Back to Journals » Drug Design, Development and Therapy » Volume 10
Acyl lipidation of a peptide: effects on activity and epidermal permeability in vitro
Authors Rocco D, Ross J, Murray P, Caccetta R
Received 12 January 2016
Accepted for publication 20 February 2016
Published 8 July 2016 Volume 2016:10 Pages 2203—2209
DOI https://doi.org/10.2147/DDDT.S104111
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Video abstract presented by Rima Caccetta.
Views: 201
Daniel Rocco,1,2 James Ross,1,2 Paul E Murray,1 Rima Caccetta1,2
1School of Pharmacy, 2Curtin Health Innovation Research Institute, Curtin University, Perth, WA, Australia
Abstract: Short-chain lipid conjugates can increase permeability of a small peptide across human epidermis; however, the emerging lipoaminoacid (LAA) conjugation technique is costly and can deliver mixed synthetic products of varied biological potential. LAA conjugation using a racemic mixture produces a mixture of D- and L-stereoisomers. Individual enantiomers can be produced at an extra cost. We investigated an affordable technique that produces only one synthetic product: short-chain (C7–C8) acyl lipidation. Acyl lipidation of Ala-Ala-Pro-Val, an inhibitor of human neutrophil elastase (HNE; believed to lead to abnormal tissue destruction and disease development), was investigated as an alternative to LAA conjugation. The current study aimed to assess the effects of acyl lipidation (either at the N-terminal or at the C-terminal) on neutrophil elastase activity in vitro and on transdermal delivery ex vivo. The inhibitory capacity of the acyl conjugates was compared to LAA conjugates (conjugated at the N-terminal) of the same peptide. The L-stereoisomer appears to rapidly degrade, but it represents a significantly (P<0.05) better inhibitor of HNE than the parent peptide (Ala-Ala-Pro-Val). Although the D-stereoisomer appears to permeate human epidermal skin sections in a better fashion than the L-stereoisomer, it is not a significantly better inhibitor of HNE than the parent peptide. Acyl lipidation (with a C7 lipid chain) at either end of the peptide substantially enhances the permeability of the peptide across human skin epidermis as well as significantly (P<0.005) increases its elastase inhibitory potential. Therefore, our current study indicates that acyl lipidation of a peptide is a more economical and effective alternative to LAA conjugation.
Keywords: topical delivery, fatty acid conjugation, skin permeability, transdermal delivery, elastase inhibition, lipopeptide
Introduction
Delivery of peptides transdermally is a particularly attractive option. Transdermal delivery of drugs avoids the first-pass metabolism and peptidases of the oral route (due to low levels of metabolic enzymes), is easily accessible, and allows for painless delivery of drugs. However, even small peptide molecules have difficulty penetrating the skin unaided due to their hydrophilic properties.1
The greatest barrier to transdermal drug delivery is the outermost layer of the epidermis, the stratum corneum.1 This highly ordered layer of skin is rich in lipids; hydrophilic molecules are therefore unable to effectively partition into it and penetrate the deeper regions of the skin. Much research is concentrated on skin permeation enhancement, and in recent years, lipidation of peptides, which increases the lipophilicity of such molecules, has emerged as a promising option.2–5
The tetrapeptide L-Ala-L-Ala-L-Pro-L-Val (L-AAPV; 356 Da) is an inhibitor of the enzyme human neutrophil elastase (HNE), which plays a part in host defense and cellular remodeling following injury.2 HNE levels are normally tightly controlled by its endogenous inhibitors, but in lesional skin of psoriasis, allergic contact dermatitis, and atopic dermatitis, HNE can be present in much greater amounts to that of normal.3,6 It is proposed that these excessive levels of HNE lead to the cellular damage and inflammation observed in these skin conditions. However, the hydrophilic nature of the unmodified peptide prevents delivery across the skin to the intended site of action.1
Previous research has indicated that the chemical modification of peptides through the conjugation of lipid groups improves the transdermal delivery characteristics of the molecule. Lipidated forms of Phe-Gly, tetragastrin, and L-AAPV have demonstrated improved transdermal delivery characteristics compared to the unmodified peptide.4,7,8
Enhancement of the transdermal delivery of L-AAPV was achieved through the attachment of lipoaminoacid (LAA) conjugates where it was shown that the lipidated form was able to cross samples of human epidermis more effectively than the unmodified form of the peptide.7 However, the permeability profiles were found to be different between the D and the L isomers, and generation of the single isomer is time-consuming and costly. Therefore, in our current work, we investigated simpler and less expensive conjugates of this peptide. Acyl lipid conjugation is threefold less expensive than synthesizing individual LAA D- or L-isomers. The current research work allowed for the comparison of acyl lipidation at either the N-terminal or the C-terminal to N-terminal LAA conjugation with regard to activity and transepidermal delivery in vitro.
Materials and methods
Materials
Customized peptides (Figure 1) L-AAPV, C7-L-Ala-L-Ala-L-Pro-L-Val (L-C7-AAPV), L-Ala-L-Ala-L-Pro-L-Val-C7 (L-AAPV-C7), and C8-L-Ala-L-Ala-L-Pro-L-Val (L-C8-AAPV) were purchased from GL Biochem (Shanghai, People’s Republic of China), and the C8LAA conjugates, L-octylglycine-L-Ala-L-Ala-L-Pro-L-Val (or L-C8LAA-AAPV) and D-octylglycine-L-Ala-L-Ala-L-Pro-L-Val (or D-C8LAA-AAPV), were purchased from NeoMPS (PolyPeptide Laboratories Group, Strasbourg, France). Propylene glycol (PG) was obtained from BDH Chemical Pty, Ltd. (Poole, UK). High-performance liquid chromatography (HPLC)-grade acetonitrile was used, and all other chemicals were of analytical grade. Phosphate-buffered saline (PBS) solution (137 mM NaCl, 2.7 mM KCl, 10.1 mM Na2HPO4, 18 mM KH2PO4) at pH 7.4, trifluoroacetic acid (TFA) and all other reagents including acetonitrile and octanol were purchased from Sigma-Aldrich (St Louis, MO, USA).
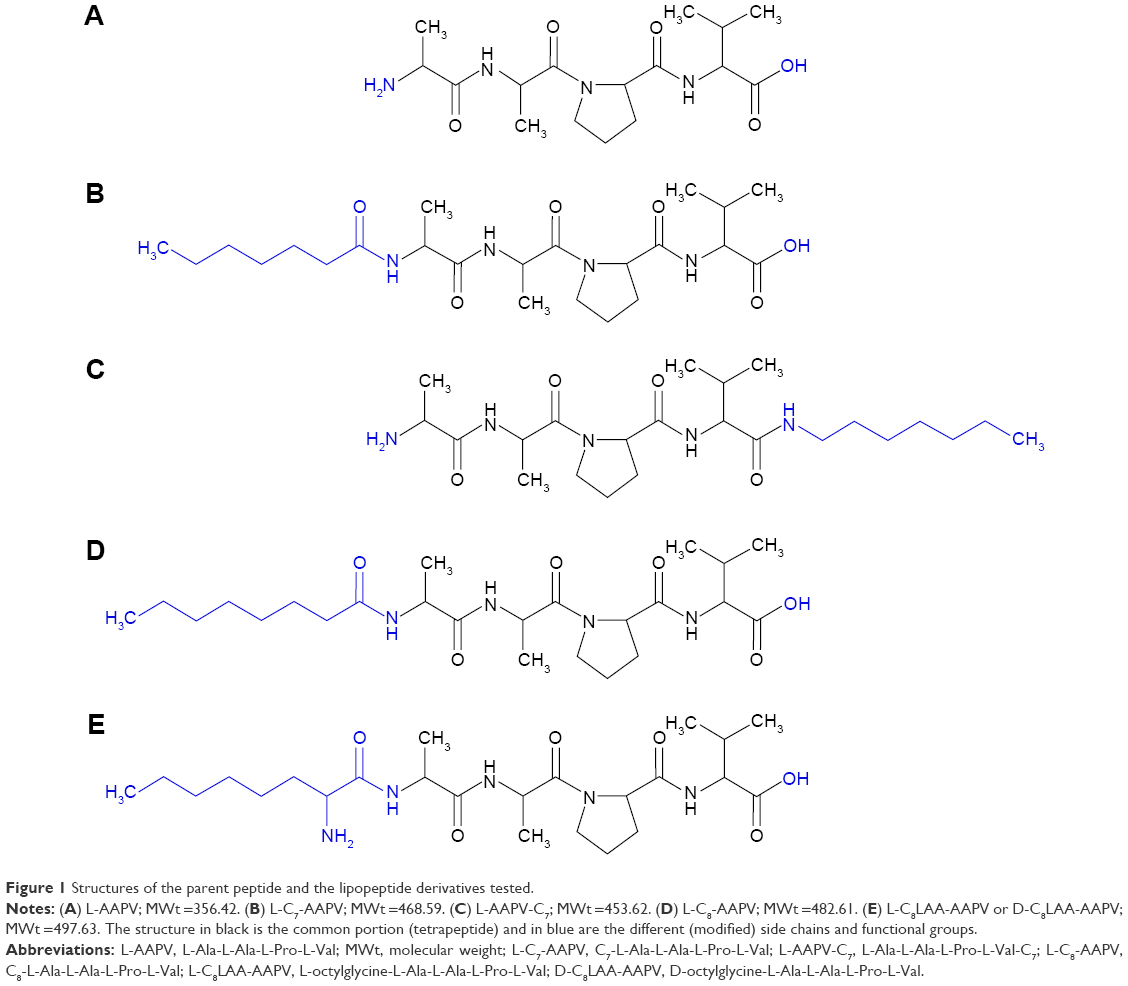 | Figure 1 Structures of the parent peptide and the lipopeptide derivatives tested. |
Determination of octanol–water partition coefficient
The partition coefficients of the parent tetrapeptide and lipopeptide derivatives were determined by using a shake-flask method adapted from the Organisation for Economic Co-operation and Development Guidelines.9 In separate vials, 2 mg of L-AAPV, L-C7-AAPV, or L-AAPV-C7 was disolved in a solution of 0.5 mL 1-octanol and 0.5 mL PBS. This procedure was performed for each peptide at three different pH levels: 4.5, 7.4, and 9 (PBS pH is 7.4; pH was adjusted to 4.5 by using HCl and 9 by using NaOH). All three peptides were completely soluble in the 1 mL volumes at each of the different pH levels. The solutions were then continually shaken for 3 hours and centrifuged to achieve phase separation. Each phase was then analyzed by the HPLC-UV method described later to determine the concentration of peptide in each phase. The concentrations in each phase obtained were used to determine the octanol–water partition coefficient (log Ko/w) using Equation 1.
|
|
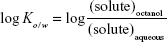
Computer estimates were obtained using ChemAxon® software.
Bioactivity assays
An EnzChek Elastase Assay Kit (E-12056) purchased from Thermo Fisher Scientific (Waltham, MA, USA) was used to carry out the in vitro inhibition assays. Fluorescence assays were carried out using a 2300 Series EnSpire Multimode Reader using the EnSpire Manager 4.10 software. The wavelength for excitation was set to 485 nm, and the wavelength for emission set to 530 nm.
The DQ elastin (bovine neck ligament elastin) stock solution (1 mg/mL), 1× reaction buffer (0.1 M Tris–HCl, pH 8.0), and porcine pancreatic elastase stock solution (100 U/mL) were prepared in accordance with the kit assay protocol. Porcine pancreatic elastase stock solution was diluted with 1× reaction buffer to give a final concentration of 0.02 U/mL of elastase working solution. The final concentration of peptides used was 1 mM. The concentration of elastase was experimentally chosen to give linear enzyme kinetics within the first 30 minutes of the reaction when using 25 μg/mL DQ elastin in each well. Fluorescence was measured after incubation at 24.3°C for 20 minutes. Inhibition percentages were relative to uninhibited reactions done in the same run. Each assessment was done in triplicate.
Permeation studies
Permeation studies were carried out using static Franz-type diffusion cells (effective diffusional area 1.1 cm2), utilizing skin samples obtained from five patients (four females and one male; mean age: 49 years old, ranging 37–61 years old) undergoing abdominoplasties in Perth Hospitals (Curtin Human Ethics Approval number PH-18-11). Written informed consent was obtained from all of the volunteers. Abdominal skin samples collected were immediately placed at 4°C. The subcutaneous fat was removed within 24 hours of surgery, and the full-thickness skin samples were stored at −20°C for no longer than 6 months. The epidermis was detached from the dermis by a standard heat separation method.10 Briefly, the epidermal layer was carefully stripped from whole-skin samples, following submersion in 60°C water for ~2 minutes, and then carefully peeling back the top epidermal layer. The epidermis was mounted to the Franz cell with the stratum corneum facing toward the donor compartment. The receptor compartment (containing a magnetic stirrer) was loaded with PBS (~3 mL at pH 7.4) and allowed to equilibrate to 35°C for 15 minutes before the donor compartment was loaded. Keeping the temperature of the receptor compartment at 35°C enabled the skin surface temperature to be kept at 32°C. Skin integrity was assessed by observing PBS movement from donor to receptor for 20 minutes before the start of experimentation. Any skin membranes suspected of having holes, before or during the experiment, by closely monitoring the donor and receptor volumes, were eliminated as they were detected.
It is important to note that there is variability between skin samples from the same individual as well as between individuals. Site differences including structural variability of skin (eg, lipid content) and previous skin care/treatment may contribute to this within the same patient as well as between patients.11 Thus, we assessed triplicate samples from five different individuals (all are abdominal sections) for each assay (ie, five separate experiments each using triplicate skin samples from a different donor each assessing the three different peptides individually within the same experiment: L-AAPV, L-C7-AAPV, and L-AAPV-C7).
The experiments began with the donor compartment loaded with 300 μL PG or 300 μL PG containing 3 mg of L-AAPV, L-C7-AAPV, or L-AAPV-C7, and sealed. The amounts were completely soluble in PG at these concentrations. Aliquots (200 μL) were withdrawn from the receptor compartment at 0, 0.5, 1, 2, 4, 8, and 24-hour time points and replaced with an equivalent amount of PBS pre-equilibrated at 35°C. The temperature of the receptor compartment was maintained at 35°C for the duration of the experiment, and this enabled a skin surface temperature of 32°C. Collected aliquots from each experiment were kept at 4°C in the injecting tray of the autosampler while awaiting analysis by HPLC-UV.
Transdermal flux was calculated as the cumulative amount in the receptor over the first 2 hours (the slope of the graphs in Figure 2) divided by the effective diffusional area of each of the skin samples.
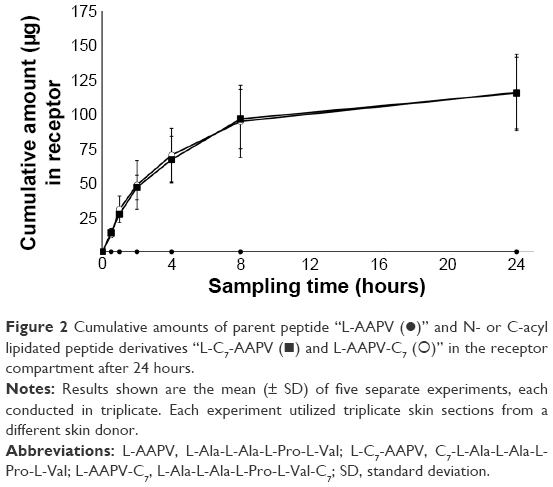 | Figure 2 Cumulative amounts of parent peptide “L-AAPV (•)” and N- or C-acyl lipidated peptide derivatives “L-C7-AAPV (◼) and L-AAPV-C7 (○)” in the receptor compartment after 24 hours. |
HPLC-UV analysis
Analysis was carried out using a previously established method1,7 with minor adjustment. Basically, the amounts of L-AAPV, L-C7-AAPV, and L-AAPV-C7 were assayed by reverse phase HPLC-UV (Agilent 1100 series) using a Phenomenex 5 μm C18 column (4.6×150 mm). Phase A was 0.1% (v/v) TFA in water, and phase B was 0.1% (v/v) TFA in acetonitrile. Elution was conducted with a flow rate of 1.0 mL/min with the column temperature held at 25°C, utilizing both isocratic and gradient elution. Elution began with 10% phase B for 5 minutes, with a linear increase from 10% to 100% phase B over 5 minutes, followed by 100% phase B holding for 2 minutes. Each sample was followed by a 5-minute wash period. Absorbance was measured at 205, 210, and 214 nm simultaneously. Calculation of analyte concentration in samples was performed with reference to external standards at 210 nm. The retention times were 3.42 minutes for the parent peptide, 10.34 minutes for the N-terminal conjugate, and 10.41 minutes for the C-terminal conjugate. The N-terminal conjugate had a limit of detection of 5 ng and a lower limit of quantitation of 15.3 ng. The C-terminal conjugate had a limit of detection of 4.5 ng and a limit of quantitation of 14.9 ng. Interday analysis of the lipopeptide standards across a concentration range of 25–50 μg/mL gave a coefficient of variation of 3.29% for the N-terminal conjugate and 3.68% for the C-terminal conjugate.
Statistical analysis
The bioactivity data were analyzed using IBM SPSS Statistics 22 software package (IBM Corporation, Armonk, NY, USA) using either analysis of variance or independent samples t-tests as appropriate. For all statistical tests, a P-value <0.05 was taken to indicate a statistically significant association.
Results
Permeability of L-AAPV versus its C7-acyl lipid conjugates
The effect of C7-acyl lipid conjugation on the N- or C-terminal (Figure 1B and C) of the parent peptide (L-AAPV; Figure 1A) on epidermal delivery was assessed in vitro. Unlike the LAA conjugates used previously,7 the HPLC-UV traces of the C7-acyl lipid conjugates of L-AAPV gave single peaks (L-C7-AAPV retention time =10.34 minutes; L-AAPV-C7 retention time =10.40 minutes) over the course of the experiment. The cumulative amount of L-AAPV and its lipidated conjugates (L-C7-AAPV or L-AAPV-C7) detected in the receptor compartment following diffusion across human epidermis is represented in Figure 2. After 24 hours, the parent peptide was not detected within the receptor, while both the N- and C-terminally conjugated lipopeptides were present in similar amounts. The permeation profile for both lipid conjugates was similar (Figure 2), and the mean cumulative amounts of the N- and C-terminal conjugates present in the receptor compartment after 24 hours (Table 1) were 3.85% and 3.87% of the applied dose, respectively, both showing a clear improvement over the parent peptide. The transdermal flux data for the lipopeptides and the parent peptide over the first hour following application to the epidermis are presented in Table 1.
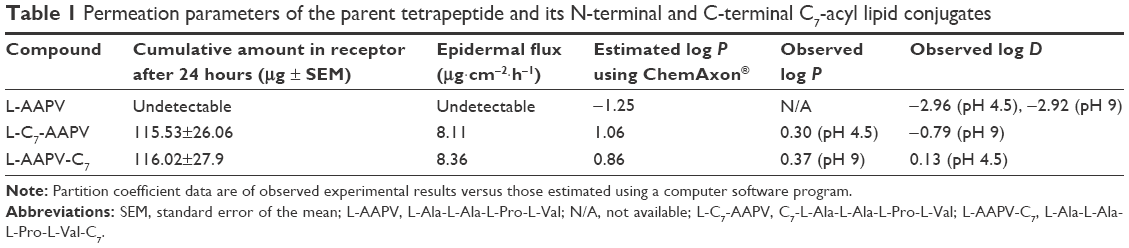 | Table 1 Permeation parameters of the parent tetrapeptide and its N-terminal and C-terminal C7-acyl lipid conjugates |
One milligram of any of the peptides (parent peptide and lipopeptides) was completely soluble in 1 mL of PBS. Concentrations up to 200 μg of peptides dissolved in 1 mL of PBS gave a linear profile when run on HPLC-UV. This amount is equivalent to 600 μg in the total receptor fluid (ie, approximately six times the maximum amounts measured at 24 hours), and thus one would expect that the experiments were performed at sufficient sink conditions.
Inhibition of porcine pancreatic elastase
The effectiveness of the different peptides, in Figure 1, to inhibit elastase was assessed. The levels of inhibition were expressed relative to uninhibited reactions run at the same time. The inhibitory activities of both the N- and C-terminal C7-acyl lipidated conjugates were significantly greater than the parent tetrapeptide (L-AAPV; Figure 3A). The inhibitory effect of L-AAPV on porcine pancreatic elastase was enhanced by the C7-acyl lipidation. The activity of C8LAA conjugates of L-AAPV (both D- and L-C8LAA-AAPV) was examined and compared to a C8-acyl derivative in vitro (Figure 3B). There was no statistically significant difference in the activity of the D-C8LAA-AAPV compared to that of the parent tetrapeptide (L-AAPV) and this had similar activity to a straight-chain C8 conjugate (L-C8-AAPV). The L-stereoisomer of the LAA conjugates (L-C8LAA-AAPV) showed significantly greater inhibition of porcine pancreatic elastase than the unmodified parent peptide.
 | Figure 3 Percent inhibition of elastase by L-AAPV, acyl conjugate (L-C8-AAPV), or lipoaminoacid (D- or L-C8LAA-AAPV) conjugates in vitro. |
Octanol–water partition coefficient of L-AAPV versus its C7-acyl lipid conjugates
L-AAPV displayed a partition coefficient value ranging from −2.92 to −2.96 over a range of pH of 4.5 and 9, demonstrating considerable hydrophilicity. Following lipidation at either the C-terminal or the N-terminal, the resulting lipopeptides displayed an increase in lipophilic nature of over 1,000-fold. Slight differences were observed in the lipophilicity of the two lipopeptides. The log P values of the C-terminal and N-terminal conjugates were 0.37 and 0.30, respectively, with their log D values being 0.13 and −0.79, respectively. This information is summarized in Table 1.
Discussion
Our current results indicate that direct coupling of C7-acyl lipid groups to either the N-terminal or the C-terminal of L-AAPV is a viable means to improve the transepidermal delivery of this potentially therapeutic tetrapeptide. This type of coupling is cheaper and less problematic than LAA conjugation. Lipidation at either end (N- or C-terminal) increases the bioactivity of this tetrapeptide; the lipopeptides examined increased the inhibitory activity of elastase compared to the parent tetrapeptide in vitro.
C7-acyl lipid conjugation at either terminal increased the amount of peptide able to cross the skin epidermis compared to that of the parent L-AAPV (Figure 2). The amount of C7-acyl lipopeptides detected in the receptor compartment 24 hours after application was 3.85% of the applied dose of the N-terminal conjugate, and 3.87% of the applied dose of the C-terminal conjugate, compared with (undetected) parent tetrapeptide. These levels of delivery can be attributed to the addition of a lipid chain to the parent peptide, which subsequently increased lipophilicity of the tetrapeptide (Table 1).
Our results are in agreement with previous studies on lipidated forms of L-AAPV.5,10 A study on the transepidermal delivery of the racemic mixture of LAA conjugates of L-AAPV by Caccetta et al showed that the D- and L-C8LAA conjugates of L-AAPV display increasing levels of delivery across human skin epidermis compared to the parent tetrapeptide L-AAPV.7 However, it was noted that the L-C8LAA-AAPV appeared to degrade over a 24-hour period of epidermal permeation with the gradual appearance, and subsequent increase, of unidentified peaks on HPLC-UV chromatograms of samples taken from the subepidermal compartment. The unidentified peaks were proposed to be degradation products of the L-stereoisomer in the diastereomeric mixture assayed. These observations were recently supported by a study investigating the epidermal permeability of the individual stereoisomers which further confirmed the stereoselective effects observed on permeation of the D-diastereomer compared to the L-diastereomer conjugate of L-AAPV.12
Unlike the LAA conjugates in previous studies,7,12 which generated racemic mixtures or are otherwise time-consuming and costly to generate the optimal stereoisomer, acyl conjugates give single products which are easier and cheaper to synthesize. Single peaks were observed on HPLC-UV chromatograms obtained using samples withdrawn from the receptor compartments.
Percentage inhibition of elastase was greater with lipidated forms of L-AAPV compared to the parent tetrapeptide. This improvement in elastase inhibition is seen with all types of lipidation of L-AAPV investigated in our current work and is in agreement with other literature.3 There is no difference between N- or C-conjugation as seen in Figure 3A, but on comparing the different lipid conjugates on the N-terminal in Figure 3B, it is clear that the L-conformer of the LAA conjugate (L-C8LAA-AAPV) has greater inhibitory activity than the straight-chain conjugate L-C8-AAPV. Although this difference is significant (P<0.05) in the inhibition bioassay, translation of its activity topically for skin conditions may be wasteful, since the L-conformer (L-C8LAA-AAPV) appears to degrade quite rapidly on epidermal permeation in vitro.7,12 Therefore, the L-stereoisomer may not be of practical therapeutic value especially with the added complications of its synthesis and cost.
While both the N-terminal (L-C7-AAPV) and the C-terminal (L-AAPV-C7) conjugates displayed increases in lipophilicity exceeding 1,000-fold, compared to the log D of the parent peptide (L-AAPV), the lipopeptides did not possess a log P value in the range between 1 and 3, which is ideal for delivery of drugs across the skin.5,13 Although the shift in log P did not reach the ideal range (log D of −2.96 for the parent to 0.30 and 0.37 for the N- and C-terminal conjugates, respectively), there was a marked enhancement in permeability with this type of lipidation which is similar to that previously reported for other lipidated conjugates of this tetrapeptide.7,12 The slight differences in log P values can be attributed to the exposed functional groups of each lipopeptide at the unacylated terminal. The exposed carboxylic acid of the N-terminal conjugate is more hydrophilic than the exposed amine of the C-terminal conjugate, resulting in a slightly lower log P value. However, at skin pH, the N-terminal conjugate is more suitable to therapeutic applications as it possesses a higher log P value at this pH compared with the C-terminal conjugate, which is ionized due to its exposed amine group, making it quite hydrophilic with a log D value of 0.13.
Our observed log P/log D values for the parent peptide are not in agreement with that recently reported by Namjoshi et al who reported a log P value of −1.272.12 In the publication, the log P was estimated using a computer program; unfortunately, the exact software used is not specified in their manuscript. Therefore, as well as determining the log P values using standard procedures in the test tube (observed values), we estimated the log P values for our peptides using a computer software program, ChemAxon® (Marvin 5.10, 2012; ChemAxon, Budapest, Hungary) for comparison (Table 1). It appears that although we could generate a similar estimated log P to that reported by Namjoshi et al using a computer software package, the observed values are very different (Table 1). It is clear from our other values that computer estimates are not in line with those observed and thus computer programs cannot be used as predictors, at this stage, for lipidated peptides unless they are further optimized for these types of molecules.
Effects of modest increases of chain length (eg, C10-acyl lipid conjugation) on measured lipophilicity and skin permeability in vitro would be of value. However, increasing the lipophilicity of the peptide may result in its transdermal delivery being impeded, as more of it may be retained within the stratum corneum,8 unable to partition into the less lipid regions of the skin. This phenomenon was observed by Yamamoto et al, where highly lipophilic derivatives of the dipeptide Phe-Gly were unable to penetrate full-thickness rat skin due to their retention within the stratum corneum.8
In summary, lipidating the parent peptide (L-AAPV) with the various conjugates examined did not reduce the elastase inhibitory activity of the peptide. In fact, in most cases, it significantly increased the inhibitory activity of the peptide. Although L-LAA conjugation may have significantly better inhibitory activity than the other conjugates, its levels appear to gradually decrease over time following epidermal permeation in vitro suggesting that this conjugate will be less available for bioactivity at its site of action. Acyl lipidation at either the N-terminal or the C-terminal significantly increased the inhibitory activity of the peptide against neutrophil elastase, and both equally enabled the tetrapeptide to permeate human epidermal skin sections. Acyl lipidation produces single products, which are less time-consuming and less costly to synthesize.
Acknowledgments
This work was supported by a Curtin Strategic Research Grant (to RC) and further funded by the School of Pharmacy at Curtin University which supported both DR and JR in the conduct of this work. E/Professor Bruce Sunderland is acknowledged for his helpful comments. The authors acknowledge the continued support and assistance of Mr Andrew Crocker, plastic and reconstructive surgeon operating in Perth Hospitals, in the acquisition of the skin samples and the generosity of the patient donors without whom this work would not have been possible.
Disclosure
The authors report no conflicts of interest in this work.
References
Benson H, Caccetta R, Chen Y, Kearns P, Toth I. Transdermal delivery of a tetrapeptide: evaluation of passive diffusion. Lett Pept Sci. 2003;10(5–6):615–620. | ||
Hornebeck W, Moczar E, Szecsi J, Robert L. Fatty acid peptide derivatives as model compounds to protect elastin against degradation by elastases. Biochem Pharmacol. 1985;34(18):3315–3321. | ||
Toth I, Christodoulou M, Bankowsky K, et al. Design of potent lipophilic-peptide inhibitors of human neutrophil elastase: in vitro and in vivo studies. Int J Pharm. 1995;125(1):117–122. | ||
Setoh K, Murakami M, Araki M, Fujita T, Yamamoto A, Muranishi S. Improvement of transdermal delivery of tetragastrin by lipophilic modification with fatty acids. J Pharm Pharmacol. 1995;47(10):808–811. | ||
Schuetz Y, Naik A, Guy R, Kalia Y. Emerging strategies for the transdermal delivery of peptide and protein drugs. Expert Opin Drug Deliv. 2005;2(3):533–548. | ||
Wiedow O, Wiese F, Christophers E. Lesional elastase activity in psoriasis: diagnostic and prognostic significance. Arch Dermatol Res. 1995;287(7):632–635. | ||
Caccetta R, Blanchfield J, Harrison J, Toth I, Benson H. Epidermal penetration of a therapeutic peptide by lipid conjugation; stereo-selective peptide availability of a topical diastereomeric lipopeptide. Int J Pept Res Ther. 2006;12(3):327–333. | ||
Yamamoto A, Setoh K, Murakami M, et al. Enhanced transdermal delivery of phenylalanyl-glycine by chemical modification with various fatty acids. Int J Pharm. 2003;250(1):119–128. | ||
OECD. Test No. 107: Partition Coefficient (n-Octanol/Water): Shake Flask Method. Paris, France: OECD Publishing; 1995. | ||
Kligman AM, Christophers E. Preparation of isolated sheets of human stratum corneum. Arch Dermatol. 1963;88:702–705. | ||
Namjoshi S, Caccetta R, Benson H. Skin peptides: biological activity and therapeutic opportunities. J Pharm Sci. 2007;97(7):2524–2542. | ||
Namjoshi S, Toth I, Blanchfield JT, Trotter N, Mancera RL, Benson HA. Enhanced transdermal peptide delivery and stability by lipid conjugation: epidermal permeation, stereoselectivity and mechanistic insights. Pharm Res. 2014;31(12):3304–3312. | ||
Flynn G. Physicochemical determinants of skin absorption. In: Gerrity T, Henry C, editors. Principles of Route-to-Route Extrapolation for Risk Assessment. New York: Elseiver Science Publishing Co., Inc; 1990:93–129. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.