the placebo arm, P<0.001.54 A follow-up study of 417 children confirmed these findings, with normalization of CRP in 99% and improvement in fevers from 54.6% of the original cohort to 5.6% of patients at 52 weeks.55 Given these results, the TENDER study, a multicenter, three-part, 5-year trial, is under way to assess the efficacy and safety of tocilizumab in sJIA.56
Novel therapeutics
While targeting IL-1 and IL-6 has shown promise in the treatment of AOSD, IL-18 is considered to be crucial in activating macrophages and favoring Th1-type cytokine production.2 Multiple studies have documented high levels of IL-18 during active AOSD, which decreases after treatment.57,58 IL-18-binding protein, tadekinig alfa, binds IL-18, preventing it from activating its receptor or stimulating IFNγ production. Though it has yet to be used in patients with AOSD, from a theoretical perspective tadekinig alfa may be a promising novel therapeutic agent. It has already been tested in Phase I studies in other diseases, and an open-label, dose-finding study in AOSD involving multiple centers in Europe is under way.59
Studies have also shown that Th17 cells, which produce IL-17, are significantly higher in patients with active AOSD than in healthy controls and correlate with disease activity.34 Therapies directed against IL-17 have been studied in rheumatoid arthritis, psoriasis, inflammatory bowel disease, and multiple sclerosis.60 In January 2015, the US FDA approved the use of secukinumab, an IL-17A-inhibiting monoclonal antibody, in the treatment of psoriasis. A recent review by Jamilloux et al has suggested potential benefit from IL-17 inhibitors in articular patterns of AOSD as well.61
Conclusion
AOSD is a rare systemic inflammatory condition that is poorly understood. It poses many diagnostic challenges, which are further magnified by the rarity of the disease and the paucity of large randomized trials. While the etiology of AOSD remains unknown, advances in immunology have enhanced our knowledge of the role of cytokines in disease pathogenesis. This, in turn, has led to the off-label use of several promising treatments originally intended for use in rheumatoid arthritis. Results from IL-1- and IL-6-inhibiting drugs have been very encouraging. With the prospect of the development of more novel therapies that target IL-17 and IL-18, we are only beginning to explore the potential of biologics in AOSD.
Disclosure
The authors report no conflicts of interest in this work.
References
Bywaters EG. Still’s disease in the adult. Ann Rheum Dis. 1971;30(2):121–133. | |
Kadavath S, Efthimiou P. Adult-onset Still’s disease-pathogenesis, clinical manifestations, and new treatment options. Ann Med. 2015;47(1):6–14. | |
Al-Homood IA. Biologic treatments for adult-onset Still’s disease. Rheumatology (Oxford). 2014;53(1):32–38. | |
Sampalis JS, Medsger TA, Jr, Fries JF, et al. Risk factors for adult Still’s disease. J Rheumatol. 1996;23(12):2049–2054. | |
Gerfaud-Valentin M, Maucort-Boulch D, Hot A, et al. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014;93(2):91–99. | |
Wouters JM, van de Putte LB. Adult-onset Still’s disease; clinical and laboratory features, treatment and progress of 45 cases. Q J Med. 1986;61(235):1055–1065. | |
Pouchot J, Sampalis JS, Beaudet F, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991;70(2):118–136. | |
Miller ML, Aaron S, Jackson J, et al. HLA gene frequencies in children and adults with systemic onset juvenile rheumatoid arthritis. Arthritis Rheum. 1985;28(2):146–150. | |
Joung CI, Lee HS, Lee SW, et al. Association between HLA-DR B1 and clinical features of adult onset Still’s disease in Korea. Clin Exp Rheumatol. 2003;21(4):489–492. | |
Correll CK, Binstadt BA. Advances in the pathogenesis and treatment of systemic juvenile idiopathic arthritis. Pediatr Res. 2014;75(1–2):176–183. | |
Pouchot J, Ouakil H, Debin ML, Vinceneux P. Adult Still’s disease associated with acute human parvovirus B19 infection. Lancet. 1993;341:1280–1281. | |
Huang SH, DeCoteau WE. Adult-onset Still’s disease: an unusual presentation of rubella infection. Can Med Assoc J. 1980;122(11):1275–1276. | |
Wouters JM, van der Veen J, van de Putte LB, de Rooij DJ. Adult onset Still’s disease and viral infections. Ann Rheum Dis. 1988;47(9):764–767. | |
Colebunders R, Stevens WJ, Vanagt E, Snoeck J. Adult Still’s disease caused by Yersinia enterocolitica infection. Arch Intern Med. 1984;144(9):1880–1882. | |
Shibuya Y, Matuo K, Kawada T, Kosugi T, Gomi T. [Adult onset Still’s disease associated esophageal cancer: a case report]. Ryumachi. 2003;43(3):577–582. Japanese. | |
Neishi J, Tsukada Y, Maehara T, Ueki K, Maezawa A, Nojima Y. Adult Still’s disease as a paraneoplastic manifestation of breast cancer. Scand J Rheumatol. 2000;29(5):328–330. | |
Yilmaz S, Karakas A, Cinar M, et al. Adult onset Still’s disease as a paraneoplastic syndrome – a case report and review of the literature. Bull Hosp Jt Dis (2013). 2013;71(2):156–160. | |
Schade L, Fritsch S, Gentili AC, de Noronha L, Azevedo VF, Paiva Edos S. Adult Still’s disease associated with ovarian cancer: case report. Rev Bras Reumatol. 2013;53(6):532–534. | |
Liozon E, Ly KH, Vidal-Cathala E, Fauchais AL. Pseudomaladie de Still et néoplasie: une observation de mélanome et revue de la littérature [Adult-onset Still’s disease as a manifestation of malignancy: report of a patient with melanoma and literature review]. Rev Med Interne. 2014;35(1):60–64. French. | |
Ahn JK, Oh JM, Lee J, Kim SW, Cha HS, Koh EM. Adult onset Still’s disease diagnosed concomitantly with occult papillary thyroid cancer: paraneoplastic manifestation or coincidence? Clin Rheumatol. 2010;29(2):221–224. | |
Otrock ZK, Hatoum HA, Uthman IW, Taher AT, Saab S, Shamseddine AI. Non-Hodgkin’s lymphoma in a woman with adult-onset Still’s disease: a case report. J Med Case Rep. 2008;2:73. | |
Mert A, Ozaras R, Tabak F, et al. Fever of unknown origin: a review of 20 patients with adult-onset Still’s disease. Clin Rheumatol. 2003;22(2):89–93. | |
Magadur-Joly G, Billaud E, Barrier JH, et al. Epidemiology of adult Still’s disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995;54(7):587–590. | |
Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still’s disease. Ann Rheum Dis. 2006;65(5):564–572. | |
Fautrel B, Le Moel G, Saint-Marcoux B, et al. Diagnostic value of ferritin and glycosylated ferritin in adult onset Still’s disease. J Rheumatol. 2001;28(2):322–329. | |
Masson C, Le Loet X, Liote F, et al. Comparative study of 6 types of criteria in adult Still’s disease. J Rheumatol. 1996;23(3):495–497. | |
Moore C, Jr., Ormseth M, Fuchs H. Causes and significance of markedly elevated serum ferritin levels in an academic medical center. J Clin Rheumatol. 2013;19(6):324–328. | |
Calabro JJ, Londino AV, Jr. Adult onset Still’s disease. J Rheumatol. 1986;13(4):827–828. | |
Cush JJ, Medsger TA, Jr, Christy WC, Herbert DC, Cooperstein LA. Adult-onset Still’s disease. Clinical course and outcome. Arthritis Rheum. 1987;30(2):186–194. | |
Goldman JA, Beard MR, Casey HL. Acute febrile juvenile rheumatoid arthritis in adults: cause of polyarthritis and fever. South Med J. 1980;73(5):555–563. | |
Reginato AJ, Schumacher HR, Jr, Baker DG, O’Connor CR, Ferreiros J. Adult onset Still’s disease: experience in 23 patients and literature review with emphasis on organ failure. Semin Arthritis Rheum. 1987;17(1):39–57. | |
Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19(3):424–430. | |
Fautrel B, Zing E, Golmard JL, et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore). 2002;81(3):194–200. | |
Chen DY, Chuang HC, Lan JL, et al. Germinal center kinase-like kinase (GLK/MAP4K3) expression is increased in adult-onset Still’s disease and may act as an activity marker. BMC Med. 2012;10:84. | |
Kim HA, An JM, Nam JY, Jeon JY, Suh CH. Serum S100A8/A9, but not follistatin-like protein 1 and interleukin 18, may be a useful biomarker of disease activity in adult-onset Still’s disease. J Rheumatol. 2012;39(7):1399–1406. | |
Jung SY, Park YB, Ha YJ, Lee KH, Lee SK. Serum calprotectin as a marker for disease activity and severity in adult-onset Still’s disease. J Rheumatol. 2010;37(5):1029–1034. | |
Efthimiou P, Kadavath S, Mehta B. Life-threatening complications of adult-onset Still’s disease. Clin Rheumatol. 2014;33(3):305–314. | |
Arlet JB, Le TH, Marinho A, et al. Reactive haemophagocytic syndrome in adult-onset Still’s disease: a report of six patients and a review of the literature. Ann Rheum Dis. 2006;65(12):1596–1601. | |
Singh S, Samant R, Joshi VR. Adult onset Still’s disease: a study of 14 cases. Clin Rheumatol. 2008;27(1):35–39. | |
Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34(5):1133–1138. | |
Forman J, Taruscio D, Llera VA, et al. The need for worldwide policy and action plans for rare diseases. Acta Paediatr. 2012;101(8):805–807. | |
Kim YJ, Koo BS, Kim YG, Lee CK, Yoo B. Clinical features and prognosis in 82 patients with adult-onset Still’s disease. Clin Exp Rheumatol. 2014;32(1):28–33. | |
Nakamura H, Odani T, Shimizu Y, Takeda T, Kikuchi H. Usefulness of tacrolimus for refractory adult-onset still’s disease: report of six cases. Mod Rheumatol. Epub 2014 July 18. | |
Fautrel B, Borget C, Rozenberg S, et al. Corticosteroid sparing effect of low dose methotrexate treatment in adult Still’s disease. J Rheumatol. 1999;26(2):373–378. | |
Giampietro C, Ridene M, Lequerre T, et al. Anakinra in adult-onset Still’s disease: long-term treatment in patients resistant to conventional therapy. Arthritis Care Res (Hoboken). 2013;65(5):822–826. | |
Lequerre T, Quartier P, Rosellini D, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis. 2008;67(3):302–308. | |
Nordstrom D, Knight A, Luukkainen R, et al. Beneficial effect of interleukin 1 inhibition with anakinra in adult-onset Still’s disease. An open, randomized, multicenter study. J Rheumatol. 2012;39(10):2008–2011. | |
Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2396–2406. | |
Kontzias A, Efthimiou P. The use of Canakinumab, a novel IL-1beta long-acting inhibitor, in refractory adult-onset Still’s disease. Semin Arthritis Rheum. 2012;42(2):201–205. | |
Charite University. Canakinumab for Treatment of Adult Onset Still’s Disease (CONSIDER). In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US); 2000 [cited September 12, 2015]. Available from: https://clinicaltrials.gov/ct2/show/NCT02204293 NLM Identifier NCT 02204293. Accessed January 1, 2016. | |
Singh JA, Wells GA, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011;(2):CD008794. | |
Suematsu R, Ohta A, Matsuura E, et al. Therapeutic response of patients with adult Still’s disease to biologic agents: multicenter results in Japan. Mod Rheumatol. 2012;22(5):712–719. | |
Puechal X, DeBandt M, Berthelot JM, et al. Tocilizumab in refractory adult Still’s disease. Arthritis Care Res (Hoboken). 2011;63(1):155–159. | |
De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2385–2395. | |
Yokota S, Itoh Y, Morio T, et al. Tocilizumab in systemic juvenile idiopathic arthritis in a real-world clinical setting: results from 1 year of postmarketing surveillance follow-up of 417 patients in Japan. Ann Rheum Dis. Epub 2015 Dec 7. | |
Yokota S, Tanaka T, Kishimoto T. Efficacy, safety and tolerability of tocilizumab in patients with systemic juvenile idiopathic arthritis. Ther Adv Musculoskelet Dis. 2012;4(6):387–397. | |
Fitzgerald AA, Leclercq SA, Yan A, Homik JE, Dinarello CA. Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 2005;52(6):1794–1803. | |
Kotter I, Wacker A, Koch S, et al. Anakinra in patients with treatment-resistant adult-onset Still’s disease: four case reports with serial cytokine measurements and a review of the literature. Semin Arthritis Rheum. 2007;37(3):189–197. | |
AB2 Bio Ltd. Therapeutic use of tadekinig alfa in adult-onset Still’s disease. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US); 2000 [cited September 12, 2015]. Available from: https://clinicaltrials.gov/ct2/show/NCT02398435 NLM Identifier NCT NCT02398435. Accessed January 16, 2016. | |
Waite JC, Skokos D. Th17 response and inflammatory autoimmune diseases. Int J Inflam. 2012;2012:819467. | |
Jamilloux Y, Gerfaud-Valentin M, Henry T, Seve P. Treatment of adult-onset Still’s disease: a review. Ther Clin Risk Manag. 2015;11:33–43. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.