Back to Journals » Biologics: Targets and Therapy » Volume 19
Adalimumab Biosimilars Demonstrate Long-Term Durability and Cost-Effectiveness in Paediatric Inflammatory Bowel Disease: A Real-World Two-Centre European Cohort Study
Authors Ancona S ![]() , Armstrong K, Longo C, Rabone R, Merrick V, Henderson P
, Armstrong K, Longo C, Rabone R, Merrick V, Henderson P ![]() , Gandullia P, Wilson DC, Arrigo S, Russell RK
, Gandullia P, Wilson DC, Arrigo S, Russell RK ![]()
Received 14 December 2024
Accepted for publication 22 February 2025
Published 29 April 2025 Volume 2025:19 Pages 265—279
DOI https://doi.org/10.2147/BTT.S511248
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Shein-Chung Chow
Silvana Ancona,1,2 Katherine Armstrong,2 Chiara Longo,3 Rosalind Rabone,2 Victoria Merrick,2 Paul Henderson,2,4 Paolo Gandullia,3 David C Wilson,2,4 Serena Arrigo,3 Richard K Russell2,4
1Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genoa, Genoa, Italy; 2Department of Paediatric Gastroenterology and Nutrition, Royal Hospital for Children and Young People, Edinburgh, UK; 3Paediatric Gastroenterology and Endoscopy Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy; 4Child Life and Health, University of Edinburgh, Edinburgh, UK
Correspondence: Richard K Russell, Royal Hospital for Children and Young People, 50 Little France Crescent, Edinburgh, EH16 4TJ, UK, Tel +44 131 312 0431, Email [email protected]
Purpose: Adalimumab biosimilars are increasingly used in paediatric Inflammatory Bowel Disease (PIBD), but data remain limited. This study assessed their durability, efficacy, safety and cost implications in PIBD.
Patients and Methods: Consecutive PIBD patients who started adalimumab biosimilars between October 2018 and December 2023 at two centres in Scotland and Italy, with at least 6 months follow-up, were included. Demographic, disease, treatment, and adverse event data were collected. Disease activity was assessed at baseline, 6, 12, 24, 36 months, and at last follow-up. Durability was evaluated using Kaplan-Meier analysis.
Results: In total 130 patients (81 males; median age 12.3 years) were included (115 Crohn’s Disease, 7 Ulcerative Colitis, 8 IBD unclassified). The biosimilars were ABP 501 (85%), GP2017 (14%), SB5 (1%); 41 (32%) patients switched from originator. After a median follow-up of 26 months, 87/130 (67%) patients remained on biosimilars, while 43 discontinued at a median of 14 months. Durability probabilities were 93%, 86%, 75%, 62%, and 57% at 6, 12, 24, 36, and 54 months, respectively. Patients previously exposed to ADA originator had a lower risk of biosimilar failure (hazard ratio, adjusted for age at diagnosis: 0.51 [95% confidence interval: 0.26– 0.99], p=0.047). Trough levels ≥ 11.6 μg/mL at 6 months were associated with greater durability (AUC=0.68, p=0.007). Adverse events occurred in 46/130 patients, mainly psoriasis and injection site reactions (13% each), with one lymphoma. Estimated cost savings were 5,030€ per patient/year.
Conclusion: This real-life study demonstrated high durability and remission rates for adalimumab biosimilars in PIBD, confirming their clinical, cost-effectiveness and safety profile in children.
Keywords: biosimilar, adalimumab, paediatric inflammatory bowel disease
Introduction
The treatment of Inflammatory Bowel Disease (IBD) has radically changed over the past few decades, driven by significant advances in the understanding of its pathogenesis and the development of targeted therapies, particularly biologic agents. Tumour necrosis factor-α (TNF-α) antagonists were the first biologic drugs approved for IBD treatment. Adalimumab (ADA) is a fully humanised IgG1 monoclonal antibody with high affinity for TNF-α. Its use has increased among IBD patients, both adults and children, in recent years.1 Although highly effective,2 biologic medications are expensive and constitute a substantial portion of healthcare costs.3 Humira® (AbbVie Biotechnology GmbH, Ludwigshafen, Germany) was the first ADA introduced to the market, also known as the “originator” or “reference medicine”, and the patent expired in Europe in October 2018, leading to the development of biosimilars. ABP 501 (Amgevita®, Amgen Europe B.V., Breda, The Netherlands) was the first ADA biosimilar approved in Europe.
To date, few observational studies have examined the use of ADA biosimilar in adult IBD patients, reporting similar safety and effectiveness to the ADA originator,4,5 even after switching.6–15 As for the paediatric population, Dipasquale et al recently described their real-life experience with the short- and medium-term effectiveness and safety of ADA biosimilar in 41 paediatric IBD (PIBD) patients.16 Their findings showed high treatment persistence rates (85% at 1 and 2 years) and a low incidence of non-serious adverse events (AEs) (10%), although with some limitations due to the small sample size and relatively short follow-up.
Given this lack of evidence, this study aimed to assess the long-term durability of ADA biosimilars in PIBD, with follow-up lasting up to 72 months, as well as their effectiveness, safety and cost implications. Durability can be considered a general marker of treatment success, as it reflects overall effectiveness, AEs, and patient acceptance.17
Materials and Methods
Study Design and Population
This was an observational, retrospective cohort (two-centre) study, conducted at the Royal Hospital for Children and Young People in Edinburgh (South East Scotland regional service, with a total population of 1.4 million and complete accrual of all PIBD diagnoses before 16 years of age) and Gaslini Children’s Hospital in Genoa (Liguria regional service, with a total population of 1.5 million and referral centre for PIBD diagnosis before 18 years of age, including referrals from other regions of Italy). We included all PIBD patients who started ADA biosimilars at any point during their disease course between October 2018 and December 2023, with a minimum follow-up period of 6 months to 30 June 2024. PIBD was diagnosed according to the Porto Criteria.18 The study end was defined as ADA biosimilar discontinuation, emigration from the region, or 30 June 2024.
Data Collection
Demographic data, including sex and age at diagnosis, and baseline disease characteristics, such as location and behaviour according to the Paris classification,19 extraintestinal manifestations, IBD-related surgeries, and previous treatments, were recorded from July to August 2024. Data on ADA biosimilar treatment included age at initiation, concomitant medications, dose escalation, and treatment duration. Disease activity was assessed at baseline, at the end of induction (if applicable), at 6, 12, 24 and 36 months, and at the end of follow-up (if not coinciding with the aforementioned timepoints). Specifically, clinical status was assessed using disease activity scores; the Paediatric Crohn’s Disease Activity Index (PCDAI)20 for patients with Crohn’s Disease (CD), and the Paediatric Ulcerative Colitis Activity Index (PUCAI)21 for those with Ulcerative Colitis (UC) and IBD unclassified (IBDU). When not reported in the electronic medical records, PCDAI and PUCAI were retrospectively calculated. Additionally, biochemical/haematological status was monitored at each follow-up through haemoglobin (Hb), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and faecal calprotectin (FC) levels. Serum ADA trough levels and anti-ADA antibodies were also collected, when available. The permitted timeframe for blood and stool tests was from 1 month before to 1 month after the specified timepoints. AEs were monitored throughout the entire follow-up period. Radiologic (abdominal ultrasound and magnetic resonance imaging), endoscopic and histologic activity data were also recorded, when available. Due to the limited availability of endoscopic data, mucosal healing (MH) rates in CD patients were estimated using the Mucosal Inflammation Non-invasive (MINI) Index, a validated non-invasive index for assessing MH in children with CD. The MINI index ranges from −3 to 25, with a value less than 8 indicating MH.22
We also evaluated drug-related costs, based on data from the Scottish Drug Tariff and Italian Medicines Agency. Costs were determined using the market value at the time of biosimilar use (Amgevita®, Hyrimoz® and Imraldi®). Cost savings were calculated by comparing the total costs of biosimilars (based on the number of pens/syringes used during the follow-up period) to the equivalent costs of the originator (for the same quantity of pens/syringes). All costs were converted to Euros, according to the exchange rate as of 30 July 2024.
ADA Trough Levels and Antibodies
In the Scottish cohort, serum ADA trough levels and anti-ADA antibodies were quantitatively measured using an Enzyme-Linked Immunosorbent Assay (Promonitor® ELISA assay, Grifols S.A., Barcelona, Spain) provided by the Scottish Biologic Therapeutic Drug Monitoring Service. The working range for ADA trough levels was <0.4 μg/mL as the lower limit and >12 μg/mL as the upper limit. Dilution of drug levels was performed upon physician request, and non-diluted ADA levels >12 μg/mL were approximated to 12 μg/mL. The ADA-antibody assay was only conducted when ADA trough levels were <5 μg/mL. The working range for the ADA-antibody assay was <10 and >200 AU/mL as the lower and upper limits, respectively; anti-ADA antibodies ≥10 AU/mL were considered positive.
In the Italian cohort, serum ADA trough levels and anti-ADA antibodies were measured at Gaslini Children’s Hospital using the Quantum Blue® kit (BÜHLMANN Laboratories AG, Schönenbuch, Switzerland). The working range for ADA trough levels was <1.3 μg/mL as the lower limit and >35 μg/mL as the upper limit. Anti-ADA antibodies were measured independently of drug levels, using a qualitative assay, with results reported as negative for concentrations <0.2 μgeq/mL or positive for concentrations ≥0.2 μgeq/mL.
Outcomes
The primary aim was to evaluate the long-term durability of ADA biosimilars in PIBD, defined as consistent treatment without the need for IBD-related surgery, switching back to the originator, discontinuing ADA biosimilars, or starting another advanced therapy (biological or small molecules). Specifically, we assessed the probability of durability at 6, 12, 24 and 36 months after the initiation of ADA biosimilars.
Secondary outcomes included: 1) the identification of predictive factors for ADA biosimilar failure; 2) an evaluation of the efficacy of ADA biosimilars in achieving clinical and luminal remission (clinical remission was defined as PCDAI or PUCAI <10, while luminal remission was defined as FC <100 μg/g combined with clinical remission); and 3) an analysis of the safety of ADA biosimilars (AEs were defined as any unwanted or harmful medical occurrences in patients following the initiation of ADA biosimilars, potentially related to the treatment).
Primary non-response was defined as the failure to achieve clinical improvement after completing the induction course in the presence of therapeutic drug levels, whereas secondary loss of response was defined as the recurrence of symptoms after an initial clinical benefit.
Statistical Analysis
Categorical variables were presented as frequencies and percentages, while continuous variables as median and interquartile range (IQR), as appropriate. Categorical variables were compared using the Chi-square test, and continuous variables were compared using the Mann–Whitney test for non-parametric data. The association between demographic and baseline characteristics and ADA biosimilar failure was investigated using univariate Cox proportional hazard regression analysis, then adjusted for age at diagnosis. The probability of durability at 6, 12, 24 and 36 months from treatment initiation was calculated using Kaplan-Meier survival curves. The Cox proportional hazards model was used to analyse differences between subgroups over time. The Schoenfeld test was applied to ensure the proportional hazards assumption was not violated. The Youden index method, derived from ROC curve analysis, was used to identify the cut-off for drug through levels predictive of ADA failure. Missing data were managed using pairwise deletion. A p-value <0.05 was considered statistically significant. All statistical analyses were performed using SPSS (version 25; IBM Corporation) and RStudio (version 2022.02.03, Build 492).
Results
One-hundred thirty-six patients started ADA biosimilars between October 2018 and December 2023; however, six patients were excluded due to a follow-up period of less than 6 months. Thus, a total of 130 patients on ADA biosimilars were included in the study: 88 (67%) from Edinburgh and 42 (32%) from Genoa. The majority of patients were male (n=81, 62%), and the median age at diagnosis was 12.3 years (IQR 10.0–14.3). Of these, 115 (88%) had CD, 7 (5%) had UC, and 8 (6%) had IBDU. Forty-one (31%) patients were on concomitant immunosuppressive treatment (thiopurines or methotrexate), and 41/130 (31%) were switched from the originator after a median time of 15 months (IQR 11.0–25.5) on the ADA originator. Among those who switched, 33 (80%) were in clinical remission, and 16 (39%) were in luminal remission. Seventeen (13%) patients had previously received infliximab (IFX), with a median IFX treatment duration of 12 months (IQR 5.0–22.0); reasons for discontinuation included primary nonresponse (n=5), secondary loss of response (n=9) (with four cases related to immunogenicity), allergy (n=1) and compliance issues (n=3) such as venous access difficulties, refusal, or travel-related concerns. Baseline characteristics are summarized in Tables 1 and previous treatments are reported in Supplementary Table 1.
The prescribed biosimilars were ABP 501 (Amgevita®) (n=110, 84%), GP2017 (Hyrimoz®, Sandoz International GmbH, Holzkirchen, Germany) (n=18, 14%) and SB5 (Imraldi®, Biogen International GmbH, Baar, Switzerland) (n=2, 1%).
 |
Table 1 Baseline Characteristics of the Study Population (n=130) |
ADA Biosimilars Durability
After a median follow-up of 26 months (IQR 12.0–50.0, maximum follow-up 72 months), 87/130 (67%) patients remained on ADA biosimilars. The probability of durability was 93% at 6 months, 86% at 12 months, 75% at 24 months, 62% at 36 months, and 57% at 54 months (Figure 1).
 |
Figure 1 Treatment persistence with Adalimumab (ADA) biosimilar estimated according to the Kaplan-Meier method. [The y-axis has been scaled to the range 0.4–1 for better visualization.]. |
Forty-three (33%) patients discontinued ADA biosimilars at a median time of 14 months (IQR 7.5–27.0). Reasons for discontinuation included primary non-response (n=6, 14%), secondary loss of response (n=16, 37%), AEs (n=14, 33%), IBD-related surgery (n=6, 14%), and severe needle phobia (n=1, 2%), which was unresponsive to intensive Play Therapy input. Among the 14 patients who discontinued ADA biosimilar due to AEs, 6 switched to the originator due to injection site reactions and/or delivery difficulties.
Using Cox regression univariate analysis adjusted for age at diagnosis, patients previously exposed to ADA originator demonstrated a lower risk of ADA biosimilar failure (adjusted HR (aHR) 0.51 [95% confidence interval (CI) 0.27–0.99], p=0.047). A similar trend was observed in those who switched from the originator, showing a likelihood predictive factor for lower failure risk (aHR 0.57 [0.30–1.10], p=0.098). No other factors, including PIBD subtype (aHR 0.72 [95% CI 0.26–2.04], p=0. 54, for CD compared to UC/IBDU), perianal disease (aHR 1.21 [95% CI 0.61–2.40], p=0.58), extraintestinal manifestations (aHR 0.81 [95% CI 0.42–1.58], p=0.54), previous exposure to IFX (aHR 1.40 [95% CI 0.63–3.13], p=0.41), and combination therapy (aHR 1.21 [95% CI 0.63–2.30], p=0.56), were identified as predictive factors for ADA biosimilar failure (Table 2).
 |
Table 2 Predictors to Adalimumab Biosimilar Failure |
Effectiveness of ADA Biosimilars
At 6 months, 92/130 (71%) patients were in clinical remission, compared to 51 (39%) patients at baseline. The clinical remission rate showed further improvement, reaching 72% (79/111 patients) at 12 months (Figure 2). Luminal remission rates also improved, increasing from 17% (22/130 patients) at baseline to 35% at both 6 months (46/130 patients) and 12 months (39/111 patients). Among the 28 patients with perianal disease, the remission rate (both clinical and radiological) remained relatively stable, with 19/28 (68%) patients in remission at 6 months, compared to 17 (61%) patients at baseline.
 |
Figure 2 Clinical remission rates during follow-up. |
During the follow-up period, patients showed statistically significant improvements in several key parameters from baseline to 6 and 36 months. Specifically, CRP levels decreased from 3.4 mg/L [1.0–18.8] at baseline to 1.0 mg/L [1.0–2.0] at 6 months and 1.0 mg/L [1.0–3.0] at 36 months (p<0.001 and p=0.004, respectively) (see Supplementary Table 2). Notably, the median FC level decreased significantly from 789 μg/g [IQR 246–1161] at baseline to 129 μg/g [IQR 38–398] at 6 months, and 76 μg/g [IQR 35–392] at 36 months (p<0.001 for both comparisons). The MINI index dropped from 12.0 [IQR 6.0–16.0] at baseline to 5.0 [IQR −2.0–8.0] at 6 months, and to 3.0 [IQR 0.0–7.7] at 36 months (p<0.001 for both) (Figure 3).
 |
Figure 3 Boxplot of clinical scores and laboratory data during follow-up (baseline, 6 months, 12 months, 24 months and 36 months). [Bold denotes significant p-value]. |
Sixty-seven (51%) patients required dose escalation at a median time of 7.5 months [IQR 3.0–21.7]. Of these, 31 (46%) patients required escalation due to low trough levels, 16 (24%) had disease relapse despite therapeutic drug concentrations, and 20 (30%) with persistently high FC levels despite clinical remission and therapeutic drug levels. Dose escalation led to an improved response in 41 (61.2%) patients overall. Twenty-two out of 31 (71%) patients who escalated due to low trough levels achieved therapeutic concentrations, 11/16 (69%) patients who escalated due to disease relapse regained clinical remission, and 8/20 (40%) patients who escalated due to persistently high FC levels were able to normalise their FC (Figure 4). However, 6/67 (9%) patients did not respond to dose escalation and subsequently discontinued ADA treatment, while 14/67 (21%) were able to successfully de-escalate after a median time of 7.0 months (IQR 4.2–11.7).
 |
Figure 4 Success rate of Adalimumab (ADA) dose escalation in all cohorts and in different groups based on the reason for escalation: low trough levels, disease relapse, and high faecal calprotectin (FC) levels. |
No statistically significant differences were observed between patients who required dose escalation and those who did not, in terms of body weight (52.4 kg [41.1–64.0] versus 53.0 kg [44.0–63,9], respectively; p=0.46), ADA dosage on a 2-weekly basis (0.76 mg/kg [0.61–0.95] versus 0.75 mg/kg [0.63–0.90], respectively; p=0.92), or concomitant immunosuppression (18/67, 27% versus 20/63, 22%, respectively; p=0.54). Unsurprisingly, ADA trough levels were significantly lower in the group requiring dose escalation compared to the non-escalation group (7.8 μg/mL [6.0–11.2], measured before dose escalation, versus 12.0 μg/mL [11.6–15.1], measured at 6 months, respectively; p<0.001).
Therapeutic Drug Monitoring and ADA Biosimilar Immunogenicity
Therapeutic drug monitoring (TDM) was conducted for all patients in the study population. Patients who experienced ADA biosimilar failure during follow-up had lower drug concentrations at 6 months compared to those who continued treatment (10.7 μg/mL [7.6–12.0] vs 12.0 μg/mL [10.4–15.9], p=0.007). Using ROC curve analysis, a cut-off of 11.6 μg/mL at 6 months was identified to differentiate patients who experienced ADA biosimilar failure (AUC 0.683, p=0.007) (Figure 5). Patients with trough levels equal to or greater than 11.6 μg/mL at 6 months had a higher probability of ADA biosimilar failure-free survival (HR 0.23 [95% CI 0.10–0.52], p<0.001) [n=88 (68%) with available levels at 6 months] (Figure 6). Importantly, these higher trough levels were not associated with an increased incidence of AEs [15 AEs (17%) in patients with trough levels <11.6 μg/mL at 6 months versus 14 (16%) in those with trough levels ≥11.6 μg/mL, p=0.15]. The same analysis was performed for post-induction trough levels measured at a median time of 7.1 weeks [IQR 5.7–10.6], and no significant cut-off was identified.
 |
Figure 5 ROC curve of Adalimumab (ADA) trough levels at 6 months in predicting ADA biosimilars failure during follow-up. |
 |
Figure 6 Kaplan-Meier curves representing different rate of Adalimumab ADA biosimilar failure in patients with ADA trough levels <11.6 μg/mL at 6 months (blue) and those with trough levels ≥ 11.6 μg/mL (red). The assumption of proportional hazard was used for analysis, and no violations were observed. |
Seven (5%) patients developed anti-ADA antibodies during follow-up. Among these, 3 patients discontinued ADA, 2 eliminated antibodies through ADA dose escalation, and in the remaining 2, the antibodies spontaneously disappeared without any intervention (see Supplementary Table 3).
ADA Biosimilar Safety
During the follow-up period, 46 (36%) patients experienced AEs, most commonly anti-TNF-α-induced psoriasis and injection site reaction (each occurring in 13% of patients) (Table 3). ADA biosimilars were discontinued due to AEs in 14 patients: 6 patients switched to the originator due to severe injection site pain, 3 discontinued due to severe psoriasis unresponsive to topical steroids, 1 stopped due to a severe infection (intracranial abscess and osteomyelitis, requiring hospitalisation and intravenous antibiotics), 1 due to alopecia, 1 due to Hodgkin Lymphoma, and 1 due to a suspected allergic reaction.
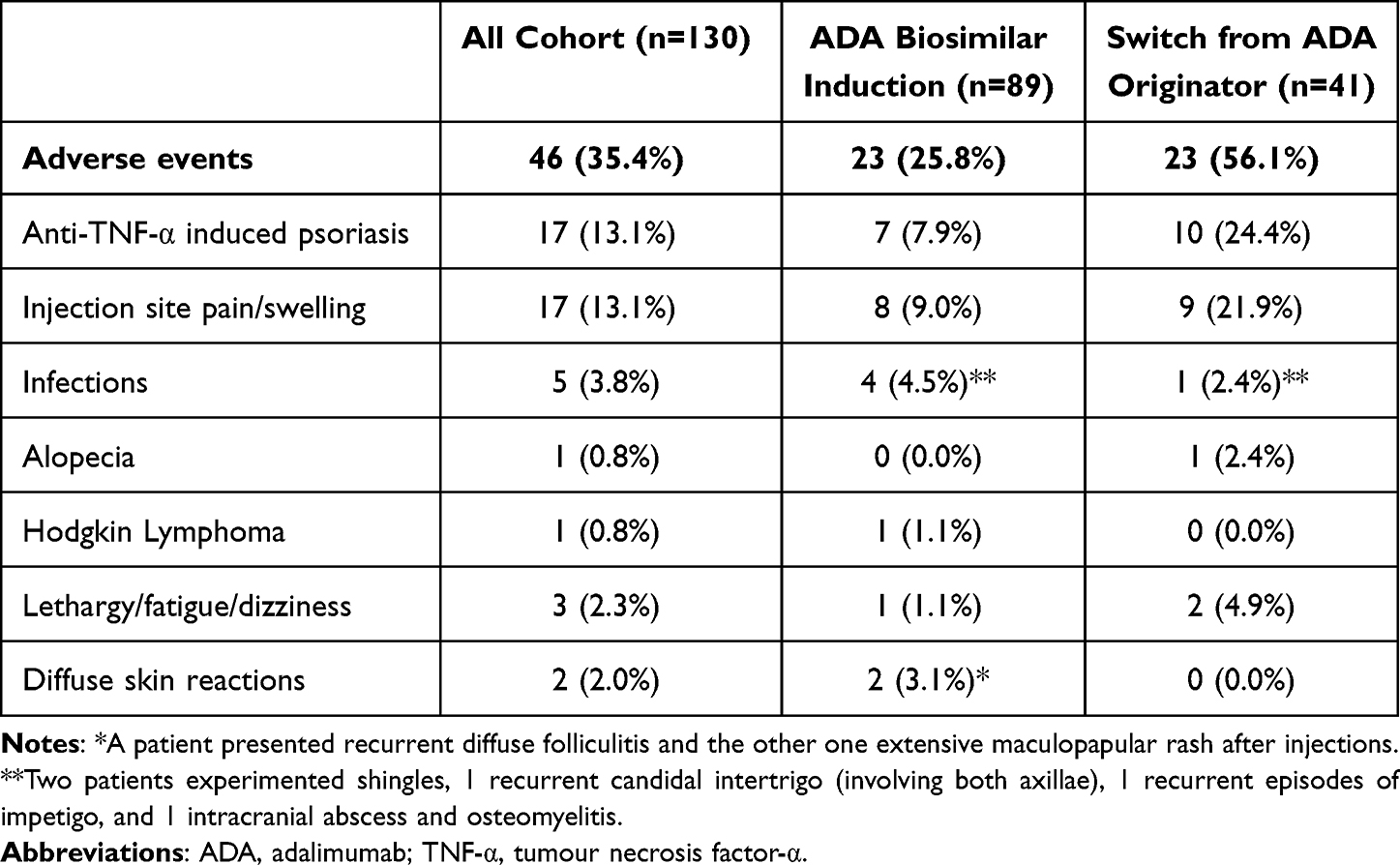 |
Table 3 Adverse Events During Follow-up |
Notably, one male patient developed Hodgkin Lymphoma 32 months after starting ADA biosimilar treatment, with the last 18 months on combination therapy with azathioprine. This patient also had a primary Epstein-Barr (EBV) infection prior to the lymphoma diagnosis. He underwent standard chemotherapy regimen and achieved complete remission of his lymphoma to date, remaining in remission of IBD off treatment for 2 years.
Among the patients who experienced injection site pain, 16/17 (94%) were on ABP 501 with 1 (6%) on GP2017. Additionally, 14 (82%) patients were using pre-filled pens, 1 (6%) was using syringes, and 2 (12%) reported pain with both devices. During the follow-up, 3 patients switched devices from pen to syringe, and 6 switched to the originator, successfully resolving the issue in both causes. Injection site pain was significantly more frequent among patients who switched from the originator (9/17, 53%) compared to those who started on ADA biosimilar (7/17, 41%) (p=0.04) (see Supplementary Table 4).
Outcomes in the Two Different Cohorts
No significant differences in ADA biosimilar durability were observed between the Scottish and Italian cohorts (HR 1.55 [95% CI 0.81–2.99], p=0.20, see Supplementary Figure 1), or in the secondary outcomes (see Supplementary Table 5). Notably, the median ADA trough levels at 6 months were similar among the two cohorts: 12.0 μg/mL [10.8–13.1] in the Scottish cohort compared to 10.2 μg/mL [7.6–14.0] in the Italian cohort (p=0.11).
Cost-Effectiveness Analysis
The estimated cost savings from using ADA biosimilars instead of the originator amounted to approximately €5,030 (£4,238) per patient per year, or €419 (£353) per patient per month. Without the use of ADA biosimilars, a minimal potential savings of €1,579,067 (£1,330,519) would have been missed.
Discussion
This study represents the first real-world assessment of the long-term durability of ADA biosimilars in PIBD.
In our cohort, 67% of patients remained on ADA biosimilar throughout the follow-up period (median follow-up of 26 months), a rate similar to that reported for ADA originator (68% of paediatric CD patients remaining on ADA after a median follow-up of 24 months).23 The treatment persistence rate is comparable to previously reported rates for ADA biosimilars: 86% at 12 months and 75% at 24 months in our study versus 85% for both the follow-ups in the Sicilian cohort.16 Dipasquale et al assessed the effectiveness and safety of ADA biosimilars in PIBD patients for the first time, however, their study was limited to 41 patients, with a short median follow-up of 11 months. The biosimilar used were ABP 501 and GP2017, 9 patients switched from the originator and no TDM data were available.16 Notably, the larger sample size and extended follow-up period of our study allowed for a novel evaluation of the longer-term durability of ADA biosimilars, which was 62% at 36 months and 57% at 54 months. Our treatment persistence rates are also similar to those found in studies on adult IBD patients; Macaluso et al reported an overall treatment persistence equal to 93% at 6 months and 86% at 12 months for ABP 501.15
Furthermore, we observed favourable clinical and biochemical remission rates amongst patients using ADA biosimilars, with statistically significant improvements in several parameters. Remarkably, there was a substantial reduction in FC levels and MINI index scores from baseline to 6 and 36 months, indicating a meaningful clinical impact of ADA biosimilars on disease activity. The MINI index can accurately assess mucosal inflammation in PIBD studies,22 especially useful when endoscopic data are lacking. At 12 months, the clinical remission rate was 71%, which is very similar to the 72% reported in the Sicilian study at the same follow-up.16
Patients previously exposed to ADA originator had a lower risk of ADA biosimilar failure, likely reflecting a more stable disease course in those who had already responded favourably to ADA. Similarly, switching from the originator appeared to have a protective role against biosimilar failure. Therefore, it is plausible that a previous exposure to ADA, rather than the switch itself, may play a more significant role in predicting greater durability.
Additionally, higher drug concentrations at 6 months were associated with greater durability, and a cut-off was identified at 11.6 µg/mL. This level exceeds the suggested levels in current ECCO-ESPGHAN guidelines24 and previous literature,25–27 but aligns with trends observed in more recent studies, such as the PANTS study, which found optimal drug concentration thresholds when predicting long-term remission to be 10.1–12.0 mg/L at week 14 and >10 mg/L at week 54.28 Similarly, Lucafò et al identified cut-off points of 7.5 and 10.5 µg/mL at week 22 as predictive of sustained response at week 52 and 82, respectively.29 Rinawi et al also observed that ADA trough levels >10 µg/mL during maintenance were associated with greater likelihoods of achieving biochemical, endoscopic and transmural remission in children with CD on ADA originator.23 These findings highlight the importance of TDM and higher doses of anti-TNF-α, especially in the paediatric population. Proactive monitoring of drug concentrations facilitates personalised treatment optimisation, enabling dose escalation or shorter intervals between injections, as needed. In our cohort, half of the patients required dose escalation during the follow-up period, with a success rate of 61% in the original reasons for escalation (low trough levels, clinical relapse, or persistently high FC levels) indicating this is a valuable clinical strategy before moving away from ADA.
AEs were reported in 36% of patients during follow-up. Injection site pain was significantly more common among patients who switched from the originator, likely due to their prior experience with different delivery devices. Similar findings have been reported in studies with adult IBD patients, where injection site pain was the most frequent cause of ADA biosimilars discontinuation after switching; previously, most commonly related to the presence of citrate in biosimilar formulations.10,12,30 However, in our cohort, all but one (94%) of the patients reporting injection site pain were receiving ABP 501, a citrate-free biosimilar. This suggests that the pain may be related to the device or technique rather than the formulation itself; indeed, factors such as delivery volume, type and needle gauge size, may influence the extent and the intensity of injection site pain.31,32 In our cohort, the majority of patients who experienced pain at injection were using a pre-filled pen device (82%) and the issue with injection pain was resolved in a significant proportion of patients by switching to the originator or to a different delivery device of the same biosimilar (pre-filled syringe instead of pre-filled pen). The greater tolerance of syringes could be related to the better control they provide over the speed and duration of injection. Injection site pain should be considered in patients on ADA biosimilars, especially after switching, as it may interfere with patients’ adherence to treatment and consequently with disease activity control. Strategies to enhance adherence should be explored by the IBD team, focusing on patients-centred selection of delivery devices, needle sizes (both diameter and length), and anatomic injection site. Additionally, patients and their carers should receive proper training on injection techniques, including the correct insertion angle and ensuring that the medication is at the appropriate temperature (kept at room temperature for approximately 30 minutes before administration).32 In refractory cases, the use of topical anesthetics could be considered. Ongoing advancements in drug delivery systems could further address these challenges,33 including the development of non-injectable subcutaneous delivery methods and novel technologies.32
Along with injection site pain, the other most common AEs in our cohort were anti-TNF induced psoriasis and psoriasiform eczema, with a prevalence of 13%, consistent with rates reported in literature.34–36 It is important to report that one male patient in our cohort developed Hodgkin Lymphoma while on combination therapy. It is well established that both PIBD and its treatments are associated with a higher risk of malignancy, including lymphoma. According to current literature, PIBD patients have a 2- to 3-fold increased cancer risk when compared to non-IBD children,37–40 independent of treatment, with a 2.7-fold increased risk for lymphoid neoplasms.39 The risk of lymphoma has been associated with thiopurine use,40–42 however data on lymphoma risk related to anti-TNF-α monotherapy remain discordant.37,38,40,43–46 Interestingly, a French population-based cohort found that lymphoma risk was higher in patients on combination therapy compared to those on thiopurine monotherapy (HR 2.35 [1.31–4.22]) or anti-TNF-α monotherapy (HR 2.35 [1.35–4.77]), both of which were also associated with an increased in lymphoma risk compared to patients unexposed to these treatments.43 Conversely, the PYRAMID registry analysed the overall safety profile of ADA in CD patients over a follow-up period of up to 6 years and did not show an increased lymphoma rate.47
In our reported case, it is difficult to determinate whether ADA, thiopurines, primary EBV infection or whether these in combination contributed most to the development of lymphoma.
Costs were more than halved with the adoption of biosimilars. This significant cost saving would have an important impact on the healthcare system, permitting earlier and wider access to biological therapy and facilitating the efficient allocation of limited financial resources.48
This study has several limitations including being a retrospective study. Due to this, few patients underwent endoscopic and radiologic reassessment, and this missing data made it impossible assessing the deep remission in a “conventional” sense. However, we attempted to estimate MH using the MINI index, which provided encouraging results. Additionally, PUCAI and PCDAI scores were retrospectively calculated when missing, but to minimise potential biases, this was done only when all required items were clearly documented in the medical records. The absence of a control group on ADA originator represents another limitation of this study, alongside with the presence of other potential confounders. These were partially mitigated through the inclusion of consecutive patients from two different European cohorts. Finally, we acknowledge that ADA trough levels were measured in the two cohorts using two different assays with different working ranges; despite this, the median drug concentrations were similar in both groups.
Conclusion
This is the first real-world study addressing the long-term durability of ADA biosimilars in PIBD patients followed in two different European PIBD centres. The high probability of durability and good rates of clinical and luminal remission, along with significant cost savings, confirm the cost-effectiveness of ADA biosimilars in children, as previously demonstrated for IFX biosimilars in PIBD.49–51
Abbreviations:
ADA, adalimumab; AEs, adverse events; aHR, adjusted hazard ratio; CD, Crohn’s Disease; CI, confidence interval; CRP, C-reactive protein; EBV, Epstein-Barr virus; ESR, erythrocyte sedimentation rate; FC, faecal calprotectin; Hb, haemoglobin; HR, hazard ratio; IBDU, Inflammatory Bowel Disease unclassified; IFX, infliximab; IQR, interquartile rangeMH – mucosal healing; MINI, mucosal inflammation non-invasive; PCDAI, paediatric Crohn’s Disease activity; PIBD, paediatric Inflammatory Bowel Disease; PUCAI, paediatric Ulcerative Colitis Activity Index; TNF-α, Tumour Necrosis Factor-α; UC, Ulcerative Colitis.
Data Sharing Statement
The data underlying this study are available in the paper and in its online supplementary material. The data will be shared on reasonable request to the corresponding author.
Ethics Statement
In line with local protocols, and as a non-interventional study, formal ethics approval was not required for the Edinburgh cohort, as this was deemed a review of service delivery and clinical practice. Specifically, the Health Research Authority (HRA) does not require a Research Ethics Committee review for clinical audits, which involve interventions already in use to inform the delivery of best care (further details are available at https://hra-decisiontools.org.uk/research/).
For the Genoa cohort, patients were enrolled in the SIGENP (Italian Society of Pediatric Gastroenterology Hepatology and Nutrition) IBD registry, established in January 2009. Informed consent was obtained from all patients and their parents, following approval by the regional Ethics Committee (Comitato Etico Regionale Liguria).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding for this study.
Disclosure
SA fellowship is supported by University of Genoa. RKR has received speaker’s fees, travel support, or has performed consultancy work with: Nestle Health Sciences, AbbVie, Pharmacosmos, Lilly, Celltrion Healthcare, Ferring, Janssen & Pfizer. The remaining authors report no conflicts of interest in this work.
References
1. Burgess CJ, Jackson R, Chalmers I, et al. The inexorable increase of biologic exposure in paediatric inflammatory bowel disease: a Scottish, population-based, longitudinal study. Aliment Pharmacol Ther. 2022;56(10):1453–1459. doi:10.1111/apt.17217
2. Hyams JS, Griffiths A, Markowitz J, et al. Safety and efficacy of adalimumab for moderate to severe Crohn’s disease in children. Gastroenterology. 2012;143(2):365–374.e2. doi:10.1053/j.gastro.2012.04.046
3. Leonard E, Wascovich M, Oskouei S, Gurz P, Carpenter D. Factors affecting health care provider knowledge and acceptance of biosimilar medicines: a systematic review. J Manag Care Spec Pharm. 2019;25(1):102–112. doi:10.18553/jmcp.2019.25.1.102
4. Wang F, Li X, Shi Y, et al. Efficacy and safety of Adalimumab biosimilar (HS016) in inflammatory bowel disease from the real-world study. Front Pharmacol. 2023:14. doi:10.3389/fphar.2023.1259183
5. Kamat N, Kedia S, Ghoshal UC, et al. Effectiveness and safety of Adalimumab biosimilar in inflammatory bowel disease: a multicenter study. Indian J Gastroenterol. 2019;38(1):44–54. doi:10.1007/s12664-018-0922-1
6. Ribaldone DG, Caviglia GP, Pellicano R, et al. Effectiveness and safety of Adalimumab biosimilar ABP 501 in Crohn’s disease: an observational study. Rev esp enferm dig. 2020:112. doi:10.17235/reed.2020.6693/2019
7. Lukas M, Malickova K, Kolar M, et al. Switching from originator adalimumab to the biosimilar Sb5 in patients with inflammatory bowel disease: short-term experience from a single tertiary clinical centre. J Crohns Colitis. 2020;14(7):915–919. doi:10.1093/ecco-jcc/jjaa001
8. Tursi A, Mocci G, Allegretta L, et al. Comparison of performances of adalimumab biosimilars SB5, ABP501, GP2017, and MSB11022 in treating patients with inflammatory bowel diseases: a real-life, multicenter, observational study. Inflamm Bowel Dis. 2023;29(3):376–383. doi:10.1093/ibd/izac092
9. Hanauer S, Liedert B, Balser S, Brockstedt E, Moschetti V, Schreiber S. Safety and efficacy of BI 695501 versus Adalimumab reference product in patients with advanced Crohn’s disease (VOLTAIRE-CD): a multicentre, randomised, double-blind, Phase 3 trial. Lancet Gastroenterol Hepatol. 2021;6(10):816–825. doi:10.1016/S2468-1253(21)00252-1
10. Derikx LAAP, Dolby HW, Plevris N, et al. Effectiveness and safety of adalimumab biosimilar sb5 in inflammatory bowel disease: outcomes in originator to SB5 switch, double biosimilar switch and bio-naïve SB5 observational cohorts. J Crohns Colitis. 2021;15(12):2011–2021. doi:10.1093/ecco-jcc/jjab100
11. Deprez N, De Somer T, Baert D, et al. Evaluation of the safety and effectiveness after switch from Adalimumab originator to biosimilar SB5 in patients with inflammatory bowel disease in a real-life setting. Acta Gastro Enterologica Belgica. 2022;85(4):557–564. doi:10.51821/85.4.10724
12. Tapete G, Bertani L, Pieraccini A, et al. Effectiveness and safety of nonmedical switch from adalimumab originator to sb5 biosimilar in patients with inflammatory bowel diseases: twelve-month follow-up from the TABLET Registry. Inflamm Bowel Dis. 2022;28(1):62–69. doi:10.1093/ibd/izab027
13. Cingolani L, Barberio B, Zingone F, et al. Adalimumab biosimilars, ABP501 and SB5, are equally effective and safe as adalimumab originator. Sci Rep. 2021;11(1):10368. doi:10.1038/s41598-021-89790-4
14. Casanova MJ, Nantes Ó, Varela P, et al. Real‐world outcomes of switching from Adalimumab originator to adalimumab biosimilar in patients with inflammatory bowel disease: the ADA-SWITCH study. Aliment Pharmacol Ther. 2023;58(1):60–70. doi:10.1111/apt.17525
15. Macaluso FS, Cappello M, Busacca A, et al. SPOSAB ABP 501: a Sicilian prospective observational study of patients with inflammatory bowel disease treated with adalimumab biosimilar ABP 501. J Gastroenterol Hepatol. 2021;36(11):3041–3049. doi:10.1111/jgh.15590
16. Dipasquale V, Pellegrino S, Ventimiglia M, et al. Adalimumab biosimilar in pediatric inflammatory bowel disease: a retrospective study from the sicilian network for inflammatory bowel disease (SN-IBD). Healthcare. 2024;12(3). doi:10.3390/healthcare12030404
17. Atia O, Friss C, Focht G, et al. Durability of adalimumab and infliximab in children with Crohn’s disease: a nationwide comparison from the epi-IIRN cohort. Inflamm Bowel Dis.2024;30:2097–104 doi:10.1093/ibd/izad301
18. Levine A, Koletzko S, Turner D, et al. ESPGHAN revised Porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J Pediatr Gastroenterol Nutr. 2014;58(6):795–806. doi:10.1097/MPG.0000000000000239
19. Levine A, Griffiths A, Markowitz J, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis. 2011;17(6):1314–1321. doi:10.1002/ibd.21493
20. Hyams JS, Ferry GD, Mandel FS, et al. Development and validation of a pediatric Crohn’s disease activity index. J Pediatr Gastroenterol Nutr. 1991;12(4):439–447.
21. Turner D, Hyams J, Markowitz J, et al. Appraisal of the pediatric ulcerative colitis activity index (PUCAI). Inflamm Bowel Dis. 2009;15(8):1218–1223. doi:10.1002/ibd.20867
22. Cozijnsen MA, Ben Shoham A, Kang B, et al. Development and validation of the mucosal inflammation noninvasive index for pediatric Crohn’s disease. Clin Gastroenterol Hepatol. 2020;18(1):133–140.e1. doi:10.1016/j.cgh.2019.04.012
23. Rinawi F, Popalis C, Tersigni C, et al. Long-term outcomes with adalimumab therapy in pediatric Crohn disease: associations with adalimumab exposure. J Pediatr Gastroenterol Nutr. 2022;74(3):389–395. doi:10.1097/MPG.0000000000003366
24. Van Rheenen PF, Aloi M, Assa A, et al. The medical management of paediatric Crohn’s disease: an ecco-espghan guideline update. J Crohns Colitis. 2021;15(2):171–194. doi:10.1093/ecco-jcc/jjaa161
25. Kim MJ, Kim E, Kang B, Choe YH. Therapeutic drug monitoring of adalimumab during long‐term follow‐up in paediatric patients with Crohn disease. J Pediatr Gastroenterol Nutr. 2021;72(6):870–876. doi:10.1097/MPG.0000000000003070
26. Mazor Y, Almog R, Kopylov U, et al. Adalimumab drug and antibody levels as predictors of clinical and laboratory response in patients with Crohn’s disease. Aliment Pharmacol Ther. 2014;40(6):620–628. doi:10.1111/apt.12869
27. Assa A, Matar M, Turner D, et al. Proactive monitoring of adalimumab trough concentration associated with increased clinical remission in children with Crohn’s disease compared with reactive monitoring. Gastroenterology. 2019;157(4):985–996.e2. doi:10.1053/j.gastro.2019.06.003
28. Chanchlani N, Lin S, Bewshea C, et al. Mechanisms and management of loss of response to anti-TNF therapy for patients with Crohn’s disease: 3-year data from the prospective, multicentre PANTS cohort study. Lancet Gastroenterol Hepatol. 2024;9(6):521–538. doi:10.1016/S2468-1253(24)00044-X
29. Lucafò M, Curci D, Bramuzzo M, et al. Serum adalimumab levels after induction are associated with long-term remission in children with inflammatory bowel disease. Front Pediatr. 2021:9. doi:10.3389/fped.2021.646671
30. Lukas M, Kolar M, Reissigova J, et al. A switch from originator-Adalimumab to the biosimilar SB5 in patients with Crohn’s disease: an analysis of two propensity score-matched cohorts. Scand J Gastroenterol. 2022;57(7):814–824. doi:10.1080/00365521.2022.2041082
31. Allegretti JR, Brady JH, Wicker A, Latymer M, Wells A. Relevance of adalimumab product attributes to patient experience in the biosimilar era: a narrative review. Adv Ther. 2024;41(5):1775–1794. doi:10.1007/s12325-024-02818-9
32. Usach I, Martinez R, Festini T, Peris JE. Subcutaneous Injection of Drugs: Literature Review of Factors Influencing Pain Sensation at the Injection Site. Adv Ther. 2019;36(11):2986–2996. doi:10.1007/s12325-019-01101-6
33. Lei P, Yu H, Ma J, et al. Cell membrane nanomaterials composed of phospholipids and glycoproteins for drug delivery in inflammatory bowel disease: a review ☆. Int J Biol Macromol. 2023;249:126000. doi:10.1016/j.cell.2022.11.029I
34. Fréling E, Baumann C, Cuny JF, et al. cumulative incidence of, risk factors for, and outcome of dermatological complications of Anti-TNF therapy in inflammatory bowel disease: a 14-Year Experience. Am J Gastroenterol. 2015;110(8):1186–1196. doi:10.1038/ajg.2015.205
35. Pugliese D, Guidi L, Ferraro PM, et al. Paradoxical psoriasis in a large cohort of patients with inflammatory bowel disease receiving treatment with anti‐TNF alpha: 5‐year follow‐up study. Aliment Pharmacol Ther. 2015;42(7):880–888. doi:10.1111/apt.13352
36. Eickstaedt J, Paller AS, Lund E, et al. Paradoxical psoriasiform eruptions in children receiving tumor necrosis factor α inhibitors. JAMA Dermatol. 2023;159(6):637. doi:10.1001/jamadermatol.2023.0549
37. Atia O, Harel S, Ledderman N, et al. Risk of cancer in paediatric onset inflammatory bowel diseases: a nation-wide study from the epi-IIRN. J Crohns Colitis. 2022;16(5):786–795. doi:10.1093/ecco-jcc/jjab205
38. Kjærgaard VS, Jensen CB, Elmahdi R, Burisch J, Allin KH, Jess T. Cancer risk in pediatric-onset inflammatory bowel disease: a population-based Danish cohort study. Gastroenterology. 2020;159(4):1609–1611. doi:10.1053/j.gastro.2020.06.030
39. Olén O, Askling J, Sachs M, et al. Childhood onset inflammatory bowel disease and risk of cancer: a Swedish nationwide cohort study 1964-2014. BMJ. 2017;358. doi:10.1136/bmj.j3951
40. Ehrström A, Jansson S, Jørgensen MH, Wewer V, Malham M. The risk of cancer in pediatric-onset immune-mediated inflammatory diseases – a nationwide study. J Autoimmun. 2024;149:103321. doi:10.1016/j.jaut.2024.103321
41. Kotlyar DS, Lewis JD, Beaugerie L, et al. Risk of lymphoma in patients with inflammatory bowel disease treated with azathioprine and 6-mercaptopurine: a meta-analysis. Clin Gastroenterol Hepatol. 2015;13(5):847–858.e4. doi:10.1016/j.cgh.2014.05.015
42. Malham M, Jansson S, Malmborg P, et al. Risk factors of cancer in pediatric-onset inflammatory bowel disease in Denmark And Finland. J Pediatr Gastroenterol Nutr. 2023;77(1):55–61. doi:10.1097/MPG.0000000000003781
43. Lemaitre M, Kirchgesner J, Rudnichi A, et al. Association between use of thiopurines or tumor necrosis factor antagonists alone or in combination and risk of lymphoma in patients with inflammatory bowel disease. JAMA. 2017;318(17):1679. doi:10.1001/jama.2017.16071
44. Dulai PS, Thompson KD, Blunt HB, Dubinsky MC, Siegel CA. Risks of serious infection or lymphoma with anti-tumor necrosis factor therapy for pediatric inflammatory bowel disease: a systematic review. Clin Gastroenterol Hepatol. 2014;12(9):1443–1451;quize88–9. doi:10.1016/j.cgh.2014.01.021
45. Burmester GR, Panaccione R, Gordon KB, McIlraith MJ, Lacerda APM. Adalimumab: long-term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn’s disease. Ann Rheum Dis. 2013;72(4):517–524. doi:10.1136/annrheumdis-2011-201244
46. Zamani M, Alizadeh‐Tabari S, Murad MH, et al. Meta analysis: risk of lymphoma in patients with inflammatory bowel disease in population based cohort studies. Aliment Pharmacol Ther. 2024;60:1264–75doi:10.1111/apt.18277
47. D’Haens G, Reinisch W, Panaccione R, et al. Lymphoma risk and overall safety profile of adalimumab in patients with Crohn’s disease with up to 6 years of follow-up in the PYRAMID registry. Am J Gastroenterol. 2018;113(6):872–882. doi:10.1038/s41395-018-0098-4
48. Deiana S, Gabbani T, Annese V. Biosimilars in inflammatory bowel disease: a review of post-marketing experience. World J Gastroenterol. 2017;23(2):197. doi:10.3748/wjg.v23.i2.197
49. Richmond L, Curtis L, Garrick V, et al. Biosimilar infliximab use in paediatric IBD. Arch Dis Child. 2018;103(1):89–91. doi:10.1136/archdischild-2017-313404
50. Gervais L, McLean LL, Wilson ML, et al. Switching from originator to biosimilar infliximab in paediatric inflammatory bowel disease is feasible and uneventful. J Pediatr Gastroenterol Nutr. 2018;67(6):745–748. doi:10.1097/MPG.0000000000002091
51. Chanchlani N, Mortier K, Williams LJ, et al. Use of infliximab biosimilar versus originator in a pediatric United Kingdom inflammatory bowel disease induction cohort. J Pediatr Gastroenterol Nutr. 2018;67(4):513–519. doi:10.1097/MPG.0000000000002011
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.