")
Back to Journals » Cancer Management and Research » Volume 13
Adrenocortical Carcinoma in Eight Children: A Report and Literature Review
Authors Li J, Zhang W, Hu H, Zhang Y, Wen Y, Huang D
Received 4 November 2020
Accepted for publication 22 January 2021
Published 11 February 2021 Volume 2021:13 Pages 1307—1314
DOI https://doi.org/10.2147/CMAR.S289191
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bilikere Dwarakanath
Jing Li, Weiling Zhang, Huimin Hu, Yi Zhang, Yuan Wen, Dongsheng Huang
Department of Pediatrics, Beijing Tongren Hospital of China Capital Medical University, Beijing, 100176, People’s Republic of China
Correspondence: Dongsheng Huang
Department of Pediatrics, Beijing Tongren Hospital of China Capital Medical University, No. 1 Dongjiao Lane, Dongcheng District, Beijing, 100730, People’s Republic of China
Tel/Fax +86 10 5826 6032
Email [email protected]
Objective: This study aimed to summarize the clinical characteristics, comprehensive treatment, and prognosis of adrenocortical carcinoma (ACC) in children.
Methods: The clinical data of eight children with definite diagnoses of ACC were retrospectively analyzed, and statistical methods were used to analyze the clinical characteristics, comprehensive treatment mode, and prognosis of these patients.
Results: (1) Clinical characteristics: two were males and six were females with the median age of onset was six-years old were involved. Four patients had a rash and precocious puberty as the symptoms of onset. European Network for the Study of Adrenal Tumors (ENSAT) staging: stage II, two patients; stage IV, six patients. (2) Comprehensive treatment: all eight patients underwent surgical treatment and received six cycles of chemotherapy: the regimen was “etoposide + pirarubicin + cisplatin + mitotane.” (3) Prognosis analysis: among these eight patients, two patients died, two patients achieved complete remission, the disease was stable in four patients, and the overall five-year survival rate was 75%. Prognosis analyzed according to ENSAT staging (stage II versus stage IV) revealed that two-year survival rates of the two groups were 100% versus 65%, respectively, without statistical significant (χ2 = 1.066, P = 0.302). Prognosis analyzed according to Weiss score (Weiss score was < 6, five patients;≥ 6, three patients) revealed That survival time of the two groups was 50± 9.52 months versus 6± 1.70 months, the two-year survival rates of the two groups were 100% versus 35%, and the difference in survival rates between these two groups was statistically significant (χ2 = 4.091, P = 0.043).
Conclusion: The Weiss score is an important prognostic factor for ACC. The chemotherapy regimen “mitotane + etoposide + adriamycin + cisplatin” is recommended.
Keywords: adrenocortical carcinoma, children, prognosis
Introduction
Adrenocortical carcinoma (ACC) is a rare malignant tumor with unknown pathogenesis and poor prognosis. Foreign data show that the incidence is approximately 0.7–2 per million and patients with ACC account for 0.05–2.00% of malignant tumor patients.1 The independent annual incidence rate of ACC in children is 0.3 per million. Due to the low incidence rate of ACC, there are few reports on it. Here, we analyze the children diagnosed with ACC who were admitted to our hospital, from September 2014 to August 2019, and the clinical characteristics, treatment, and outcome are analyzed and summarized.
Methods
Subjects
The clinical data of eight children with a definite diagnosis of ACC who were treated in our hospital from September 1, 2014 to August 31, 2019 were retrospectively analyzed. Parents were invited to sign an informed consent form for all examinations and treatments that has been reviewed and approved by the Ethics Committee of our hospital.
Diagnosis and Staging
Diagnostic Criteria
All patients received surgical resection of a tumor or biopsy and were diagnosed with ACC by two or more consulting pathologists.
Clinical staging (based on the criteria formulated by the European Network for the Study of Adrenal Tumors [ENSA]):2 There are four stages (the details are shown in Table 1).
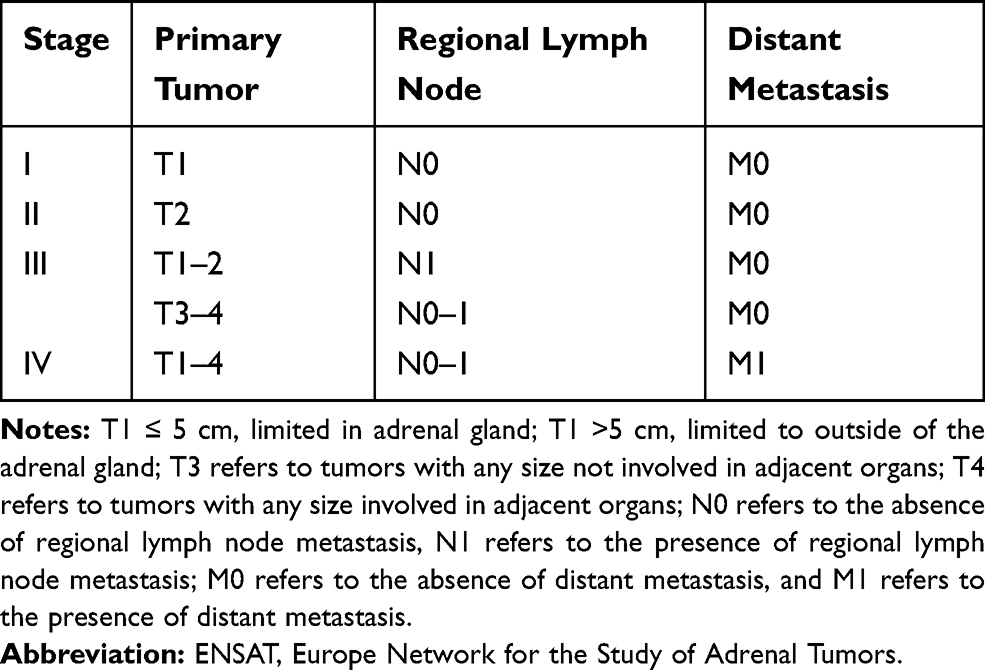 |
Table 1 ENSAT Stage of ACC Children |
Weiss Score
Histologically, the diagnostic criteria summarized by Weiss3 in 1984 are usually applied: (1) Nuclear grade III or IV; (2) Mitotic rate > 5/50 high power fields (HPF); (3) Pathological type: karyokinesis; (4) Tumor cells with eosinophilic cytoplasm account for more than 75% of the total cells; (5) Diffuse growth; (6) Extensive necrosis; (7) Invasion of vein structure; (8) Invasion of the structure of sinusoid space; and (9) Invasion of the capsule. Each item scores 1 point.
Comprehensive Treatment
Following imaging and other examinations, if it was estimated that the tumor could be removed completely, the patient underwent surgery first. Biopsy alone or adjuvant chemotherapy was performed in patients for whom complete resection presented difficulties, and the operation was performed after the tumor shrank. If surgery was performed, chemotherapy started within seven days after the operation. All the patients received six courses of chemotherapy. If the disease was stable, chemotherapy could be stopped.
Prognosis Evaluation
Complete Remission (CR)
All lesions had completely disappeared for more than four weeks.
Partial Remission (PR)
The primary tumor reduced by ≥ 64%, the metastatic tumor reduced by ≥ 30%, there were no new lesions.
Disease Progression (PD)
The primary tumor increased by ≥40% compared to its size at the initial diagnosis, the metastasis tumor increased by ≥20%, new lesions appeared.
Stable Disease (SD)
Tumor volume was between PD and PR.
Recurrence Assessment
Recurrence refers to the recurrence of local or metastatic tumor lesions one month after the complete disappearance of the tumor.
Statistical Analysis
Statistical analysis was conducted using the statistical software GraphPad Prism 8.0. The Kaplan–Meier method was used for prognosis analysis. P < 0.05 was considered statistically significant.
Results
Clinical Characteristics
Among these eight patients, two were males and six were females. The age of onset was 1.6–12 years old, and the median age of onset was six years old. The patients with an age of onset under five years old accounted for 37.5% (3/8). Four patients had rash and precocious puberty as the symptoms of onset, two patients had abdominal pain as the symptom of onset, one patient had headache and hypertension as the symptoms of onset, and one patient had precocious puberty and hypertension as the symptoms of onset. Serum testosterone and estradiol (E2) were increased in three patients, cortisol (COR) and adrenocorticotropic hormone (ATCH) were increased in three patients, and the neuron-specific enolase (NSE) level was increased in four patients. Four children were treated with chemotherapy firstly because they were not suitable for surgery, and resection was performed after the tumor shrank. According to the ENSAT standard stages, two patients were at stage II and six patients were at stage IV. Lung metastasis was found in six patients, liver metastasis was found in three patients, and lymph node metastasis was found in one patient. Clinical characteristics and pathological and laboratory features are presented in Tables 2 and 3.
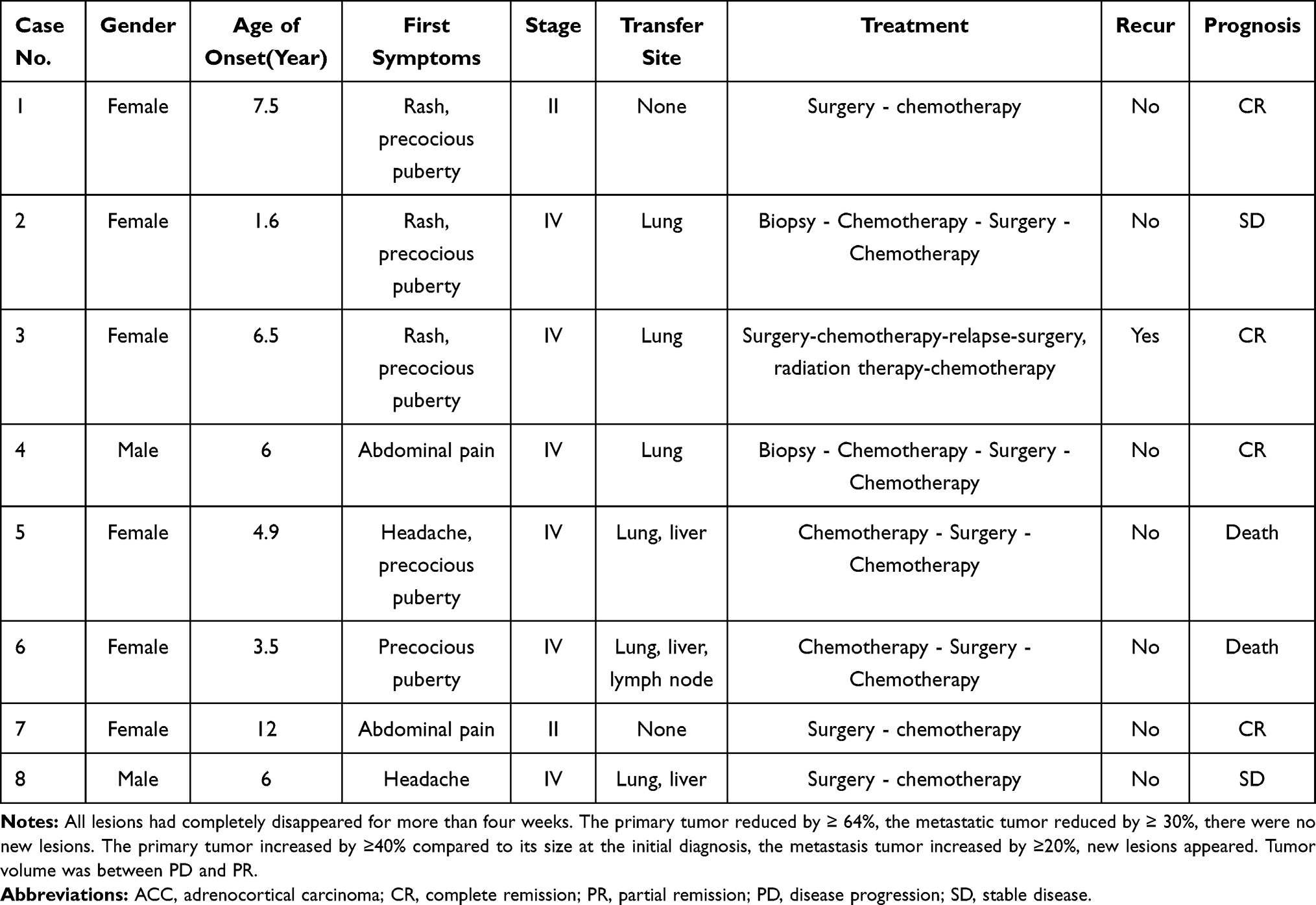 |
Table 2 Clinical Characteristics of Eight ACC Patients |
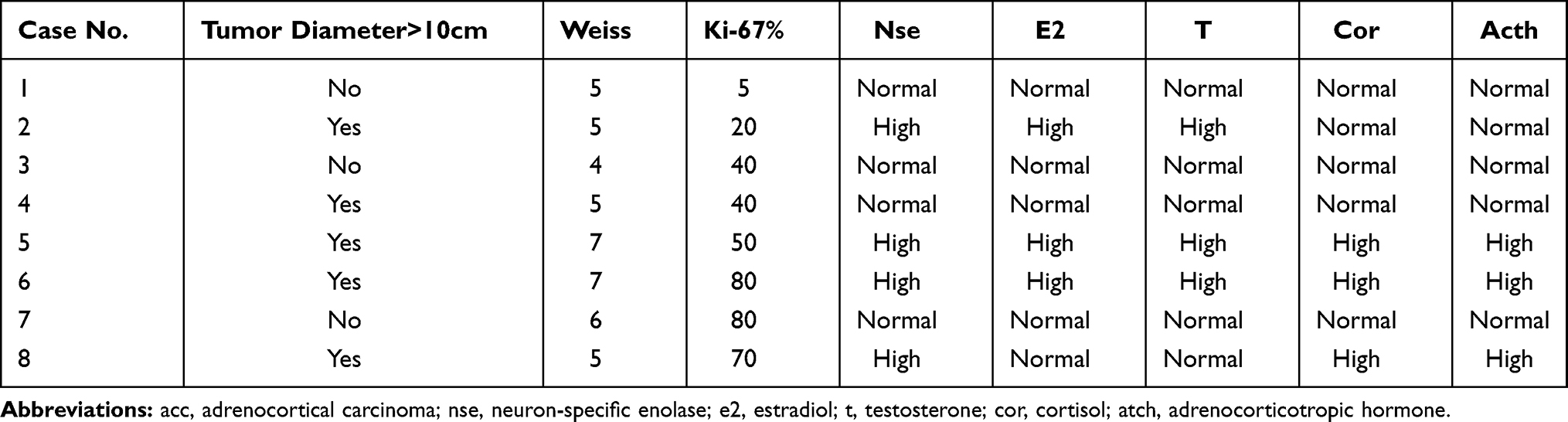 |
Table 3 Pathological and Laboratory Features of Eight acc Patients |
Comprehensive Treatment Plan
All the eight children underwent tumor resection. One patient had positive surgical margins (case 6), and in the six patients at stage IV the metastatic tumors were not resected. Chemotherapy was given to all patients after the definite diagnosis. After surgery, six cycles of chemotherapy were given. The regimen was “etoposide + pirarubicin + cisplatin + mitotane.” If the effect was not good, the regimen “streptozotocin + mitotane” was used instead. One child (case 3) was initially assessed as stage II. After complete resection, the patient received six cycles of the regimen “cisplatin + cyclophosphamide + vincristine + etoposide” and then the treatment was stopped. One year later, the tumor recurred with pulmonary metastasis. The recurrent lesion was resected, the metastatic lesion was treated with local radiotherapy, and the patient was treated with the regimen “etoposide + pirarubicin + cisplatin + mitotane.” After regular chemotherapy, the tumor visibly shrank, and the tumor thrombus in the inferior vena cava was significantly reduced, as shown in Figure 1.
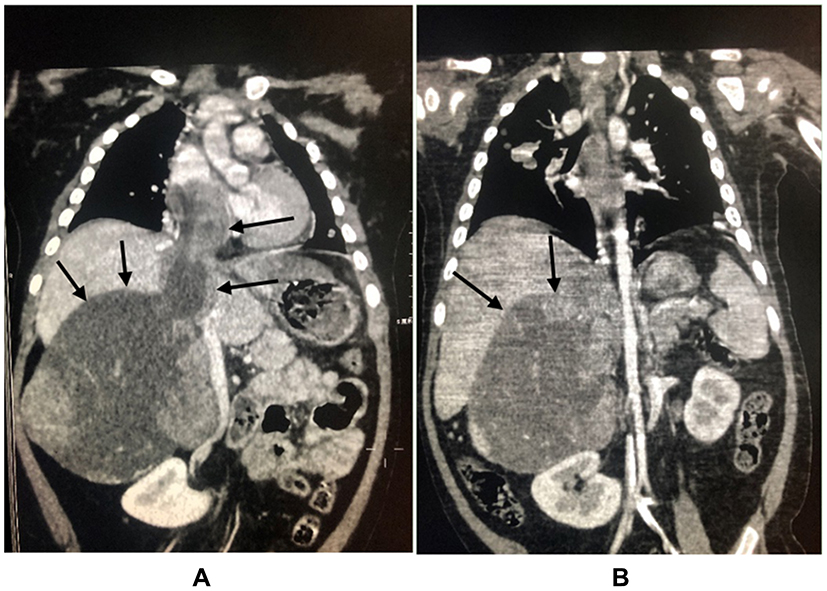 |
Figure 1 Comparison of abdominal CT before and after chemotherapy (the patient was a 2-year-old girl, the main complain was progressive aggravation of facial rash, the patient visited our hospital in January 2019, the clinical diagnosis was adrenal space occupying, the tumor was punctured and biopsied, ACC was confirmed by biopsy, chemotherapy was given first because of the intolerance to operation. (A) is abdominal CT at onset, the size of adrenal masses was 85 × 71 × 110mm, tumor thrombi in inferior vena cava could be seen. (B) indicates that the tumor shrank to 65 × 65 × 100mm after four cycles of chemotherapy, and the tumor thrombi in inferior vena cava were significantly reduced.) Decreased size of the adrenal cortex lesions of the same child in the same position after chemotherapy was indicated in arrows. |
Prognosis
Overall Prognosis
As of December 31, 2019, the follow-up duration was 3–61 months and the median duration of follow-up was 14.5 months. Among these eight patients, two patients had died, and six patients had survived. Four of these patients achieved CR, and two achieved SD. The overall two-year survival rate was 75%. The survival curve of the eight children is shown in Figure 2.
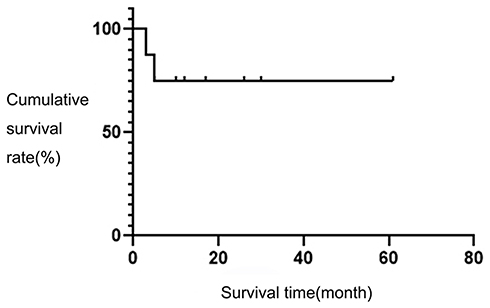 |
Figure 2 The survival curves of eight children. |
Prognosis Analysis of Patients at Different Stages
According to the ENSAT stage, two patients were at stage II and six patients were at stage IV. The two-year survival rates of the two groups were 100% versus 65%, and the difference in survival rates was not statistically significant (χ2 = 1.066, P = 0.302). The survival curve is shown in Figure 3.
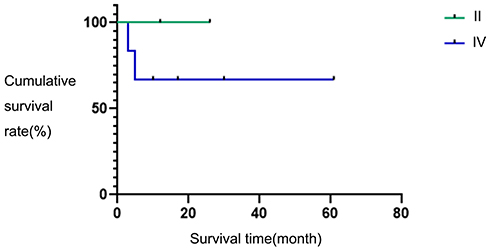 |
Figure 3 Survival curves in different stages. |
Weiss Score and Prognosis
In the patients in this study, the Weiss score was <6 in five patients, and ≥6 in three patients. The survival time of the two groups was 50 ± 9.526 months versus 6 ± 1.70 months, the two-year survival rates of the two groups were 100% versus 35%, and the difference in survival rate between these two group was statistically significant (χ2 = 4.091, P = 0.043). The survival curves of these two groups are shown in Figure 4.
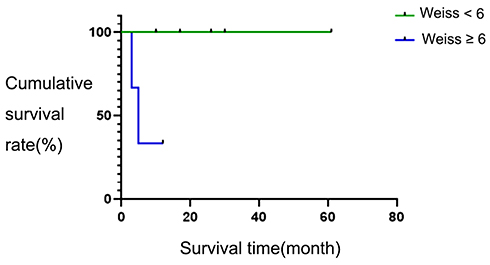 |
Figure 4 Survival curves based on different Weiss values. |
Discussion
Adrenocortical carcinoma is a rare malignant tumor with unknown pathogenesis and poor prognosis. Foreign data show that the incidence is approximately 0.7–2 per million and patients with ACC account for 0.05–2.00% of malignant tumor patients.1 The age of onset of the disease is bimodal: the disease mainly occurs in children under five years old and adults of 40–50 years old. The independent annual incidence rate of ACC in children is 0.3 per million, but the incidence rate in southern Brazil is relatively high, ranging from 3.4–4.2 per 100 million, approximately 10 times the global average.4 The disease usually occurs in girls: the male-to-female ratio is approximately 1:5. In the present study, two patients were boys (2/8), and six patients were girls (6/8). The age of onset was 1.6–12 years old, and the median age of onset was six years old. The patients with an age of onset under five years old accounted for 37.5% (3/8).
According to the presence of endocrine function, primary ACC can be divided into two categories. Functional ACC mainly produces COR, aldosterone, and sex hormones. When the tumor volume is small, the patients present obvious endocrine disorders and often visit a doctor at the early stage of the tumor. Common laboratory tests are as follows: low dose COR inhibition test, serum COR, E2, testosterone, aldosterone, and plasma renin activity. Virilization is the most common symptom in children, accounting for 84.2% of symptoms, manifesting as the appearance of pubic hair, hypertrophy of the clitoris, enlargement of the penis, facial acne, voice breaking, hirsutism, enhancement of muscle development, and acceleration of growth and development. Patients with only virilization account for 55%,5 and patients with feminization are rare. The patients with a main manifestation of Cushing’s syndrome in the infant age group account for approximately 5%. Symptoms of Cushing’s syndrome include the following: a moon-shaped face, centripetal obesity, polycythemia, and hypertension. Hypertension is the most common symptom.
Non-functional ACC does not secrete corticosteroids, so the patients usually have local symptoms and systemic symptoms caused by the tumor. The main symptoms are low back pain, abdominal pain, abdominal mass, loss of appetite, emaciation, anemia, low fever, bone pain, and other non-specific symptoms. At diagnosis, the stage is relatively late, with a high degree of malignancy and a poor prognosis, and the natural course of the disease is short. In children, non-functional ACC accounts for 10%, while in adults it accounts for 38%.6,7 In the present study, 75% (6/8) of the tumors had endocrine function, so tumors without endocrine function accounted for 25% (2/8). Hypertension was the main symptom in 25% of patients (2/8), patients with abnormal sexual signs as the main symptom accounted for 62.5% (5/8), and 25% (2/8) of patients had abdominal pain as the main symptom when they visited the doctor. Laboratory test results were as follows: three patients had increased testosterone and E2, three patients had increased COR and ATCH, and four patients had increased NSE. Because laboratory tests are not specific, it is necessary to exclude pheochromocytoma, neuroblastoma, and other malignant tumors when hormone levels are abnormal. Furthermore, laboratory examination of endocrine function cannot be used as the main index to differentiate benign and malignant tumors as some laboratory indicators change with age. This can obscure whether the changes are caused by a tumor, which creates difficulties in making a clinical judgment. Therefore, laboratory examination is only for reference.
The staging system of ACC can be used to evaluate the prognosis and adjust the treatment plan. In 2004, the Union for International Cancer Control (UICC) and the World Health Organization (WHO) proposed a staging system based on MacFarlane, but its effect in determining prognosis was not good. Therefore, the ENSAT staging system was proposed in 2009. The new staging system plays an important role in judging the prognosis of patients.8 The five-year survival rates of patients at the four stages were 73.9%, 63.8%, 44.1%, and 6.9%, respectively.9 The patients in this study were staged according to the ENSAT standard: two patients were at stage II, and six patients were at stage IV. The two-year survival rates of the two groups were 100% versus 65%, and the difference in survival rate was not statistically significant (χ2 = 1.066, P = 0.302). Considering that the follow-up duration may have been short, and the number of cases was small, it is necessary to extend the follow-up duration and increase the sample size. The lung is the most frequent site of ACC metastasis, followed by the liver and the retroperitoneal space. In the patients in this study, patients with lung metastasis accounted for 75% (6/8), patients with liver metastasis accounted for 37.5% (3/8), and patients with retroperitoneal lymph node metastasis accounted for 12.5% (1/8).
Surgery is the only radical treatment at present.10,11 Drug therapy or local radiotherapy is recommended for patients who have an unresectable tumor, high risk factors, or positive surgical margins. Theoretically, the operation should completely remove all tumors visible to the naked eye and include the removal of surrounding fat tissue and areas suspected of tumor invasion.12 For patients with distant metastasis at stage IV, it is worth discussing whether to perform surgery. However, some scholars consider that resection of the primary tumor can improve prognosis and reduce hormone secretion.8 Pathology is the gold standard of diagnosis, but no antibody is specific for the diagnosis of ACC. Histologically, the diagnostic criteria summarized by Weiss in 1984 are usually applied.3 Previous studies have shown that the sensitivity and specificity of the Weiss scoring system in the diagnosis of malignant cortical carcinoma are 100% and 96%, respectively. In addition, the Weiss score is related to the overall prognosis. The overall survival time of patients with a Weiss score of <6 is significantly longer than that of patients with a Weiss score of ≥ 6 (P < 0.001).13,14 In the patients in this study, five patients had a Weiss score of <6, and three patients had a Weiss score of ≥6. The five-year survival rates of the two groups were 100% versus 35%: the difference was statistically significant (χ2 = 4.091, P = 0.043). It was also found that the overall survival time of ACC patients with a Ki-67 index >10% was significantly worse (P < 0.01).15 However, the Ki67 index of all patients in this study was high, so comparison was not carried out. The malignancy of an adrenal tumor may be positively correlated with its size: in the patients in this study, the two-year survival rate was 50% in patients whose tumor size was >10 cm, and 100% in patients whose tumor size was <10 cm.
The effect of adjuvant chemotherapy is not ideal in treating ACC because in adrenal cortical cancer cells multidrug-resistance (MDR) genes are often expressed, so the tumor cells are not sensitive to common chemotherapy drugs. The existing chemotherapy regimens are based on mitotane. Fassnacht et al16 conducted a control study on 149 patients with stage II ACC: 35 patients were treated with mitotane, and 114 patients were not treated, as a control group. The results revealed that the five-year survival rate of the treatment group was 87%, while that of the control group was only 53%, suggesting that mitotane can significantly improve the survival rate of stage II ACC patients. To improve the effect of adjuvant chemotherapy and reduce the side-effects—such as nausea, vomiting, drowsiness, headache, hypothyroidism, and liver and kidney damage—other common chemotherapy drugs are often used in combination. At present, the commonly used chemotherapy regimens include etoposide + doxorubicin + cisplatin + mitotane (EDP/M) and streptozotocin + mitotane (SZ/M).17 Berruti et al18 conducted a concurrent, multicenter study, which consisted of a Phase II clinical trial on 72 patients with poor radiotherapy effect. The regimen was EDP-mitotane, and it was completely effective in five patients and partially effective in 30 patients. The remission period was 18 months, the effectiveness rate was 49%. The survival time of these patients was longer than that of patients without post-operative adjuvant therapy (P = 0.01): 36% (8/22) patients had complete or partial remission. The two-year and five-year survival rates were 70% and 33%, respectively, and the prognosis of patients with distant metastasis was poor (P = 0.02).
In the patients in this study, the regimen “etoposide + pirarubicin + cisplatin + mitotane” was used for all patients, if the effect was not good, the regimen “streptozotocin + mitotane” was used instead. The patients in this study had good response to chemotherapy. After surgery, six cycles of chemotherapy were given. If the condition was stable, the treatment stopped. At the end of the follow-up period, two of the eight children had died, four patients had achieved CR, and two patients did not undergo metastatic lesion resection and achieved SD.
With the development of the study of the molecular mechanism of ACC, a variety of cytokines or their receptors have been found to play an important role in tumor growth and proliferation. Therefore, the research of targeted drugs has become a hot topic in recent years. In 2007, Samnotra et al19 conducted a phase II trial of gefitinib in the treatment of unresectable ACC: the results were not ideal. Later, Quinkler et al20 used erlotinib combined with gemcitabine in the treatment of advanced ACC, but this also failed to achieve the expected goal. O’Sullivan et al21 gave axitinib to 13 patients with advanced or local ACC: no remission was achieved.
Based on the recent clinical research results, patients with ACC are advised to remove the tumor as completely as possible, and mitotane or other chemotherapeutic drugs are used after the operation. For patients who are not suitable for surgery, mitotane combined with etoposide, Adriamycin, and cisplatin is recommended as the first-line chemotherapy. Due to the limited effect of the existing treatment plan for ACC, it is urgent to develop new treatment strategies.
Ethics Approval and Consent to Participate
I confirm that I have read the Editorial Policy pages. This study was conducted with approval from the Ethics Committee of Beijing Tongren Hospital of China Capital Medical University. This study was conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from guardians of all participants.
Consent for Publication
All patient guardians signed a document of informed consent.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Allolio B, Fassnacht M. Clinical review: adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91(6):2027–2037. doi:10.1210/jc.2005-2639
2. Fassnacht M, Johanssen S, Quinkler M, et al. Limited prognostic value of the 2004 international union against cancer staging classification for adrenocortical carcinoma: proposal for a revised TNM classification. Cancer. 2009;1(15):243–250. doi:10.1002/cncr.24030
3. Weiss LM. Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am J Surg Pathol. 1984;8(3):163–169. doi:10.1097/00000478-198403000-00001
4. Ribeiro R, Figueiredo B. Childhood adrenocortical tumours. Eur J Cancer. 2004;40(8):1117–1126. doi:10.1016/j.ejca.2004.01.031
5. Gundgurthi A, Kharb S, Dutta MK, et al. Childhood adrenocortical carcinoma: case report and review. Indian J Endocrinol Metab. 2012;16(3):431–435.
6. Ng L, Libertino JM. Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol. 2003;169:5211. doi:10.1016/S0022-5347(05)64023-2
7. Han W, Li J, Qin H, et al. Diagnosis and treatment of adrenal cortical carcinoma in children. Chin J Pediatr Surg. 2015;36(1):44–48.
8. Ye JN, Wang WQ. Progress in pharmaceutical treatment for adrenocortical carcinoma. Chin J Endocrinol Metab. 2011;027(1):91–94.
9. Lughezzani G, Sun M, Perrotte P, et al. The European network for the study of adrenal tumors staging system is prognostically superior to the international union against cancer-staging system: a North American validation. Eur J Cancer. 2010;46(4):713–719.
10. Conzo G, Gambardella C, Candela G, et al. Single center experience with laparoscopic adrenalectomy on a large clinical series. BMC Surg. 2018;18(1):2. doi:10.1186/s12893-017-0333-8
11. Conzo G, Pasquali D, Gambardella C, et al. Long-term outcomes of laparoscopic adrenalectomy for cushing disease. Int J Surg. 2014;12(Suppl 1):S107–S111. doi:10.1016/j.ijsu.2014.05.036
12. Liu T, Jia H, Kong CZ. Diagnosis and treatment of primary nonfunctioning adrenal cortical carcinoma. J Mod Oncol. 2011;03:95–97.
13. Niu S, Zhang YS. Advance in diagnosis and treatment of adrenocortical tumor. Chin J Endocr Surg. 2016;10(3):248–251.
14. Xu LY, Xu YZ, Lian JP, et al. The diagnostic and prognostic value of EGFR, IGF1R, Ki67 and weiss score for adrenocortical tumors. J Mod Urol. 2016;21:417.
15. Nakamura Y, Yamazaki Y, Felizola SJ, et al. Adrenocortical carcinoma: review of the pathologic features, production of adrenal steroids, and molecular pathogenesis. Endocrinol Metab Clin North Am. 2015;44(2):399–410. doi:10.1016/j.ecl.2015.02.007
16. Fassnacht M, Allolio B. What is the best approach to an apparently nonmetastatic adrenocortical carcinoma? Clin Endocrinol. 2010;73:561. doi:10.1111/j.1365-2265.2010.03867.x
17. Skogseid B, Bandin E, Fassnacht M et al. Experience from an ongoing Phase III study: FIRM-ACT.
18. Berruti A, Terzolo M, Sperone P, et al. Etoposide doxorubicin and cisplatin plus mitotane in the treatment of advanced adrenocortical carcinoma: a large prospective phase II trial. Endocr Relat Cancer. 2005;12:657–666. doi:10.1677/erc.1.01025
19. Samnotra V, Vassilopoulou—Sellin R, Fojo AT, et al. A phase II trial of gefitinib monotherapy in patients with unresectable adrenocortical carcinoma(ACC). J Clin Oncol. 2007;25(Suppl 18):15527.
20. Quinkler M, Hahner S, Wortmann S, et al. Treatment of advanced adrenocortical carcinoma with erlotinib plus gemcitabine. J Clin Endocrinol Metab. 2008;93(6):2057–2062. doi:10.1210/jc.2007-2564
21. O’Sullivan C, Edgerly M, Velarde M, et al. The VEGF inhibitor axitinib has limited effectiveness as a therapy for adrenocortical cancer. J Clin Endocrinol Metab. 2014;99(4):1291–1297. doi:10.1210/jc.2013-2298
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.