Back to Journals » The Application of Clinical Genetics » Volume 16
Adeno-Associated Virus (AAV) - Based Gene Therapies for Retinal Diseases: Where are We?
Authors Ail D, Malki H ![]() , Zin EA
, Zin EA ![]() , Dalkara D
, Dalkara D
Received 16 February 2023
Accepted for publication 19 May 2023
Published 30 May 2023 Volume 2023:16 Pages 111—130
DOI https://doi.org/10.2147/TACG.S383453
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Divya Ail, Hugo Malki, Emilia A Zin, Deniz Dalkara
Sorbonne Université, INSERM, CNRS, Department of Therapeutics, Institut de la Vision, Paris, 75012, France
Correspondence: Divya Ail, Department of Therapeutics, Institut de la Vision, 17 Rue Moreau, Paris, 75012, France, Tel +33 153462648, Email [email protected]
Abstract: Owing to their small size and safety profiles, adeno-associated viruses (AAVs) have become the vector of choice for gene therapy applications in the retina. In addition to the naturally occurring AAVs, several engineered variants with enhanced properties are being developed for experimental and therapeutic applications. Nonetheless, there are still some challenges impeding successful application of AAVs for a broader range of retinal gene therapies. The small size of AAV particles ensures efficient tissue transduction but also limits the packaging capacity to a few kilobases. Further, AAV’s ability to cross retinal barriers is still an obstacle to pan-retinal transduction of the outer retina with tolerable doses. Lastly, despite overall safety, there have been recent reports of immune responses to AAVs in the eye. Hence, evaluation and prediction of immune responses to AAVs has come to be considered an integral part of future clinical success. This review focuses on the use of AAV in clinical trials for retinal diseases, and discusses developments of variants and novel strategies to overcome immune responses to AAVs.
Keywords: retinal gene therapy, adeno-associated virus, AAV, clinical trials, capsid variants, immune responses
Introduction
AAVs are non-pathogenic viruses belonging to the Parvoviridae family under the genus Dependoparvovirus. They can infect humans but are non-pathogenic and are known to have low immunogenicity. They are naturally replication-deficient and hence termed as ‘helper-dependent virus’ as they depend on the presence of adenovirus, herpesvirus or papilloma virus as helpers to replicate.1–3 In the absence of a ‘helper virus’ AAV remains latent by integrating into the host genome or remaining as extrachromosomal episomes. AAVs are small, non-enveloped viruses with a capsid size of 25nm and package single-stranded DNA of approximately 4700 base pairs.4 There are 13 naturally occurring AAV serotypes (AAV1 – AAV13) and 100s of AAV variants have been identified or engineered. These AAV serotypes possess distinct tropisms that are tissue specific and species specific. The tissue tropism/specificity of AAVs is determined by the proteins on the surface of capsids. The capsid proteins bind specific cell-surface receptors which in turn recruit co-receptors leading to internalization by endocytosis into the cell.5 Following internalization AAVs escape the endosomes and gain entry into the nucleus.6 AAV relies on the host cell mechanisms for the second strand synthesis.7
For experimental and clinical applications recombinant AAVs (rAAV) are created by replacing the viral genome by the transgene of interest while retaining the ITRs. The ITRs flank the transgene and ensure its encapsidation within the AAV. The other structural (capsid proteins) and non-structural (replication and helper) genes are supplied separately.4,8 rAAVs rarely integrate into the host cell genome, thereby reducing the risk of insertional mutagenesis following gene therapy.
Gene therapy refers to the introduction of genetic material into tissues to rectify the effects of disease-causing mutations. Inherited diseases caused by gene mutations resulting in deficiency or malfunction of proteins involved in cellular functions are ideal targets for gene therapy. Such diseases can be treated by replacement of the entire defective gene, silencing of the mutation or correction/editing of the mutation. The therapeutic gene is packaged into AAVs and delivered to target tissue by local injections.9 AAV delivery to the retina is normally done by two routes: subretinal and intravitreal. Subretinal injection (SRI) involves injection between the Retinal Pigment Epithelium (RPE) and the photoreceptors resulting in efficient AAV delivery to the outer retina. Intravitreal injection (IVT) involves AAV delivery into the vitreous space thereby causing transduction of the Ganglion Cell Layer (GCL) and some other cell types of the inner retina.10 The retina is an ideal target for gene therapy due to two main reasons: First, many gene mutations responsible for Inherited Retinal Diseases (IRDs) have been identified,11 and second, the retina is enclosed within the eye in a relatively immune-privileged space. The retina is shielded by a blood-retinal barrier and also benefits from the unique intraocular microenvironment.12 This prevents the occurrence of a strong systemic effect to the AAV-mediated ocular gene therapy.
Clinical Trials Using AAVs for Retinal Diseases
Over the last five decades, substantial efforts into understanding the basic biology and engineering of AAV have resulted in clinical success of AAVs. The first FDA approved retinal gene therapy called Luxturna developed by Spark therapeutics is for a retinal disease called Leber Congenital Amaurosis type 2 (LCA2). LCA2 is an autosomal recessive disease caused by mutations in the RPE65 gene present in the RPE layer of the retina.13 The therapy aims at replacing the RPE65 gene by delivering a healthy cDNA copy using AAV2 injected directly adjacent to the RPE layer by SR injections. The proof-of-concept for vision restoration was provided in a canine model.14 This led to clinical trials involving LCA2 patients who received SR injections of RPE65 packaged in AAV2 via SR delivery. The trials reported improvement in visual acuity, pupillary reflex, light sensitivity as well as the patients’ ability to navigate in low light.15–17 Furthermore, it was also demonstrated that it was safe and effective to inject the contralateral eye.18 However, long-term follow-up in 2 out of 3 clinical trials reported a decline in the visual gains in adult patients, indicating that the treatment was unable to halt the degeneration.15,19 This highlights two important aspects: the level of AAV transduction required for therapeutic benefits in humans may be higher and the timing of therapeutic intervention is crucial (earlier interventions before the onset of degeneration will be more beneficial).
The earliest clinical trials used AAV2 and this serotype accounts for 53% of all trial for retinal diseases. Most of the current clinical trials (about 81%) continue to use naturally occurring serotypes. Only 19% of the current clinical trials use engineered AAVs including, AAV2tYF (generated by rational design), AAV2-7m8 and 4D-R100 (generated by directed evolution) (Figure 1A and B). Interestingly, the newly developed variant AAV-4D-R100 is being used to deliver genes to the photoreceptors and the RPE by IVT injections. It is worth noting that all the completed trials used naturally occurring serotypes, whereas among the ongoing trials 34% (10 out of 29) use engineered serotypes (Table 1).
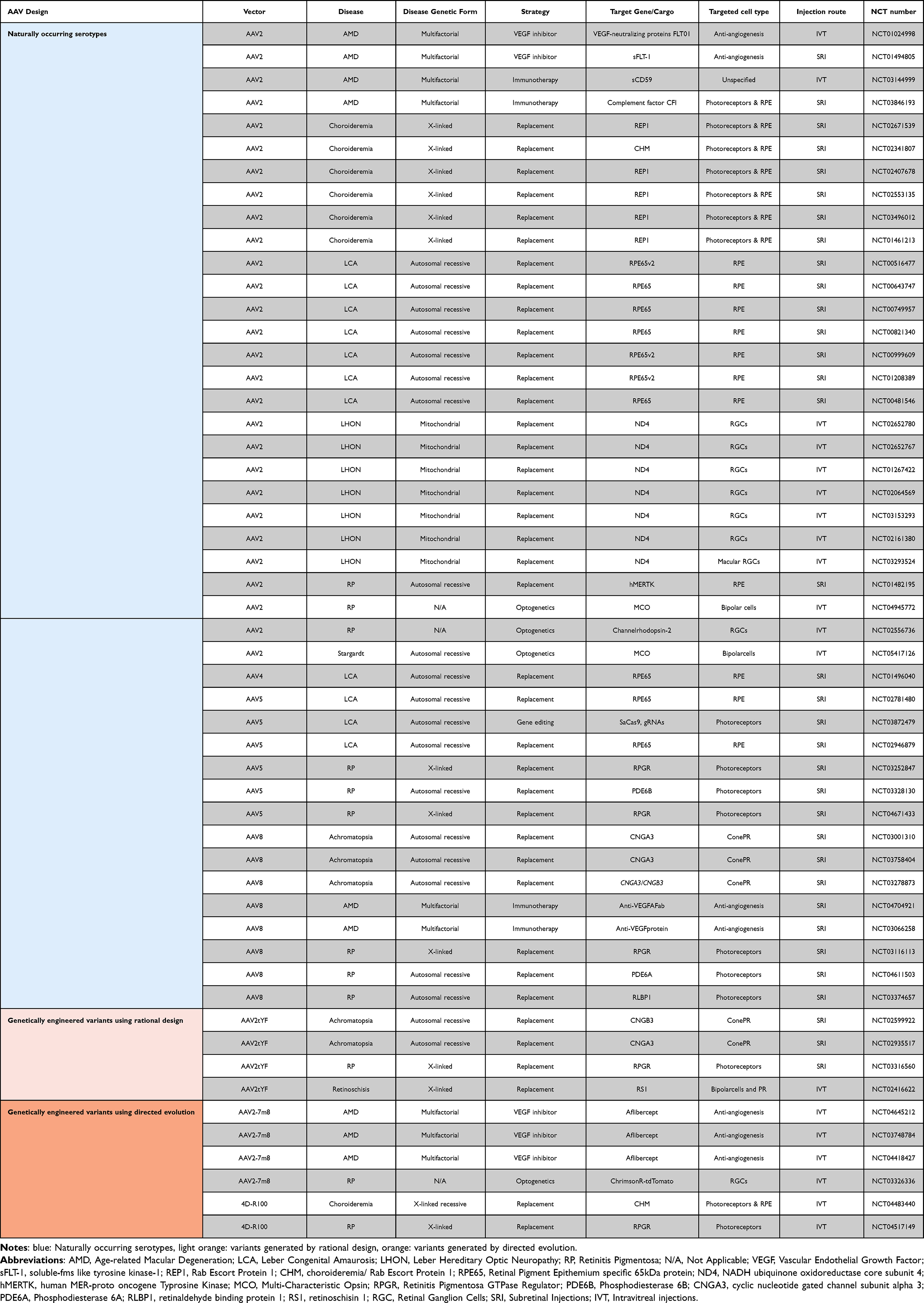 |
Table 1 Naturally Occurring and Engineered AAVs Used in Clinical Trials |
 |
Figure 1 Distribution of (A) Naturally occurring AAVs (blue) and engineered AAVs (Orange) used in 53 clinical trials for retinal diseases; (B) distribution of AAV serotypes being used in clinical trials with the naturally occurring ones (AAV2, AAV4, AAV5 and AAV8) in shades of blue and the engineered variants (AAV2tYF, 4D-R100 and AAV2-7m8) in shades of Orange; (C) distribution of the therapeutic strategy used in clinical trials; (D) distribution of clinical trials using the subretinal delivery route for outer retina or other regions of the retina; (E) distribution of clinical trials by injection route including subretinal (Orange) and intravitreal (blue) injections; (F) distribution of trials using intravitreal delivery routes and targeting the inner, outer or other regions of the retina. |
The early clinical trial success has paved the way for many other clinical trials (Table 1 and Table 2). The majority of current gene therapies focus on gene replacement for diseases caused by recessive mutations (Figure 1C). These are straightforward targets since the replacement provides the protein and restores the missing function in target tissue. However, for dominant mutations first the malfunctioning gene has to be suppressed and then the correct gene needs to be supplemented. Another widely tested therapy is the anti-VEGF therapy for wet-AMD (Age-related Macular Degeneration) (Figure 1C). AMD has a global prevalence of close to 200 million and this number is bound to increase with an increase in elderly population overtime, making the development of therapy urgent.20 AMD has two forms: dry and wet AMD. It is estimated that about 15% of the patients develop what is known as the wet form of the disease, wherein neovascularization occurs and the poorly formed blood vessels result in blood leakage. Vascular Endothelial Growth Factor (VEGF) has been recognized as an important factor involved in this process of neovascularization and the present clinical practice involves the repeated injection of anti-VEGF antibodies or soluble receptor inhibitors.21 However, patients need frequent (often monthly) anti-VEGF injections, which are expensive.22 Hence, the anti-VEGF gene therapies aim to have stable expression of VEGF inhibitors, offering an alternative long-term solution.
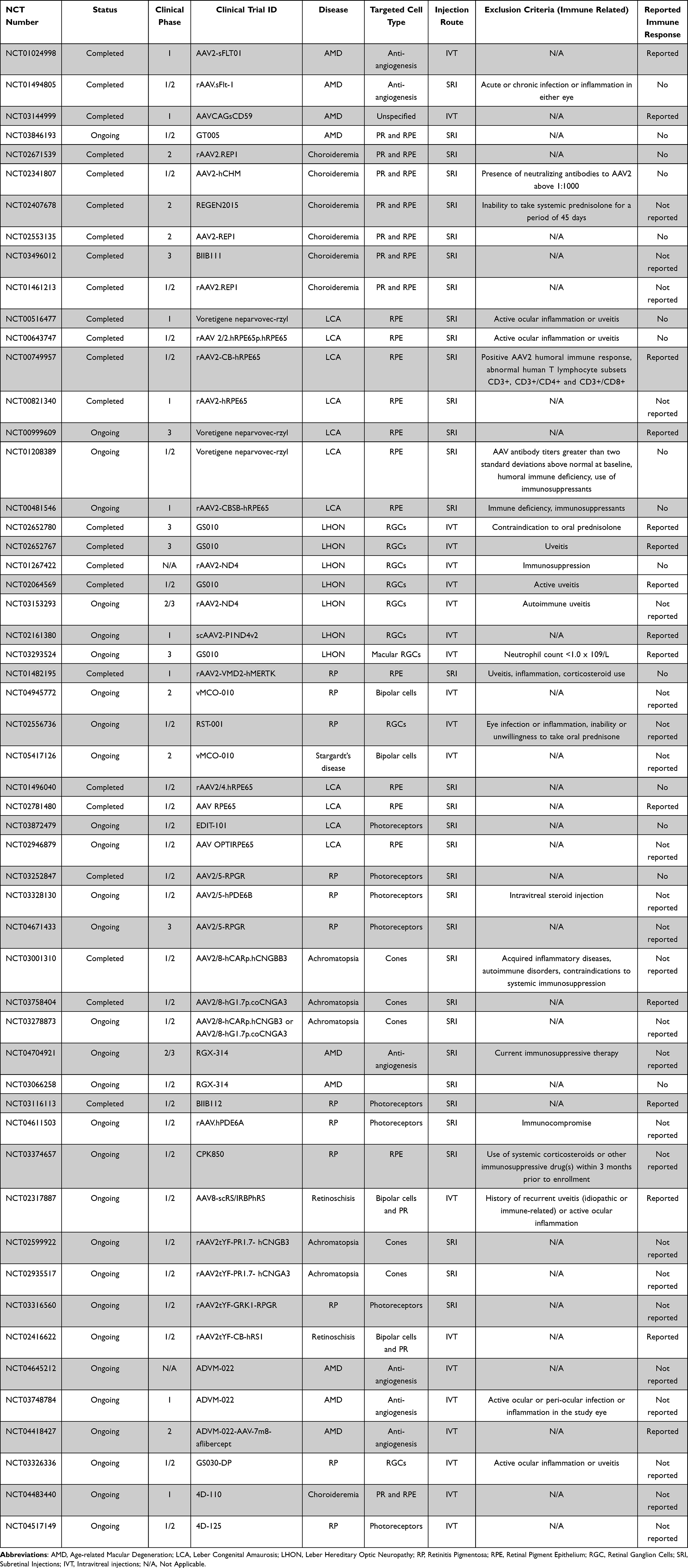 |
Table 2 Exclusion Based on Pre-Existing Immunity and Immune Responses in Clinical Trials |
All deliveries to the retina in clinical trials are by SRI or IVT routes (Figure 1D–F). Therapies that target the cell types of the inner retina can be supplied by IVT injections. Further, anti-VEGF therapy can also be provided intravitreally as no cell-type is targeted, rather it acts to curb a symptom (angiogenesis). Contrary to these, most of the replacement-based therapies are provided by SRI as the disease-causing mutations mostly occur in photoreceptors or RPE.
As mentioned earlier, one important caveat for the success of gene therapy is early intervention, since the cells where the gene correction or supplementation is required have to be present. In late stages of retinal degeneration, when the photoreceptors are mostly lost, correction of gene mutations will not provide any therapeutic effect. To circumvent this issue mutation-independent optogenetic therapy is now being developed and tested. Following the loss of photoreceptors, optogenetic therapy aims at converting the surviving retinal neurons into photosensitive cells. This is achieved by the expression of light-sensitive microbial opsins in the remaining cell types of the retina.23 A recent study delivered the light-sensing Channelrhodopsin protein - ChrimsonR packaged in an engineered vector (AAV2-7m8) by IVT injection to mainly target the foveal retinal ganglion cells of a blind patient. In the study, the patient was reported as being able to perceive, locate, count and touch different objects using the treated eye alone. Furthermore, during visual perception, object-related activity was observed above the visual cortex.24 This success has encouraged further trials involving microbial opsins (Table 1). Optogenetic therapy can benefit patients with end-stage degeneration who presently have no therapeutic options. In the near future, it might be extended to target more upstream retinal neurons with more sensitive G protein coupled opsins for better visual outcomes.25
Challenges with AAVs
The small size of AAV is an advantage because it allows efficient diffusion through many tissue barriers. However, smaller size also limits the genome size. Hence, transgenes including their promoters and regulatory sequences have to be below 4.7kb in size. This is a limiting factor in gene therapies that aim at replacement of defective genes, as some of the disease-causing genes are larger than 4.7kb in size. Moreover, gene replacement is not a viable option for IRDs caused by autosomal dominant mutations displaying dominant negative or toxic gain-of-function mechanisms.26 For these reasons, the field of gene therapy is moving towards gene editing using the power of editing tools such as CRISPR-Cas9, Base editors (BEs) and Prime editors for correcting gene mutations.27–29 Some Cas9 proteins such as saCas9 fit into AAV along with the target guide RNA, whereas the most commonly used one - spCas9 does not. But, base and prime editors are too large to fit into a single AAV.30 Although gene-editing techniques are precise and efficient, they are still known to have off-target effects.31,32 Newer technologies are being developed to circumvent this issue, but none of them guarantee complete avoidance of off-targets.33 Hence, it is important to limit unwanted editing in cell-types that do not need correction. This can be done by using cell-specific promoters. The challenge of using these editing tools gets more complex when they need to be expressed in a cell-specific manner, which is achieved by using cell-specific promoters.34 But, in most cases, these promoters occupy significant space out of the 4.7kb available in AAVs alongside polyadenylation sequences and other potential cis regulatory elements that are necessary for optimal transgene expression.
Although the early success and clinical usage of AAV-based gene therapy was achieved in the retina, there are still certain challenges that remain. As mentioned earlier, with SRIs transduction of the photoreceptors can be achieved. However, SRIs are limited to a small region of the retina and with this route of injection a pan-retinal transduction is not achieved. Additionally, this is an invasive technique and is likely to damage an already fragile and degenerating retina.35 The IVT route of injection results in a pan-retinal transduction which is limited to the ganglion cells or some inner retinal cells. Certain engineered variants have been developed which, when delivered by the IVT route, can cross all retinal barriers and transduce the photoreceptors of the mouse retina.36–38 A variant (AAV2-7m8) developed in mice did not translate well for non-human primate applications.37 Species-specific directed evolution was performed to identify variants for NHPs but these had limited success due to the more prominent barriers to AAV transduction in NHPs compared to mice.38 One of the major barriers, the Inner Limiting Membrane (ILM) is thicker in the primate and human retina compared to rodents.39 Although IVT injections can be less invasive, there is a higher probability of immune response due to the presence of immune cells in the vitreous fluid.40
AAVs are non-pathogenic and do not cause any disease in humans. But they are ubiquitously present and about 50–90% of the human population has already been exposed to AAVs.41,42 Such exposure initiates an immune response and results in the development of anti-AAV antibodies. The anti-AAV antibodies are of two types – Total or Binding antibodies (BAbs) and a subset of BAbs called Neutralizing antibodies (NAbs). BAbs are all the antibodies that are produced against a specific AAV serotype and upon repeat exposure to the same serotype BAbs can trigger a stronger immune response that can potentially result in inflammation. A subset of BAbs called NAbs are capable of neutralizing AAV thereby reducing the efficacy of gene therapy.43 Hence, prior exposure to AAVs results in the presence of pre-existing antibodies and when a patient with high pre-existing antibodies receives AAV-based gene therapy, it compromises both the efficacy and the safety of the therapy. These pre-existing BAbs and NAbs are serotype, species, age and population specific.44 The highest seroprevalence in the human population was for anti-AAV2 antibodies, while anti-AAV8 was the lowest.41,42 Studies in non-human primates reported the highest seroprevalence was against AAV8 and AAV9, while AAV5 was the lowest.45,46 Irrespective of the pre-existing immunity, AAVs can trigger an immune response in the eye. Even though the eye is considered immune privileged, reports from clinical trials and experiments have provided plenty of evidence to the contrary (Table 2).47
Strategies to Overcome the AAV Size Restriction
To overcome the size limitation of AAVs, new strategies including the use of simultaneous dual or triple AAVs are being developed, extending total AAV cargo capacity to 9kb and 14kb, respectively.48,49 This approach is based on concatemerization and/or homologous recombination of AAV vectors that each contains a distinct part of the transgene, flanked by inverted terminal repeat sequences (ITRs) and/or transgene homologies. With these strategies, the vectors can reconstitute the full transgene coding sequence.49,50 Thus, it provides an interesting approach to treat autosomal recessive diseases with genes longer than 5kb. Unfortunately, some of the more than 280 genes involved in IRDs would require gene replacement that exceed the packaging capacity of AAVs (https://web.sph.uth.edu/RetNet/). Recently, in vivo proof-of-concept of dual AAVs therapies have been reported by successfully delivering large therapeutically relevant genes involved in IRDs pathologies such as MYO7A (6.7kb) or ABCA4 (6.8kb).51–53 Moreover, an evaluation of dual-AAV strategies studied the in vivo efficiency of this approach to deliver transgenes to retinal cells. The authors compared the efficiency of the three main strategies of dual-AAV reconstitution: trans-splicing (ITRs concatemerization), overlapping (homologous recombination) and the hybrid method (concatemerization or homologous recombination). They showed that the hybrid method was the most relevant when compared to the other two in the mouse retina, confirming results from previous studies showing the hybrid approach to be efficient in vivo. The authors also demonstrated about 10% reconstitution efficiency based on protein expression compared to unique AAV reporter control.54 Even if the reconstitution efficiency is low, phenotypic improvement in disease models have been reported in previous studies.52,54,55
Furthermore, some genes involved in genetic disease are still larger than dual-AAVs capacity. In a recent study, Maddalena et al performed triple-AAV-based gene delivery of a reporter transgene (Fusion of EGFP and DsRed that was split into 3 vectors) and therapeutic transgenes (CDH23; ALMS1). Interestingly, when considering the reporter construct, a low percentage of full fusion protein expression was observed in mouse retina with ~2% expression of the single control AAV encoding the entire reporter, whereas a higher efficiency was achieved in pig retina at ~39%, suggesting structural and molecular interspecies specificities impacting viral transduction and intracellular reconstitution.48
Gene editing has been proposed as an alternative to gene replacement as it allows to correct the genetic defect in situ in the chromosome. This strategy has rapidly moved towards clinical application after pre-clinical proof of concept studies. In 2019, Maeder et al delivered a single AAV-SaCas9 construct with two different guide RNAs to excise an aberrant splice donor in the 7kb gene CEP290 in mouse and non-human primate, removing the ISV26 disease-causing mutation for LCA type 10, thus avoiding technical genetic supplementation with multiple vectors. With this strategy, they could avoid the technical challenge of genetic supplementation with multiple vectors.56 However, this elegant therapeutic strategy depends on mutational context. To overcome such obstacles, some groups are developing multiple AAV strategies to deliver new gene editing tools that usually exceed AAV cargo capacity, such as prime editors or base editors.57 BEs are DNA editing tools that use a dead Cas9 (dCas9; no/residual catalytic activity) fused with cytidine (CBEs) or adenine (ABEs) deaminases allowing single base conversion of C:G>T:A or A:T> G:C.27,58 This strategy is particularly relevant in both monogenic autosomal recessive and autosomal dominant diseases contexts for repairing pathogenic variants. BEs were used recently in several cell types/organs, including the retina, and were delivered by dual-AAVs approaches.27,59 The delivery methods are usually based on trans-splicing inteins, where each part of the transgene containing split protein fused to N-terminal and C-terminal intein sequences. Thus, reconstitution occurs at the protein level.60,61 Two recent studies showed interesting results in rd12 mice, an LCA model involving the RPE65 gene with nonsense mutation c.130C>T, p.(R44X).28,62 Both teams performed dual-AAV delivery of split-ABEs to correct point mutation and reached 3% and 6% A>G editing at the DNA level in RPE cells, leading to retinal function improvements.
Dual or triple AAV approaches offer new perspectives that would not be possible with other available viral vectors, especially in the retina. Despite their greater packaging capacity, other vectors such as adenovirus and lentivirus have variable immunogenicity and transduction efficiencies/specificities.63,64 Nevertheless, these multiple-AAV approaches imply co-transduction of the same cell by 2 or 3 vectors, decreasing the probability of full reconstitution. One of the problems with dual/triple AAV strategies is the risk of producing truncated proteins, which have been reported previously.48,51,55 As it raises safety concerns, these issues must be addressed for future clinical applications. Finally, to circumvent the use of multiple vectors, a new trend is the development of size-minimized tools, such as ABE involving a smaller Cas9 protein that thus can fit in a single AAV.65,66
Depending on the targeted cell type, dominant or recessive form of the disease, therapeutic aims, and desired cargo, using AAVs is not always the most straightforward choice. Further, other drawbacks of AAVs have to be considered despite the lack of data such as the consequences of delivery in post-mitotic cells in which the viral genome can persist for years leading to long-term endonuclease expression;17,67–69 and immune response against the transgene;44 and potential genotoxicity.70 Due to such considerations, over the last few years, non-viral vectors were increasingly studied and their therapeutic potential has been demonstrated.71–73 These delivery approaches offer some advantages such as relatively low immunogenicity, no integration risks and delivery of a large range of components like DNA, RNAs or proteins.74 Non-viral vectors are a heterogenous family including various components such as lipid nanoparticles (LNPs), polymer-based vectors, or inorganic materials.75,76 Nonetheless, when it comes to the retina, those vectors, which are bigger (~100–200nm) than AAVs (~25nm), may face numerous physical and physiological barriers such as limiting membranes, hyper structure of photoreceptors outer segments, RPE phagocytosis activity, tight cell junctions and low extracellular space volume.77–81 Moreover, other variables have to be considered such as the stability of the cargos that could be degraded in the extracellular space.81 Despite these difficulties, some studies reported retinal delivery of DNA plasmid or minicircle in mouse models of disease-associated genes like RPE65.82,83 These vectors also offer an opportunity to deliver gene-modifying tools in mRNA or proteins form, limiting their intracellular presence and potentially reducing the risk of off-targets effects.70,84,85 So far, CRISPR/Cas9 transfer into the retina in its ribonucleoprotein (RNP) conformation has been used to target the VEGFA gene, whose overexpression is involved in AMD. Recent studies delivered RNP complexes using LNPs and achieved significant editing in RPE cells.86,87 Finally, despite interesting characteristics, their efficacy is lower compared to AAV viral vectors. For example, in two different studies, the same group was able to introduce ABEs to the retina as RNPs by LNPs, as well as via AAV, showing 1.8% and 6% efficiency, respectively, leading to phenotype improvement only with AAV-ABEs suggesting a better clinical relevance.62,88 These advances are encouraging and their development in parallel with that of AAV approaches is crucial to develop safer therapeutic methods.
Strategies to Engineer AAVs with Enhanced Properties for Retinal Gene Therapy
The search for AAV variants with improved tropism and infection profiles has walked hand in hand with the development of AAV as vectors for gene therapy. In the retina, the natural serotypes of AAV have broad tropism for all cell types, and infection is dependent on route of delivery. SRIs permit infection of mainly RPE and photoreceptors, while IVTs lead to transduction in retinal ganglion cells and amacrine cells.89–91 SRI, while currently the method of choice in clinical trials aiming to reach the outer retina, results in retinal detachment due to the formation of the surgical bleb, an undesirable prospect for patients with retinas already undergoing degeneration. Formation of the bleb under the fovea has also resulted in retinal thinning.18 Moreover, AAV has limited reach outside of the bleb due to restricted lateral spread.92,93 While IVTs permit AAV to reach a larger area of the tissue, they nevertheless require vitrectomies and result in poor transduction of the inner and outer retina due to physical barriers such as the ILM.89 The ILM is coated in a thick layer of glycans, of which heparan-sulfate (HS) is essential for AAV2 binding. While binding to HS reduces AAV2’s dilution in the vitreous, it also leads to reduced penetration of the retinal tissue. For almost two decades now there has been a search for AAV variants capable of overcoming these obstacles and show increased transduction efficiencies to assist the delivery of a lower AAV dose. Different methods have been developed for identifying such variants, and many are now the vectors of choice in clinical trials (Table 1).
With crystallography of natural occurring serotypes and the understanding of the role of different amino acids on the capsid surface, researchers have explored a rational design approach. By swapping amino acids important for HS and receptor binding (R484, R487, K532, R585, and R588) or ubiquitination pathways (substitution of threonine, tyrosine or serine residues), it is possible to modify tropism or to increase transduction efficiencies in different tissues, including the retina.36,94–96 As such, the variant AAV2tYF (three tyrosine to phenylalanine substitutions, Y444F, Y500F, Y730F) has shown improved transduction profiles in canine animal models by evading degradation through the proteasome complex.97,98 AAV2tYF is currently in use in four clinical trials (Table 1), both through SR and IVT injections. However, rational design has its limitations and extensive pan retinal expression still has not been shown with this method.
Along similar lines to rational design, targeting AAVs to known cell receptors has recently been explored with the use of nanobodies.99 By harnessing the desirable characteristics of camelid antibodies, a fragment of the heavy-chain variable domain can be attached to the AAV capsid, conferring the desired cell tropism by targeting known receptors or other membrane proteins. Incredibly small and with high specificity, these fragments, also known as nanobodies, have been successfully incorporated onto the capsids of different AAV serotypes (AAV1, 2, 8 and 9) and they have also been targeted to different cell proteins (a single-pass transmembrane protein, a multi-pass ion channel, and a glycosylphosphatidylinositol-anchored ectoenzyme) expressed in HEK cell membranes. While not yet tested in vivo, these AAVs targeted via nanobodies greatly enhanced their transduction efficiencies and present a potential future for solving issues of transduction in specific cell types from different tissues, including the retina.
With the development of technologies capable of reconstructing ancestral proteins, libraries of hypothetical ancestral AAV capsids can also be manufactured and screened. By looking at the node Anc80, the theoretical ancestor of AAV1, 2, 3, 6, 7, 8 and 9, a library of potential ancestors was developed in silico and tested in vitro and in vivo.100 The variant Anc80L65 outperformed all others in its photoreceptor infectivity profile, and, interestingly, showed low development of an immune response. So far, this vector has been tested in mouse and NHP retinas, and confirms the promising possibilities of capsid development in silico.101
Directed evolution has become an important, high-throughput method for the development of new AAV variants. Mutations to the cap gene of naturally occurring serotypes are introduced, and new variants are identified after several rounds of selection. There are several methods to generate highly diverse libraries, including the insertion of random peptide sequences into a defined location on the capsid, error-prone PCR and site directed mutagenesis, gene shuffling, and insertion of peptide sequences in random locations.102–105 Selective pressure is applied, such as reaching the outer retina through IVT route, and variants that fulfill the desired tasks are identified. The first AAV variant capable of overcoming the ILM barrier was found through directed evolution and is called AAV2-7m8, a variant that has a seven-amino acid insert at R587.37 This mutation confers the ability to partially evade HS binding and the “sink” effect of these proteoglycans at the ILM. Therefore, AAV2-7m8 is capable of passing through the physical barrier of the ILM and subsequently infecting all retinal layers through an IVT route achieving pan retinal expression. This is not only true in animal models such as mice, which have thinner ILMs, but also in sheep and NHPs,37,106 as well as in patients (Table 1). However, in animal models with more complex and thicker ILMs, AAV2-7m8 does not yield a uniform pattern of expression. In the search for variants with more efficient profiles in human patients, other AAV variants have been identified in the last few years with improved infectivity or transduction characteristics after IVT injections in NHPs.38,107,108 Of these, 4D-R100 is currently being used in two clinical trials (Table 1). Directed evolution has also been employed to develop AAV variants that are capable of evading immune responses (described in the next section).
With the identification of several new AAV variants, accurate and quantitative methods to compare their efficiencies and profiles are required. Traditionally, such comparisons required large numbers of animals and variations between injections and individual animals can lead to unreliable results. This is undesirable, especially when working with large animal models. With this in mind, methods such as scAAVengr were developed, whereby barcoded libraries of known AAV variants allow for the comparison of different naturally occurring or engineered capsids side by side.107 By using single-cell RNA-seq, it is possible to compare expression patterns of different variants in parallel within not only the same retina, but also within individual cells. This method is especially valuable for comparing different variants in NHP models in vivo or in human retinal explants ex vivo, indicating AAV reliability for future clinical trials.109
As a high-throughput method that uses next-generation sequencing, directed evolution of AAV capsids now generates large data sets that can be further explored through artificial intelligence (AI) and machine learning. AI-based algorithms can aid in the development of new diverse libraries and in the selection of variants that perform the required tasks and increase the success rate in finding relevant AAVs. Recently, computational models have predicted AAV viability in silico, which was further tested with AAV libraries in vitro.110,111 Different methods to identify variants after initial screening rounds include calculations of enrichment factors and credibility scores.112 Moreover, computational methods have recently helped guide capsid engineering and the understanding of AAV biology.113 Currently, it is possible to develop AAV variants with high viability using in silico design, and in future, with the study of complex directed evolution datasets, machine learning and AI can aid research groups in predicting and designing AAV variants with improved infectivity and transduction characteristics for uses in the retina.
Strategies to Overcome Immune Responses
Adverse immune outcomes due to AAV delivery to the eye include inflammation, reduced efficacy and clearance of transduced cells. The most common strategy to prevent or manage the immune response in clinical trials or clinics is by immunosuppression (Table 2). Non-specific immunosuppressive agents are provided to patients before, during and after the AAV injections. These corticosteroids treatments vary from one study to another and systematic comparisons evaluating their effects are lacking. Immunosuppression is also routinely used in large animal models such as canines and non-human primates. But these are provided as per the veterinarian’s recommendation and have not been systematically evaluated for retinal studies and there are not any standard recommendations available.
Several studies have shown that the dose of the vector administered influences the immune outcome. A study analyzing safety thresholds of AAV2 and AAV8 in NHPs reported anti-AAV NAB production and anti-transgene T-cell response at the highest doses tested.114 A recent study on 41 NHPs concluded that systemic levels of both anti-AAV BABs and NABs vary in a dose-dependent manner post ocular AAV injections.46 A study comparing two engineered variants (AAV2-7m8 and AAV8BP2) reported that AAV2-7m8 was more efficient in transducing most retinal cell types, whereas AAV8BP2 had a better safety profile at higher doses.115 There are multiple factors such as the capsid, transgene or promoter that influence the immune response and with increased dose the effect is cumulative. Promoter-associated ocular toxicity and reduced function have been reported in mice. A comparison CAG (ubiquitous) and Rho (cell-specific) promoters reported reduction in retinal function (measured by ERG) with ubiquitous promoter.116 Another study compared the effect of many ubiquitous (CAG, CMV, UbiC) and cell-specific (RedO, Rho, CAR, GRK, Best1) promoters. They reported higher toxicity to both retina and RPE by ubiquitous promoters. They further reported higher microglial activation and migration caused by ubiquitous (CMV) promoter compared to cell-specific (RedO) promoter. They reported higher innate immune responses with ubiquitous promoters, as measured by levels of TNF-α, IL-1β, IL-6 and IL-γ.117 More recently, we compared serum antibody levels after ocular delivery of AAV-mediated gene delivery in nonhuman primates (NHPs). We showed that NHPs receiving ubiquitous (CAG) promoter had higher serum antibody levels compared to the ones that received cell-specific (SNCG) promoter.46 Hence, using lower doses can ensure lower immune responses. This is particularly important when there is a requirement for readministration of AAV injections to maintain or augment therapeutic benefits.18,19 Readministration may be required because of the bilateral nature of the diseases affecting the eyes, wherein the contralateral eye may need therapy months or years after the first eye. Such readministration has been tested in non-human primates and also reported in clinical trials.18,118 Furthermore, presently efficient delivery to the photoreceptors is achieved by subretinal injections in clinical settings. This mode of delivery transduces only a small part of the retina, thus readministration to target other areas at later time points may be necessary to increase the therapeutic benefits.119
It would be highly desirable to have AAV variants that can evade immune responses and there have been attempts to engineer such viruses by rational design or directed evolution.120 A study employed the directed evolution strategy to develop variants that can escape immune response. A library of AAV2 variants was generated by error-prone PCR and then these variants were screened for their ability to transduce cells in the presence of serum antibodies. The best variants identified in this screen had 10 to 100 fold higher resistance to neutralizing antibodies.103 Another study used the capsid shuffling method (wherein the capsid sequences of known AAVs are fragmented and reassembled to generate variant sequences) to generate AAV variants library and screen these to find a less immunogenic variant. A novel variant identified from this screen named chimeric 1829 showed better transduction and lower reactivity to anti-AAV antibodies.121 A study identified the antibody binding regions on the AAV capsid using structural information acquired by cryo-electron microscopy. They further mutated these regions and reported that the variants thus obtained were more resistant to antibody neutralization.122 Apart from these attempts to engineer variants with desirable properties, another approach is to biomine variants to which humans would have no/low exposure. A study identified an ancestral AAV called Anc80 by in silico sequence reconstruction and reported this variant to be less immunogenic.100 A recent study isolated 4 new AAVs originating from bats and reported these variants as being more efficient in evading neutralizing antibodies. However, the overall transduction efficiencies of these variants were lower than the ones currently used.123
Another strategy to prevent the recognition of AAVs by NABs is to mask the antibody binding regions on the capsid surface by chemical modification. A polymer called Polyethylene Glycol (PEG) was conjugated with AAV to form PEGylated AAVs. These were reported to have higher resistance to NABs and result in enhanced transduction in the presence of NABs.124–126 However, when the ratio of PEG/AAV was increased the transduction efficiency reduced most likely because excessive coverage with PEGs reduced the exposure of the AAV capsid regions essential for infectivity.124 Some researchers have also attempted encapsulating the AAV capsid within gel beads or polymeric particles with the intention of providing initial protection against NABs and then progressively degrading the polymers and releasing AAVs.127 Although this strategy can be useful for lentivirus to some extent, it proved inefficient for AAVs as the release and spread of AAVs is dependent on diffusion mechanisms.128
Another strategy utilizes empty capsid (capsid decoys) that bind the anti-AAV antibodies, leaving the AAVs carrying the therapeutic gene intact. This was achieved by mutating the receptor binding site of AAVs and generating empty capsids. The empty capsids were injected along with the AAV capsid containing the transgene of interest. A higher transduction and low immunogenicity was reported when delivered systemically.129 Another study used a similar strategy with additional inhibition of macrophages reporting improved transduction.130 In these studies, the AAV delivery was systemic, wherein, on one hand, the threat and severity of immune response is higher but, on the other hand, higher doses can be tolerated. The eye is relatively immune privileged, but the dose and volume of AAV that can be tolerated is limited. The feasibility and usefulness of this technique for applications in the eye has not yet been tested.
Increased understanding of mechanisms by which AAV capsid and DNA interact with immune cells has enabled designing some recent immune evasion strategies. Immune cells have surface receptors called Toll-like Receptors (TLR) that sense DNA from pathogenic viruses containing unmethylated CpG motifs. CpG binding to TLR promotes its dimerization and activates TLR9 signaling. This leads to innate immune responses eventually recruiting other immune cells to the site of infection and further triggers adaptive immune responses.131 In 2019, two groups independently reported that the antibody response to AAV is mediated by toll-like receptor 9 (TLR9). They showed that TLR9 was responsible for activating T-cells against an AAV epitope and TLR9 also induces a cytotoxic T-cell response against the transgene resulting in loss of expression.132,133 Another study identified TLR9 inhibitory sequences (TLRi) that were 12–24 nucleotides in size. They designed the transgene sequences to include the TLRi sequences and reported lower immune responses and higher transduction efficiency in mouse and pig models.134 A study evaluated immune responses in Tlr null mutant (Tlr9-/-) and wild-type mice after injections of an immunogenic AAV variant (AAVrh32.33). They reported inhibition of IFN-γ T-cell responses toward capsid and transgene resulting in minimal cellular infiltrate and stable transgene expression.135 As mentioned earlier, immune responses occur upon interaction of CpG motifs on viral DNA with TLR receptors on immune cells. CpG islands are clustered regions rich in CG motifs that are common in AAV vector sequences and promoter sequences. Hence, in addition to modifying TLR9 receptors, attempts have been made to optimize the CpG content of the AAV genome that was reported to reduce T-cell expansion, lowering the immune response.135,136 The ITRs of AAVs are rich in CpG motifs. A recent study modified the ITRs and generated AAVs that contained CpG-free ITRs. They reported that the elimination of CpG motifs from the ITRs did not affect the biological activity of AAVs.137 The use of these strategies, either independently or in combination, can result in improved gene delivery in the presence of antibodies and facilitate repeat AAV injections.
Conclusion and Future Perspectives
In the context of immune responses, it is important to make a distinction between tolerable immune responses and detrimental immune responses. When any substance is introduced in the ocular space, an immune response can occur, which indicates a normal and expected reaction. However, when this immune response escalates, spreads outside the area of the intervention and cannot be managed, it can be referred to as a detrimental immune response. It becomes a cause of concern for ocular gene therapy applications when the inflammation does not subside over time, resulting in worse visual outcomes and clearance of transduced cells. Hence, while designing and optimizing different parameters of the gene therapy, one must not only consider the type of immune response that may occur, but more importantly, if these responses will adversely affect the therapeutic outcome. So far, most studies observe or measure distinct aspects of the innate and adaptive immune system, and report the presence or absence of the immune response. Future studies can benefit from a combination of immunomonitoring tools that can provide an accurate account of the situation. Another aspect that is lacking is studies investigating cellular and molecular mechanisms of the immune responses in the ocular space. These can provide valuable insights for effective evasion and management of immune responses.
While discussing “where are we now?” with AAVs it is also important to consider “where we are going?”. The present clinical trials mostly attempt to use a “replacement strategy” to deliver a small transgene (below 4.7kb) for monogenic diseases with recessive mutations. But, as the strategies get more complicated – involving large editing tools (CRISPR, Prime and BE) and large genes (above 4.5kb) – alternatives to AAVs will be required. Although the feasibility of double and triple AAVs has been reported, presently the efficiency is not sufficient for therapeutic benefits.49 Hence, there is a worldwide quest to develop non-viral vectors with the intention of delivering large cargo and to avoid viral components. Non-viral delivery can be as simple as delivering naked DNA or more sophisticated like using Virus-Like Particles (VLPs). There have been attempts of direct delivery of plasmid DNA,138 but this results in transient expression and is prone to being easily degraded by host cellular mechanisms. Several studies have also explored deliveries of liposomes, polymers and nanoparticles conjugated with DNA or protein.91 Some of these result in stable particles but the lack of cell-specificity in most cases and large size in some cases restricts their use. Recently, created synthetic vectors called VLPs are nanoscale structures made up of one or more different molecules with the ability to self-assemble, mimicking the form and size of a virus particle, but lacking the viral genetic material and being non-infectious.139 These new developments are interesting but none of these have efficiencies comparable to AAVs, thereby preventing their usage in vivo for retinal gene therapy applications. Moving forward, the field will benefit from development of both novel AAV vectors with enhanced properties as well as non-viral vectors that can carry larger or different cargo (DNA, mRNA or protein) while having AAVs’ desirable diffusion and cell entry properties.
Disclosure
Dr Deniz Dalkara reports grants from Foundation Fighting Blindness, USA and European Research Council, during the conduct of the study; Dr Deniz Dalkara is a co-inventor on patent #9193956 (Adeno-associated virus virions with variant capsid and methods of use thereof), with royalties paid to Adverum Biotechnologies and on pending patent applications on noninvasive methods to target cone photoreceptors (EP17306429.6 and EP17306430.4) licensed to Gamut Tx now Sparting- Vision. Dr Deniz Dalkara also has personal financial interests in Tenpoint Tx. and SparingVision, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. McPherson RA, Rosenthal LJ, Rose JA. Human cytomegalovirus completely helps adeno-associated virus replication. Virology. 1985;147(1):217–222. doi:10.1016/0042-6822(85)90243-0
2. Weindler FW, Heilbronn R. A subset of herpes simplex virus replication genes provides helper functions for productive adeno-associated virus replication. J Virol. 1991;65(5):2476–2483. doi:10.1128/jvi.65.5.2476-2483.1991
3. Cotmore SF, Agbandje-McKenna M, Canuti M, et al. ICTV virus taxonomy profile: parvoviridae. J Gen Virol. 2019;100(3):367–368. doi:10.1099/jgv.0.001212
4. Hastie E, Samulski RJ. Adeno-associated virus at 50: a golden anniversary of discovery, research, and gene therapy success—a personal perspective. Hum Gene Ther. 2015;26(5):257–265. doi:10.1089/hum.2015.025
5. Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral vector platforms within the gene therapy landscape. Sig Transduct Target Ther. 2021;6(1):53. doi:10.1038/s41392-021-00487-6
6. Bartlett JS, Wilcher R, Samulski RJ. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J Virol. 2000;74(6):2777–2785. doi:10.1128/JVI.74.6.2777-2785.2000
7. Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. 1996;70(5):3227–3234. doi:10.1128/jvi.70.5.3227-3234.1996
8. Samulski RJ, Chang LS, Shenk T. A recombinant plasmid from which an infectious adeno-associated virus genome can be excised in vitro and its use to study viral replication. J Virol. 1987;61(10):3096–3101. doi:10.1128/jvi.61.10.3096-3101.1987
9. Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Prog Retin Eye Res. 2021;100975. doi:10.1016/j.preteyeres.2021.100975
10. Martin KRG, Klein RL, Quigley HA. Gene delivery to the eye using adeno-associated viral vectors. Methods. 2002;28(2):267–275. doi:10.1016/S1046-2023(02)00232-3
11. Schneider N, Sundaresan Y, Gopalakrishnan P, et al. Inherited retinal diseases: linking genes, disease-causing variants, and relevant therapeutic modalities. Prog Retin Eye Res. 2022;89:101029. doi:10.1016/j.preteyeres.2021.101029
12. Willett K, Bennett J. Immunology of AAV-mediated gene transfer in the eye. Front Immunol. 2013;4. doi:10.3389/fimmu.2013.00261
13. Pierce EA, Bennett J. The status of RPE65 gene therapy trials: safety and efficacy. Cold Spring Harb Perspect Med. 2015;5(9):a017285. doi:10.1101/cshperspect.a017285
14. Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28(1):92–95. doi:10.1038/ng0501-92
15. Bainbridge JWB, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–2239. doi:10.1056/NEJMoa0802268
16. Jacobson SG. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130(1):9. doi:10.1001/archophthalmol.2011.298
17. Testa F, Maguire AM, Rossi S, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with leber congenital amaurosis type 2. Ophthalmology. 2013;120(6):1283–1291. doi:10.1016/j.ophtha.2012.11.048
18. Bennett J, Wellman J, Marshall KA, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on Phase 1 trial. Lancet. 2016;388(10045):661–672. doi:10.1016/S0140-6736(16)30371-3
19. Jacobson SG, Cideciyan AV, Roman AJ, et al. Improvement and decline in vision with gene therapy in childhood blindness. N Engl J Med. 2015;372(20):1920–1926. doi:10.1056/NEJMoa1412965
20. Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–e116. doi:10.1016/S2214-109X(13)70145-1
21. Amoaku WM, Chakravarthy U, Gale R, et al. Defining response to anti-VEGF therapies in neovascular AMD. Eye. 2015;29(6):721–731. doi:10.1038/eye.2015.48
22. Ricci F, Bandello F, Navarra P, Staurenghi G, Stumpp M, Zarbin M. Neovascular age-related macular degeneration: therapeutic management and new-upcoming approaches. IJMS. 2020;21(21):8242. doi:10.3390/ijms21218242
23. Lindner M, Gilhooley MJ, Hughes S, Hankins MW. Optogenetics for visual restoration: from proof of principle to translational challenges. Prog Retin Eye Res. 2022;91:101089. doi:10.1016/j.preteyeres.2022.101089
24. Sahel JA, Boulanger-Scemama E, Pagot C, et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat Med. 2021;27(7):1223–1229. doi:10.1038/s41591-021-01351-4
25. Simon CJ, Sahel JA, Duebel J, Herlitze S, Dalkara D. Opsins for vision restoration. Biochem Biophys Res Commun. 2020;527(2):325–330. doi:10.1016/j.bbrc.2019.12.117
26. Meng D, Ragi SD, Tsang SH. Therapy in rhodopsin-mediated autosomal dominant retinitis pigmentosa. Mol Ther. 2020;28(10):2139–2149. doi:10.1016/j.ymthe.2020.08.012
27. Levy JM, Yeh WH, Pendse N, et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat Biomed Eng. 2020;4(1):97–110. doi:10.1038/s41551-019-0501-5
28. Choi EH, Suh S, Foik AT, et al. In vivo base editing rescues cone photoreceptors in a mouse model of early-onset inherited retinal degeneration. Nat Commun. 2022;13(1):1830. doi:10.1038/s41467-022-29490-3
29. Suh S, Choi EH, Raguram A, Liu DR, Palczewski K. Precision genome editing in the eye. Proc Natl Acad Sci USA. 2022;119(39):e2210104119. doi:10.1073/pnas.2210104119
30. Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–191. doi:10.1038/nature14299
31. Grünewald J, Zhou R, Garcia SP, et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569(7756):433–437. doi:10.1038/s41586-019-1161-z
32. Pan X, Qu K, Yuan H, et al. Massively targeted evaluation of therapeutic CRISPR off-targets in cells. Nat Commun. 2022;13(1):4049. doi:10.1038/s41467-022-31543-6
33. Vicente MM, Chaves-Ferreira M, Jorge JMP, Proença JT, Barreto VM. The off-targets of clustered regularly interspaced short palindromic repeats gene editing. Front Cell Dev Biol. 2021;9:718466. doi:10.3389/fcell.2021.718466
34. Buck T, Wijnholds J. Recombinant Adeno-Associated Viral Vectors (rAAV)-vector elements in ocular gene therapy clinical trials and transgene expression and bioactivity assays. IJMS. 2020;21(12):4197. doi:10.3390/ijms21124197
35. Pfeiffer RL, Marc RE, Jones BW. Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog Retin Eye Res. 2020;74:100771. doi:10.1016/j.preteyeres.2019.07.004
36. Petrs-Silva H, Dinculescu A, Li Q, et al. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol Ther. 2009;17(3):463–471. doi:10.1038/mt.2008.269
37. Dalkara D, Byrne LC, Klimczak RR, et al. In vivo–directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5(189). doi:10.1126/scitranslmed.3005708
38. Byrne LC, Day TP, Visel M, et al. In vivo–directed evolution of adeno-associated virus in the primate retina. JCI Insight. 2020;5(10):e135112. doi:10.1172/jci.insight.135112
39. Peynshaert K, Devoldere J, Minnaert AK, De Smedt SC, Remaut K. Morphology and composition of the inner limiting membrane: species-specific variations and relevance toward drug delivery research. Curr Eye Res. 2019;44(5):465–475. doi:10.1080/02713683.2019.1565890
40. Boehm M, Oellers P, Thanos S. Inflammation and immunology of the vitreoretinal compartment. IADT. 2011;10(4):283–309. doi:10.2174/187152811796117717
41. Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno‐associated viruses. J INFECT DIS. 2009;199(3):381–390. doi:10.1086/595830
42. Boutin S, Monteilhet V, Veron P, et al. Prevalence of serum IgG and neutralizing factors against Adeno-Associated Virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–712. doi:10.1089/hum.2009.182
43. Fitzpatrick Z, Leborgne C, Barbon E, et al. Influence of pre-existing anti-capsid neutralizing and binding antibodies on AAV vector transduction. Mol Ther. 2018;9:119–129. doi:10.1016/j.omtm.2018.02.003
44. Ren D, Fisson S, Dalkara D, Ail D. Immune responses to gene editing by viral and non-viral delivery vectors used in retinal gene therapy. Pharmaceutics. 2022;14(9):1973. doi:10.3390/pharmaceutics14091973
45. Wang D, Zhong L, Li M, et al. Adeno-associated virus neutralizing antibodies in large animals and their impact on brain intraparenchymal gene transfer. Mol Ther. 2018;11:65–72. doi:10.1016/j.omtm.2018.09.003
46. Ail D, Ren D, Brazhnikova E, et al. Systemic and local immune responses to intraocular AAV vector administration in non-human primates. Mol Ther. 2022;24:306–316. doi:10.1016/j.omtm.2022.01.011
47. Bucher K, Rodríguez-Bocanegra E, Dauletbekov D, Fischer MD. Immune responses to retinal gene therapy using adeno-associated viral vectors – implications for treatment success and safety. Prog Retin Eye Res. 2021;83:100915. doi:10.1016/j.preteyeres.2020.100915
48. Maddalena A, Tornabene P, Tiberi P, et al. Triple vectors expand AAV transfer capacity in the retina. Mol Ther. 2018;26(2):524–541. doi:10.1016/j.ymthe.2017.11.019
49. Tornabene P, Trapani I. Can adeno-associated viral vectors deliver effectively large genes? Hum Gene Ther. 2020;31(1–2):47–56. doi:10.1089/hum.2019.220
50. Ghosh A, Duan D. Expanding adeno-associated viral vector capacity: a tale of two vectors. Biotechnol Genet Eng Rev. 2007;24(1):165–178. doi:10.1080/02648725.2007.10648098
51. Dyka FM, Boye SL, Chiodo VA, Hauswirth WW, Boye SE. Dual adeno-associated virus vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum Gene Ther Methods. 2014;25(2):166–177. doi:10.1089/hgtb.2013.212
52. Trapani I, Colella P, Sommella A, et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med. 2014;6(2):194–211. doi:10.1002/emmm.201302948
53. McClements ME, Barnard AR, Charbel Issa P, MacLaren RE. Assessment of AAV dual vector safety in theAbca4-/- mouse model of stargardt disease. Transl Vis Sci Technol. 2020;9(7):20. doi:10.1167/tvst.9.7.20
54. Carvalho LS, Turunen HT, Wassmer SJ, et al. Evaluating efficiencies of dual AAV approaches for retinal targeting. Front Neurosci. 2017;11:503. doi:10.3389/fnins.2017.00503
55. Trapani I, Toriello E, de Simone S, et al. Improved dual AAV vectors with reduced expression of truncated proteins are safe and effective in the retina of a mouse model of Stargardt disease. Hum Mol Genet. 2015;24(23):6811–6825. doi:10.1093/hmg/ddv386
56. Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019;25(2):229–233. doi:10.1038/s41591-018-0327-9
57. Pulman J, Sahel JA, Dalkara D. New editing tools for gene therapy in inherited retinal dystrophies. CRISPR J. 2022. doi:10.1089/crispr.2021.0141
58. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–471. doi:10.1038/nature24644
59. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38(7):824–844. doi:10.1038/s41587-020-0561-9
60. Truong DJJ, Kühner K, Kühn R, et al. Development of an intein-mediated split–Cas9 system for gene therapy. Nucleic Acids Res. 2015;43(13):6450–6458. doi:10.1093/nar/gkv601
61. Yee T, Wert KJ. Base and prime editing in the retina-from preclinical research toward human clinical trials. Int J Mol Sci. 2022;23(20):12375. doi:10.3390/ijms232012375
62. Jo DH, Jang HK, Cho CS, et al. Visual function restoration in a mouse model of Leber congenital amaurosis via therapeutic base editing. Mol Ther Nucleic Acids. 2023;31:16–27. doi:10.1016/j.omtn.2022.11.021
63. Puppo A, Cesi G, Marrocco E, et al. Retinal transduction profiles by high-capacity viral vectors. Gene Ther. 2014;21(10):855–865. doi:10.1038/gt.2014.57
64. Han IC, Burnight ER, Ulferts MJ, et al. Helper-dependent adenovirus transduces the human and rat retina but elicits an inflammatory reaction when delivered subretinally in rats. Hum Gene Ther. 2019;30(11):1371–1384. doi:10.1089/hum.2019.159
65. Davis JR, Wang X, Witte IP, et al. Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors. Nat Biomed Eng. 2022;6(11):1272–1283. doi:10.1038/s41551-022-00911-4
66. Zhang H, Bamidele N, Liu P, et al. Adenine base editing in vivo with a single adeno-associated virus vector. GEN Biotechnol. 2022;1(3):285–299. doi:10.1089/genbio.2022.0015
67. Rivera VM, Gao G, Grant RL, et al. Long-term pharmacologically regulated expression of erythropoietin in primates following AAV-mediated gene transfer. Blood. 2005;105(4):1424–1430. doi:10.1182/blood-2004-06-2501
68. Penaud-Budloo M, Le Guiner C, Nowrouzi A, et al. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J Virol. 2008;82(16):7875–7885. doi:10.1128/JVI.00649-08
69. Sehara Y, Fujimoto K, Ikeguchi K, et al. Persistent expression of dopamine-synthesizing enzymes 15 years after gene transfer in a primate model of parkinson’s disease. Hum Gene Ther Clin Dev. 2017;28(2):74–79. doi:10.1089/humc.2017.010
70. Hanlon KS, Kleinstiver BP, Garcia SP, et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat Commun. 2019;10(1):4439. doi:10.1038/s41467-019-12449-2
71. Anselmo AC, Mitragotri S. Nanoparticles in the clinic: an update post COVID‐19 vaccines. Bioeng Transl Med. 2021;6(3). doi:10.1002/btm2.10246
72. Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385(6):493–502. doi:10.1056/NEJMoa2107454
73. Thi TTH, Suys EJA, Lee JS, Nguyen DH, Park KD, Truong NP. Lipid-based nanoparticles in the clinic and clinical trials: from cancer nanomedicine to COVID-19 vaccines. Vaccines. 2021;9(4):359. doi:10.3390/vaccines9040359
74. Ramamoorth M. Non viral vectors in gene therapy- an overview. JCDR. 2015. doi:10.7860/JCDR/2015/10443.5394
75. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15(8):541–555. doi:10.1038/nrg3763
76. Zu H, Gao D. Non-viral vectors in gene therapy: recent development, challenges, and prospects. AAPS J. 2021;23(4):78. doi:10.1208/s12248-021-00608-7
77. Benedicto I, Lehmann GL, Ginsberg M, et al. Concerted regulation of retinal pigment epithelium basement membrane and barrier function by angiocrine factors. Nat Commun. 2017;8(1):15374. doi:10.1038/ncomms15374
78. Kuo SP, Chiang PP, Nippert AR, Newman EA. Spatial organization and dynamics of the extracellular space in the mouse retina. J Neurosci. 2020;40(41):7785–7794. doi:10.1523/JNEUROSCI.1717-20.2020
79. Chien Y, Hsiao YJ, Chou SJ, et al. Nanoparticles-mediated CRISPR-Cas9 gene therapy in inherited retinal diseases: applications, challenges, and emerging opportunities. J Nanobiotechnol. 2022;20(1):511. doi:10.1186/s12951-022-01717-x
80. Xu L, Wang X, Liu Y, Yang G, Falconer RJ, Zhao CX. Lipid nanoparticles for drug delivery. Adv Nanobiomed Res. 2022;2(2):2100109. doi:10.1002/anbr.202100109
81. Salman A, Kantor A, McClements ME, Marfany G, Trigueros S, MacLaren RE. Non-viral delivery of CRISPR/Cas cargo to the retina using nanoparticles: current possibilities, challenges, and limitations. Pharmaceutics. 2022;14(9):1842. doi:10.3390/pharmaceutics14091842
82. Rajala A, Wang Y, Zhu Y, et al. Nanoparticle-assisted targeted delivery of eye-specific genes to eyes significantly improves the vision of blind mice in vivo. Nano Lett. 2014;14(9):5257–5263. doi:10.1021/nl502275s
83. Gallego I, Villate-Beitia I, Martínez-Navarrete G, et al. Non-viral vectors based on cationic niosomes and minicircle DNA technology enhance gene delivery efficiency for biomedical applications in retinal disorders. Nanomedicine. 2019;17:308–318. doi:10.1016/j.nano.2018.12.018
84. Chen Y, Liu X, Zhang Y, et al. A self-restricted CRISPR system to reduce off-target effects. Mol Ther. 2016;24(9):1508–1510. doi:10.1038/mt.2016.172
85. Petris G, Casini A, Montagna C, et al. Hit and go CAS9 delivered through a lentiviral based self-limiting circuit. Nat Commun. 2017;8:15334. doi:10.1038/ncomms15334
86. Kim K, Park SW, Kim JH, et al. Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Res. 2017;27(3):419–426. doi:10.1101/gr.219089.116
87. Holmgaard AB, Askou AL, Jensen EG, et al. Targeted knockout of the vegfa gene in the retina by subretinal injection of RNP complexes containing Cas9 protein and modified sgRNAs. Mol Ther. 2021;29(1):191–207. doi:10.1016/j.ymthe.2020.09.032
88. Jang HK, Jo DH, Lee SN, et al. High-purity production and precise editing of DNA base editing ribonucleoproteins. Sci Adv. 2021;7(35):eabg2661. doi:10.1126/sciadv.abg2661
89. Dalkara D, Kolstad KD, Caporale N, et al. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther. 2009;17(12):2096–2102. doi:10.1038/mt.2009.181
90. Lipinski DM, Thake M, MacLaren RE. Clinical applications of retinal gene therapy. Prog Retin Eye Res. 2013;32:22–47. doi:10.1016/j.preteyeres.2012.09.001
91. Trapani I, Puppo A, Auricchio A. Vector platforms for gene therapy of inherited retinopathies. Prog Retin Eye Res. 2014;43:108–128. doi:10.1016/j.preteyeres.2014.08.001
92. Khabou H, Garita-Hernandez M, Chaffiol A, et al. Noninvasive gene delivery to foveal cones for vision restoration. JCI Insight. 2018;3(2):e96029. doi:10.1172/jci.insight.96029
93. Boye SL, Choudhury S, Crosson S, et al. Novel AAV44.9-based vectors display exceptional characteristics for retinal gene therapy. Mol Ther. 2020;28(6):1464–1478. doi:10.1016/j.ymthe.2020.04.002
94. Boye SL, Bennett A, Scalabrino ML, et al. Impact of heparan sulfate binding on transduction of retina by recombinant adeno-associated virus vectors. J Virol. 2016;90(8):4215–4231. doi:10.1128/JVI.00200-16
95. Zhong L, Li B, Mah CS, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci USA. 2008;105(22):7827–7832. doi:10.1073/pnas.0802866105
96. Petrs-Silva H, Dinculescu A, Li Q, et al. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol Ther. 2011;19(2):293–301. doi:10.1038/mt.2010.234
97. Song C, Dufour VL, Cideciyan AV, et al. Dose range finding studies with two RPGR transgenes in a canine model of X-linked retinitis pigmentosa treated with subretinal gene therapy. Hum Gene Ther. 2020;31(13–14):743–755. doi:10.1089/hum.2019.337
98. Dufour VL, Cideciyan AV, Jie YG, et al. Toxicity and efficacy evaluation of an adeno-associated virus vector expressing codon-optimized RPGR delivered by subretinal injection in a canine model of X-linked retinitis pigmentosa. Hum Gene Ther. 2020;31(3–4):253–267. doi:10.1089/hum.2019.297
99. Eichhoff AM, Börner K, Albrecht B, et al. Nanobody-enhanced targeting of AAV gene therapy vectors. Mol Ther. 2019;15:211–220. doi:10.1016/j.omtm.2019.09.003
100. Zinn E, Pacouret S, Khaychuk V, et al. In silico reconstruction of the viral evolutionary lineage yields a potent gene therapy vector. Cell Rep. 2015;12(6):1056–1068. doi:10.1016/j.celrep.2015.07.019
101. Carvalho LS, Xiao R, Wassmer SJ, et al. Synthetic adeno-associated viral vector efficiently targets mouse and nonhuman primate retina in vivo. Hum Gene Ther. 2018;29(7):771–784. doi:10.1089/hum.2017.154
102. Müller OJ, Kaul F, Weitzman MD, et al. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol. 2003;21(9):1040–1046. doi:10.1038/nbt856
103. Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24(2):198–204. doi:10.1038/nbt1182
104. Perabo L, Goldnau D, White K, et al. Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J Virol. 2006;80(14):7265–7269. doi:10.1128/JVI.00076-06
105. Grimm D, Lee JS, Wang L, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. 2008;82(12):5887–5911. doi:10.1128/JVI.00254-08
106. Ross M, Obolensky A, Averbukh E, et al. Outer retinal transduction by AAV2-7m8 following intravitreal injection in a sheep model of CNGA3 achromatopsia. Gene Ther. 2022;29(10–11):624–635. doi:10.1038/s41434-021-00306-1
107. Öztürk BE, Johnson ME, Kleyman M, et al. scAAVengr, a transcriptome-based pipeline for quantitative ranking of engineered AAVs with single-cell resolution. eLife. 2021;10:e64175. doi:10.7554/eLife.64175
108. Kotterman M, Beliakoff G, Croze R, et al. Directed evolution of AAV targeting primate retina by intravitreal injection identifies R100, a variant demonstrating robust gene delivery and therapeutic efficacy in non-human primates. Bioengineering. 2021. doi:10.1101/2021.06.24.449775
109. Xi Z, Öztürk BE, Johnson ME, Turunç S, Stauffer WR, Byrne LC. Quantitative single-cell transcriptome-based ranking of engineered AAVs in human retinal explants. Mol Ther. 2022;25:476–489. doi:10.1016/j.omtm.2022.04.014
110. Bryant DH, Bashir A, Sinai S, et al. Deep diversification of an AAV capsid protein by machine learning. Nat Biotechnol. 2021;39(6):691–696. doi:10.1038/s41587-020-00793-4
111. Sinai S, Jain N, Church GM, Kelsic ED. Generative AAV capsid diversification by latent interpolation. Synth Biol. 2021. doi:10.1101/2021.04.16.440236
112. Nemoto T, Ocari T, Planul A, et al. In-silico monitoring of directed evolution convergence to unveil best performing variants with credibility score. Bioinformatics. 2023. doi:10.1101/2023.01.03.522172
113. Ogden PJ, Kelsic ED, Sinai S, Church GM. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science. 2019;366(6469):1139–1143. doi:10.1126/science.aaw2900
114. Vandenberghe LH, Bell P, Maguire AM, et al. Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci Transl Med. 2011;3(88):88ra54. doi:10.1126/scitranslmed.3002103
115. Ramachandran PS, Lee V, Wei Z, et al. Evaluation of dose and safety of AAV7m8 and AAV8BP2 in the non-human primate retina. Hum Gene Ther. 2017;28(2):154–167. doi:10.1089/hum.2016.111
116. Khabou H, Cordeau C, Pacot L, Fisson S, Dalkara D. Dosage thresholds and influence of transgene cassette in adeno-associated virus–related toxicity. Hum Gene Ther. 2018;29(11):1235–1241. doi:10.1089/hum.2018.144
117. Xiong W, Wu DM, Xue Y, et al. AAV cis -regulatory sequences are correlated with ocular toxicity. Proc Natl Acad Sci USA. 2019;116(12):5785–5794. doi:10.1073/pnas.1821000116
118. Amado D, Mingozzi F, Hui D, et al. Safety and efficacy of subretinal readministration of a viral vector in large animals to treat congenital blindness. Sci Transl Med. 2010;2(21):21ra16. doi:10.1126/scitranslmed.3000659
119. Weed L, Ammar MJ, Zhou S, et al. Safety of same-eye subretinal sequential readministration of AAV2-hRPE65v2 in non-human primates. Mol Ther Methods Clin Dev. 2019;15:133–148. doi:10.1016/j.omtm.2019.08.011
120. Selot R, Hareendran S, Jayandharan G. Developing immunologically inert Adeno-Associated Virus (AAV) vectors for gene therapy: possibilities and limitations. CPB. 2014;14(12):1072–1082. doi:10.2174/1389201015666140327141710
121. Li W, Asokan A, Wu Z, et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther. 2008;16(7):1252–1260. doi:10.1038/mt.2008.100
122. Tse LV, Klinc KA, Madigan VJ, et al. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc Natl Acad Sci USA. 2017;114(24). doi:10.1073/pnas.1704766114
123. Li Y, Li J, Liu Y, et al. Bat adeno-associated viruses as gene therapy vectors with the potential to evade human neutralizing antibodies. Gene Ther. 2019;26(6):264–276. doi:10.1038/s41434-019-0081-8
124. Lee GK, Maheshri N, Kaspar B, Schaffer DV. PEG conjugation moderately protects adeno-associated viral vectors against antibody neutralization. Biotechnol Bioeng. 2005;92(1):24–34. doi:10.1002/bit.20562
125. Le HT, Yu QC, Wilson JM, Croyle MA. Utility of PEGylated recombinant adeno-associated viruses for gene transfer. J Control Release. 2005;108(1):161–177. doi:10.1016/j.jconrel.2005.07.019
126. Yao T, Zhou X, Zhang C, et al. Site-specific PEGylated adeno-associated viruses with increased serum stability and reduced immunogenicity. Molecules. 2017;22(7):1155. doi:10.3390/molecules22071155
127. Jang JH, Schaffer DV, Shea LD. Engineering biomaterial systems to enhance viral vector gene delivery. Mol Ther. 2011;19(8):1407–1415. doi:10.1038/mt.2011.111
128. Madrigal JL, Shams S, Stilhano RS, Silva EA. Characterizing the encapsulation and release of lentivectors and adeno-associated vectors from degradable alginate hydrogels. Biomater Sci. 2019;7(2):645–656. doi:10.1039/C8BM01218K
129. Mingozzi F, Anguela XM, Pavani G, et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Transl Med. 2013;5(194). doi:10.1126/scitranslmed.3005795
130. Aalbers CJ, Broekstra N, van Geldorp M, et al. Empty capsids and macrophage inhibition/depletion increase rAAV transgene expression in joints of both healthy and arthritic mice. Hum Gene Ther. 2017;28(2):168–178. doi:10.1089/hum.2016.036
131. Huérfano S, Šroller V, Bruštíková K, Horníková L, Forstová J. The interplay between viruses and host DNA sensors. Viruses. 2022;14(4):666. doi:10.3390/v14040666
132. Ashley SN, Somanathan S, Giles AR, Wilson JM. TLR9 signaling mediates adaptive immunity following systemic AAV gene therapy. Cell Immunol. 2019;346:103997. doi:10.1016/j.cellimm.2019.103997
133. Herzog RW, Cooper M, Perrin GQ, et al. Regulatory T cells and TLR9 activation shape antibody formation to a secreted transgene product in AAV muscle gene transfer. Cell Immunol. 2019;342:103682. doi:10.1016/j.cellimm.2017.07.012
134. Chan YK, Wang SK, Chu CJ, et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci Transl Med. 2021;13(580):eabd3438. doi:10.1126/scitranslmed.abd3438
135. Faust SM, Bell P, Cutler BJ, et al. CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Invest. 2013;123(7):2994–3001. doi:10.1172/JCI68205
136. Xiang Z, Kurupati RK, Li Y, et al. The effect of CpG sequences on capsid-specific CD8+ T cell responses to AAV vector gene transfer. Mol Ther. 2020;28(3):771–783. doi:10.1016/j.ymthe.2019.11.014
137. Pan X, Yue Y, Boftsi M, et al. Rational engineering of a functional CpG-free ITR for AAV gene therapy. Gene Ther. 2022;29(6):333–345. doi:10.1038/s41434-021-00296-0
138. Oliveira AV, Rosa da Costa AM, Silva GA. Non-viral strategies for ocular gene delivery. Mater Sci Eng C. 2017;77:1275–1289. doi:10.1016/j.msec.2017.04.068
139. Banskota S, Raguram A, Suh S, et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell. 2022;185(2):250–265.e16. doi:10.1016/j.cell.2021.12.021
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.