")
Back to Journals » Journal of Pain Research » Volume 14
Acute Treatment of Migraine with Celecoxib Oral Solution: Results of a Randomized, Placebo-Controlled Clinical Trial
Authors Lipton RB , Munjal S , Dodick DW , Tepper SJ, Serrano D, Iaconangelo C
Received 19 October 2020
Accepted for publication 16 January 2021
Published 25 February 2021 Volume 2021:14 Pages 549—560
DOI https://doi.org/10.2147/JPR.S287571
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Robert B. Raffa
Richard B Lipton,1 Sagar Munjal,2 David W Dodick,3 Stewart J Tepper,4 Daniel Serrano,5 Charlie Iaconangelo5
1Neurology, Epidemiology and Population Health, and Psychologyand Behavioral Sciences Departments, Albert Einstein College of Medicine, Bronx, NY, USA; 2Clinical Development, Operations, and Medical Affairs, Dr. Reddy’s Laboratories, Princeton, NJ, USA; 3Neurology Department, Mayo Clinic, Scottsdale, AZ, USA; 4Neurology Department, Geisel School of Medicine at Dartmouth, Hanover, NH, USA; 5Biostatistics Department, Pharmerit Inc., Bethesda, MD, USA
Correspondence: Sagar Munjal
Dr. Reddy’s Laboratories, 107 College Road East, Princeton, NJ, 8540, USA
Tel +1 609-375 9900
Email [email protected]
Background: Nonsteroidal anti-inflammatory drugs are widely used for migraine, but gastrointestinal tolerability limits use. We previously reported results from the first treatment period of this 2-period, randomized, controlled study comparing DFN-15—an oral, ready-made liquid solution of a selective cyclo-oxygenase-2 inhibitor celecoxib—with placebo for the acute treatment of a moderate-severe migraine attack. Herein, we report the effects of treatment for the second treatment period.
Methods: In the first treatment period of this trial, adults with migraine were randomized to double-blind trial treatment of attacks of moderate or severe pain with DFN-15,120 mg or placebo. For the second treatment period, reported herein, participants were re-randomized to treat an attack of any baseline pain intensity (mild, moderate, or severe). Co-primary efficacy endpoints specified for the first attack were not specified for the second attack.
Results: Of the 531 patients who completed the first treatment period, 491 (n = 243 DFN-15; n = 248 placebo; 87% female, mean age 41 years) were re-randomized into the second double-blind treatment period. Baseline pain intensity was mild in 17.2% (85/493) of patients, moderate in 58.4% (288/493) of patients, and severe in 22.9% (113/493) of patients. At 2 hours post-dose, DFN-15 was superior to placebo for freedom from pain (46.2% [110/238] vs 31.1% [76/244], p ≤ 0.001) and the most bothersome symptom (63.4% [121/191] vs 50.0% [98/196], p = 0.010). Treatment-emergent adverse events (TEAEs) occurred in 7.1% (35/493) of patients (DFN-15: 6.1% [15/244]; placebo 8.0% [20/249]). Study drug-related TEAEs occurred in 5.1% (25/493) of patients (DFN-15: 4.5% [11/244]; placebo 5.6% [14/249]); nausea (1% [5/493]) and dysgeusia (0.8% [4/493]) were most common. No serious TEAEs, severe TEAEs, or TEAEs leading to study drug termination were reported.
Conclusions: DFN-15 was superior to placebo for pain freedom and freedom from the most bothersome symptom when patients treat a migraine attack of any baseline pain intensity. Rates of TEAEs did not differ between treatment groups.
Keywords: migraine, acute treatment, celecoxib, oral liquid
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are effective and widely used for migraine,1,2 but gastrointestinal tolerability and safety limit usage by many patients.3–5 Celecoxib (Celebrex; Pfizer Inc., New York, NY, USA), the selective cyclo-oxygenase (COX)-2 inhibitor, has better gastrointestinal tolerability than ibuprofen, naproxen, or diclofenac and better renal safety than ibuprofen.6–9
Celecoxib oral capsules have a long median time to peak plasma concentration (Tmax) of 2.5 hours.10 This long Tmax,, thought to be a predictor of slow onset, has been regarded as a limitation in migraine therapy, where rapidity of pain relief is a frequently cited patient priority.11–13 To address this limitation, an oral liquid solution of celecoxib (DFN-15, Dr. Reddy’s Laboratories, Princeton NJ, USA) was developed for the acute treatment of migraine. In healthy fasted adults, DFN-15 120 mg had a shorter Tmax than a 400 mg dose of celecoxib oral capsules (≤1 hour vs 2.5 hours) and a 44% greater relative bioavailability.14 In a dose-ranging study comparing DFN-15 with placebo in the acute treatment of migraine,15 a 120 mg dose (50 mg/mL) outperformed placebo and, because its efficacy was similar to DFN-15 240 mg, was chosen for further development. Development was guided by the hypotheses that a short Tmax would lead to rapid onset of treatment effects and that the lower dose would lead to improved GI safety and tolerability.
In the first treatment period of this randomized, double-blind, placebo-controlled clinical trial,16 patients used a 120 mg dose of DFN-15 to treat a single migraine attack of moderate to severe pain intensity at baseline. Results showed that DFN-15 was superior to placebo on the co-primary efficacy endpoints of pain freedom (35.6% [98/275] vs 21.7% [57/263], p < 0.001) and freedom from the most bothersome symptom (MBS) associated with migraine (ie, nausea, photophobia, phonophobia) at 2 hours post-dose (57.8% [134/232] vs 44.8% [104/232], p = 0.007). The earliest time point at which DFN-15 was significantly superior to placebo was at 1 hour post-dose on the pain relief endpoint (56.8% [155/273] vs 45.1% [116/257], p = 0.009). The most frequently reported treatment-emergent adverse events (TEAEs) related to treatment, dysgeusia, and nausea, were only slightly more common with DFN-15 than with placebo.16
Patients who completed the first treatment period were re-randomized to treat a second migraine attack with DFN-15 or placebo. We hypothesized that outcomes might be improved if, rather than waiting until headache pain was moderate or severe before taking study medication, patients decided when they wanted to treat, including while pain was mild. Therefore, the objectives of this study were to compare the efficacy, tolerability, and safety of 120 mg DFN-15 with placebo for the acute treatment of a single migraine attack of any pain intensity.
Materials and Methods
With the exception of the ability to treat attacks when they were mild, the methods in this study are identical to those used in previously published research.16
Ethics
The conduct of this study adhered to the Declaration of Helsinki (WMA Brazil, 2013), the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines for current Good Clinical Practice, the US Food and Drug Administration’s Code of Federal Regulations, and other applicable regulatory requirements. The protocol and other relevant study documentation were reviewed and approved by the Copernicus Group IRB (Durham, NC, USA), and patients provided written informed consent before any study-related examinations or activities were performed.
Study Design
This randomized, double-blind, placebo-controlled, multicenter study (ClinicalTrials.gov Identifier: NCT03006276) enrolled adults aged 18 to 75 years (inclusive) with at least a 1-year history of migraine17 (2–8 attacks and ≤14 headache days per month), no medication overuse, and 48 hours of headache-free time between migraine attacks. Patients satisfying inclusion criteria and completing the first double-blind treatment period returned to the study site (listed in the Supplement) within 2 to 7 days (minimum washout ≥3 days) for re-randomization to DFN-15 or matching placebo to treat a single migraine attack within 1 hour of the onset of migraine headache pain of mild, moderate, or severe intensity (Figure 1). They used an electronic diary to record treatment effects on pain, associated symptoms, functional disability, and satisfaction.
 |
Figure 1 Study schemaa. aThe red dotted box signifies that data are presented for Period 2 only; refer to Lipton et al16 for detailed methods and the results of Period 1. |
Patients
Patients had at least a 12-month history of migraine characterized by 2 to 8 attacks per month, with no more than 14 monthly headache days and at least 48 hours of headache-free time between attacks, an age of onset before age 50 years, and a usual untreated migraine pain intensity of moderate or severe (ie, 2 or 3 on a scale of 0 to 3). For the duration of the study, patients had to be able to evaluate and record pain, migraine symptoms, and study drug effectiveness information, as well as each instance of the use of study drug and rescue medication, in real-time using an electronic diary and comply with all other study procedures and scheduling requirements; be able to read, speak, and understand English proficiently; provide written informed consent; and be male or female, 18 to 75 years of age, inclusive, at screening. For the duration of the study, females could not be pregnant, and all patients had to practice a reliable form of contraception or abstinence or be sterile.
Patients were excluded if they had taken opioids, opioid-barbiturate fixed combinations, triptans, or ergot alkaloids on at least 10 days or NSAIDs or other simple analgesics on at least 15 days per month during the 90 days before screening; had been treated with onabotulinumtoxinA for migraine within 4 months before screening; had changes in the dose or type of preventive medications they used for migraine within 30 days prior to screening or during the screening phase; had taken mini-prophylaxis for menstrual migraine; or were on chronic warfarin sodium or equivalent. Patients were also excluded if they had a history of any condition that might interfere in any way with the study conduct, outcomes, or interpretation of results.
Treatments
Each patient was given a single-dose bottle of DFN-15120 mg or matching placebo containing 4.8 mL liquid. Bottles had identical labels and were assigned unique numbers by an interactive web response system. Site staff and investigators were blinded, as the kits were assembled centrally (The Coghlan Group, Bastrop, TX, USA).
Patients were permitted to take rescue medication at least 2 hours after treating and only after completing a 2-hour post-dose assessment. Rescue medication was prospectively chosen in consultation with the study clinician.
Patients treated a single migraine attack as soon as possible after pain onset, and no more than 1 hour after the onset of migraine pain of mild, moderate, or severe pain intensity. The electronic diary required that patients confirm the attack was migraine before they could record treatment response.
Assessments
Efficacy
Efficacy endpoints included 2-hour pain freedom and freedom from the MBS designated at the screening visit. We also assessed pain freedom; MBS freedom; freedom from nausea, photophobia, and phonophobia; and pain relief at 15, 30, and 45 minutes and 1, 1.5, 2, 4, and 24 hours post-dose and change in functional disability score at 2, 4, and 24 hours post-dose. Other endpoints included sustained pain relief and sustained pain freedom at 2 to 24 hours post-dose (defined as a positive response at 2 hours post-dose with no rescue medication use or relapse within 2 to 24 hours post-dose); use of rescue medication from 2 to 24 hours post-dose; treatment satisfaction at 2 and 4 hours post-dose; and treatment satisfaction at 24 hours post-dose as measured by the Patient Perception of Migraine Questionnaire-Revised (PPMQ-R).
Pain relief was defined as a reduction from moderate or severe pain to mild or no pain or from mild pain to no pain. Functional disability was measured on a scale where 0 = no disability and 3 = severely impaired. Treatment satisfaction with pre-study migraine treatment and with study treatment was measured on a scale where 1 = very satisfied and 7 = very dissatisfied; side effects were scored on a scale where 1 = not at all and 5 = extremely. Subscale scores and total scores (except the total raw score) were transformed to a 0 to 100 range by subtracting the lowest possible scale score, dividing by the range of the scale, and multiplying by 100. Missing responses were considered non-evaluable. Sustained pain freedom and pain relief at 2 to 24 hours post-dose were defined as pain freedom or pain relief at 2 hours post-dose, with no use of rescue medication and no recurrence (freedom) or worsening (relief) within 24 hours post-dose.
Safety
Safety endpoints/assessments included AEs; concomitant medication review; physical examinations and suicidality check; pregnancy tests; vital signs (blood pressure, pulse rate, and body temperature); clinical laboratory examinations (hematology, chemistry, and urinalysis); and 12-lead electrocardiogram. All AEs were coded using the Medical Dictionary for Regulatory Activities, version 19.0.
Statistical methods
The safety population included patients who received at least 1 dose of study treatment. The efficacy-evaluable population included patients who took at least 1 dose of study treatment and had at least 1 post-baseline efficacy assessment for migraine pain or the MBS.
For this second attack in a 2-attack study, patients were re-randomized between the first and second attack to make the treated attacks as independent as possible. Treatment of the second attack was intended to explore the benefits of treatment during mild pain. Therefore, unlike the first attack, which specified the co-primary endpoints of pain freedom and MBS freedom at 2 hours post-dose, the statistical analysis plan did not designate primary endpoints for the second attack. In keeping with US Food and Drug Administration guidance, as well as the statistical analysis plan for the first attack, we considered pain freedom and MBS freedom at 2 hours post-dose the most important efficacy endpoints. Statistical testing and confidence intervals (CIs) were 2-sided and performed using a significance (alpha) level of 0.05. Statistical analyses were conducted with SAS version 9.3 (Cary, NC, USA) and R statistical software version 3.4.4.18
Efficacy
The efficacy analysis was nonhierarchical and not adjusted for multiple comparisons. Missing data were imputed using last observation carried forward. For the analysis of freedom from the MBS, the symptom identified as the MBS at screening had to be present at baseline. Patients who took rescue medication before 2 hours post-dose were excluded.
P-values for freedom from nausea, photophobia, and phonophobia; pain relief; pain freedom; and freedom from the screening MBS were computed from Fisher’s exact test. For functional disability, the Wilcoxon rank-sum test was used for the comparison between treatment groups and the Wilcoxon signed-rank test for the change from baseline (−3 to +3-point change) within each treatment group. Treatment satisfaction at 2 and 4 hours post-dose and the baseline PPMQ-R response for the same question were compared for the DFN-15 group only. The Wilcoxon rank-sum test was used for this comparison, as well as for the comparison in patient-rated treatment satisfaction at 24 hours post-dose and in baseline and 24-hour post-dose total scores and total raw scores for the DFN-15 group only. The Wilcoxon signed-rank test was used to analyze the difference between baseline and post-baseline PPMQ-R for the DFN-15 group only, as well as the change from baseline to 24 hours for each subscale score, each global item score, the total score, and the total raw score.
Exploratory Analyses of the Effects of Baseline Pain Intensity on 2-Hour Freedom from Pain and the MBS
Exploratory analyses were conducted stratifying on pre-dose headache pain intensity of mild, moderate, or severe and examining the proportion of subjects who were pain-free or MBS-free at 2 hours. The corresponding p-values from Fisher’s exact test were computed for the comparisons between treatment groups. A patient’s pre-dose MBS was obtained from the electronic diary.
Power Calculations
Power calculations were conducted to size the study for the first attack. We assumed 2-hour pain-free rates of 17.6% for placebo-treated patients and 29.2% for DFN-15-treated patients and a 15% dropout rate. Under those assumptions, 600 patients would provide 88% power to detect a difference between DFN-15 and placebo at a 5% (2-sided) level of significance.
Post Hoc Analysis of Carry-Over Effects
Because this trial involved 2 independently randomized double-blind treatment periods, a post hoc analysis of the pre-specified co-primary efficacy endpoints for the first treatment period was assessed to look for carry-over effects on the second treatment period. The design yielded 4 randomized exposure patterns across the first and second double-blind treated attacks: DFN-15/DFN-15, DFN-15/placebo, placebo/DFN-15, and placebo/placebo. The potential effect of first attack treatment arm and first attack treatment response on second attack treatment efficacy was examined. The estimates were generated by fitting logistic generalized linear mixed models to each second double-blind period co-primary endpoint (2-hour freedom from pain and the MBS) separately. These models were parameterized with a random effect for sites. Predictors in these models included the following fixed effects: second double-blind period treatment arm, first double-blind period treatment arm, and first double-blind response status, as well as all interactions of the fixed effects. These models were designed to assess whether there was a meaningful carry-over effect.
In the event that a carry-over effect was detected, a second model was planned for adjusting for the effect of the first double-blind period on the second double-blind period. In this model, the first and second double-blind periods were analyzed simultaneously via generalized linear mixed models. The estimates were generated by fitting logistic generalized linear mixed models to co-primary endpoints (2-hour freedom from pain and the MBS) separately. This permitted adjusting the second double-blind period treatment effect for carry-over effects. These models were parameterized with a random effect for sites plus a random effect for subjects. The subject random effect was used to adjust for carry-over effects. Fixed effects used in these models included double-blind period, treatment arm assignment, and the interaction between double-blind period and treatment arm assignment. P-values from the z-tests were reported.
Results
Patients
The first patient was enrolled on 13 December 2016, and the last patient completed the study on 6 October 2017. In total, 535 patients were re-randomized into the second double-blind treatment period, 493 were analyzed for safety (n = 244 DFN-15; n = 249 placebo), and 491 were analyzed for efficacy (n = 243 DFN-15; n = 248 placebo) and completed the study. The washout between study treatment taken in the first and second treatment periods averaged 17 days. Thirty-six patients discontinued, most often because they did not experience a migraine attack (2.1% [11/493]). Of those who withdrew after taking DFN-15, 0.4% (2/493) cited an AE as the reason. Patient disposition is shown in Table 1.
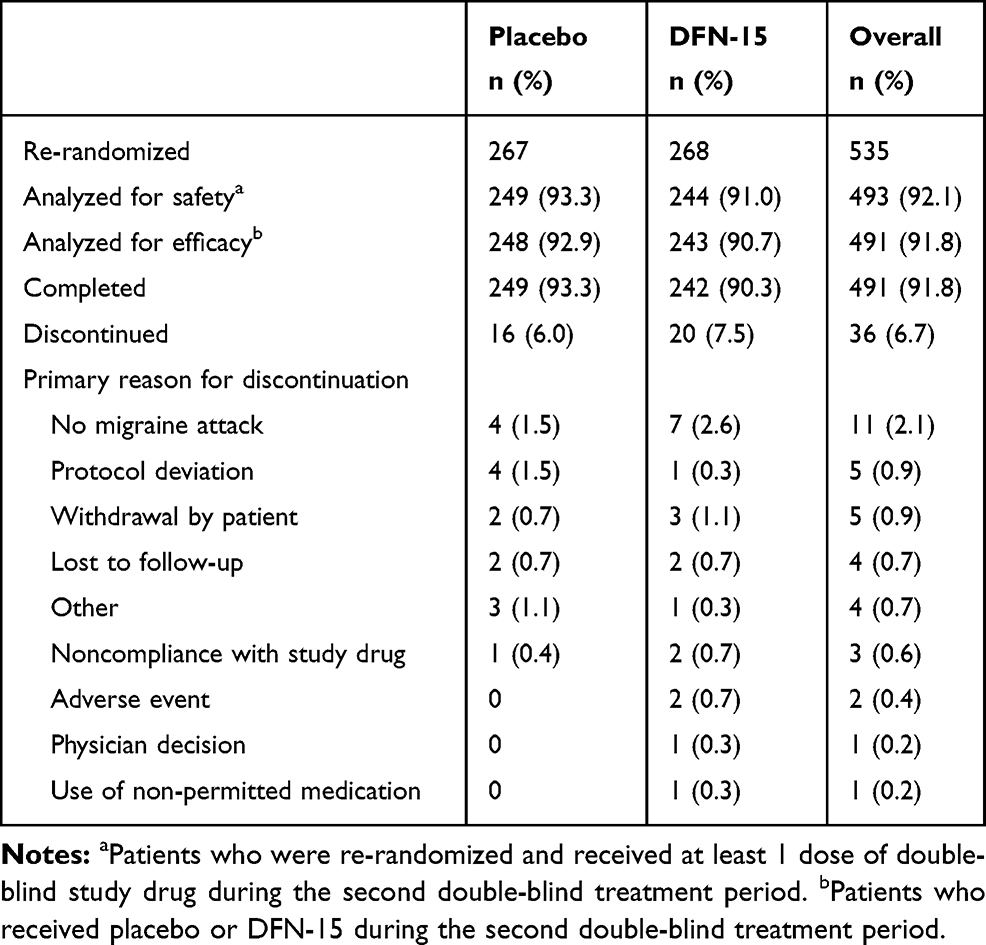 |
Table 1 Disposition of Patients |
Most patients (87% [429/493]) were female, and the population had a mean (SD) age of 40.3 (12.03) years, a mean (SD) weight of 84.0 (22.65) kg, and a mean (SD) body mass index of 30.5 (8.00) kg/m2 (Table 2). Most patients (76.2%, [435/493]) had never smoked or used nicotine products. Pre-dose levels of pain intensity in patients treated with DFN-15 were mild in 19.3% (47/243), moderate in 56.4% (137/243), and severe in 23.0% (56/243); the pre-dose levels of pain intensity in placebo-treated patients were 14.9% (37/248) mild, 60.9% (151/248) moderate, and 23.0% (57/248).
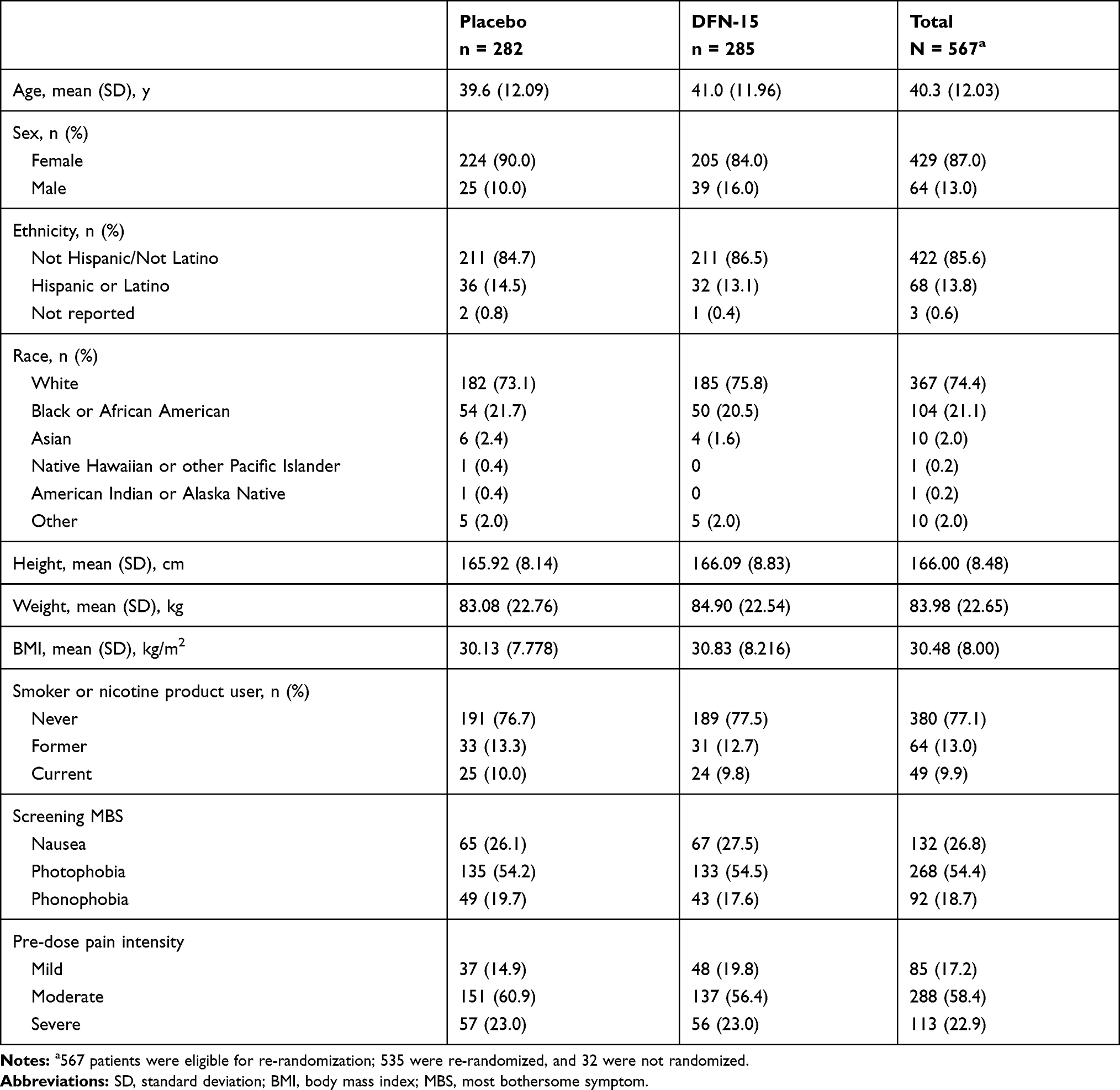 |
Table 2 Demographics and Baseline Characteristics |
Efficacy
2-Hour Freedom from Pain and the Most Bothersome Symptom
At 2 hours post-dose (Figure 2), the percentage (95% CI) of patients with pain freedom was significantly higher for DFN-15-treated patients than for placebo-treated patients (46.2% [39.8, 52.8] vs 31.1% [25.4, 37.4], p < 0.001). The percentage (95% CI) of patients with freedom from the MBS at 2 hours post-dose was also significantly higher for patients in the DFN-15 group than for those in the placebo group (63.4% [56.1, 70.2] vs 50.0% [42.8, 57.2], p < 0.001) (Figure 2).
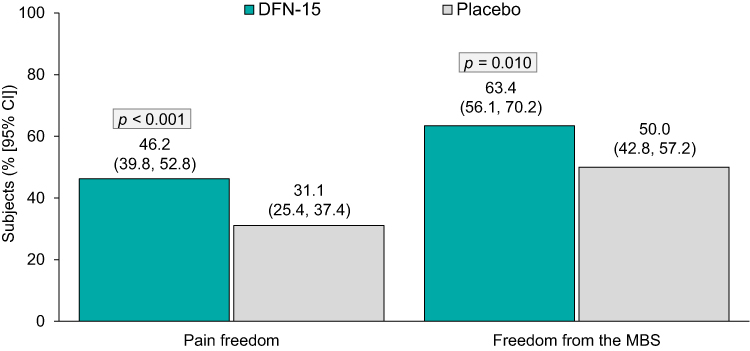 |
Figure 2 DFN-15 efficacy versus placebo on the co-primary endpoints. Abbreviations: CI, confidence interval; MBS, most bothersome symptom. |
Other 2-Hour Endpoints
The percentage (95% CI) of patients with pain relief at 2 hours post-dose with DFN-15 was significantly superior to placebo (74.4% [68.3, 79.8] vs 60.7% [54.2, 66.8], p < 0.001).
The mean (SD) change in functional disability from baseline in patients treated with DFN-15 was superior to those who were treated with placebo at 2 hours (−0.9 [0.87] vs −0.6 [0.83], p < 0.001).
At 2 hours post-dose, DFN-15 was similar to placebo for freedom from nausea (69.8% [60.1, 78.3] vs 66.9% [57.8, 75.2], p = 0.670), significantly superior to placebo for freedom from photophobia (64.1% [57.0, 70.8] vs 46.1% [39.1, 53.2], p < 0.001), and significantly superior to placebo for freedom from phonophobia (67.9% [60.2, 74.9] vs 48.4% [40.2, 56.6]). Photophobia was the most often selected as the MBS.
Early and Sustained Effects
An early onset of effects following treatment with DFN-15 was observed across multiple endpoints, including pain freedom beginning at 1 hour post-dose (23.2% [55/237] vs 15.8% [38/240], p =0.049), pain relief beginning at 45 minutes post-dose (47.7% [113/237] vs 37.1% [89/240], p = 0.021), and both photo- and phonophobia beginning at 30 minutes post-dose (21.9% [43/196] vs 14.4% [29/202], p = 0.052 and 27.4% [45/64] vs 15.6% [24/154], p = 0.014, respectively). The persistence of DFN-15 efficacy was demonstrated by its superiority to placebo for sustained pain freedom from 2 to 24 hours (40.2% [76/189] vs 27.2% [52/191], p = 0.009) and sustained pain relief from 2 to 24 hours (63.0% [119/189] vs 51.3% [8/191], p = 0.023). Table 3 summarizes the 2-hour, early, and sustained efficacy of DFN-15 relative to placebo.
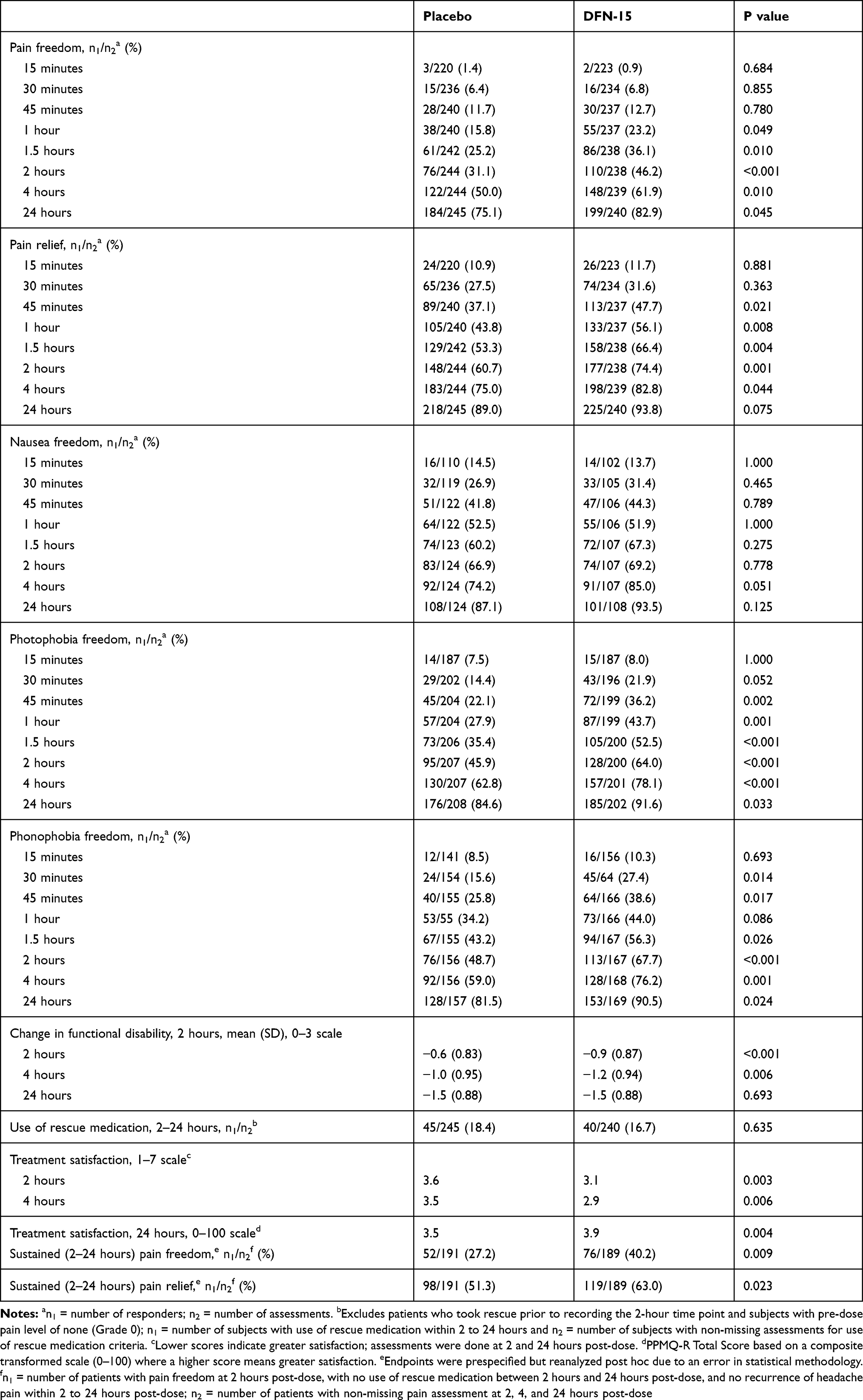 |
Table 3 DFN-15 Efficacy versus Placebo |
Satisfaction with Treatment
Patients who were treated with DFN-15 had higher overall mean treatment satisfaction scores (lower scores indicate greater satisfaction) than those who received placebo at 2 hours post-dose (3.1 vs 3.6, p = 0.003) and at 4 hours post-dose (2.9 vs 3.5, p = 0.006). At 2 hours post-dose, more DFN-15-treated patients than placebo-treated patients were satisfied/neither versus dissatisfied at 2 hours (75.3% satisfied/neither and 21.8% dissatisfied vs 65.3% satisfied/neither and 30.6% dissatisfied, p = 0.023) and at 4 hours post-dose (77.0% satisfied/neither and 18.9% dissatisfied vs 65.3% satisfied/neither and 30.2% dissatisfied, p = 0.004).
Carry-Over Analysis
A responder carry-over effect across double-blind treatment periods was detected.
Response in the first double-blind treatment period, regardless of treatment arm assignment, predicted response in the second double-blind treatment period.
For example, the probability of achieving 2-hour pain freedom in the second double-blind treatment period was 571% greater among patients who responded to treatment in the first double-blind treatment period than among those who did not respond to treatment in the first double-blind period (odds ratio [OR] = 6.71, p < 0.001). However, after adjusting for the carry-over effect across treatment periods, DFN-15 was significantly superior to placebo for both 2-hour pain freedom (OR = 2.50, p = 0.001) and 2-hour MBS freedom (OR = 2.01, p = 0.008). Importantly, the interaction of treatment by treatment period was not statistically significant for pain (p = 0.935) or MBS (p = 0.765), indicating that the treatment effect did not vary across double-blind periods after adjusting for carry-over effects. The statistically significant treatment effect estimated in the adjustment stage suggests that regardless of the carry-over effects, receiving DFN-15 resulted in a higher probability of endpoint achievement in each double-blind period.
Tolerability and Safety
As shown in Table 4, 7.1% (35/493) of patients (DFN-15: 6.1% [15/244]; placebo 8.0% [20/249]) reported a TEAE. Study drug-related TEAEs were reported by 5.1% (25/493) of patients (DFN-15: 4.5% [11/244]; placebo 5.6% [14/249]), with nausea (1% [5/493]) and dysgeusia (0.8% [4/493]) the most common among them. No serious TEAEs or TEAEs leading to study drug termination were reported, and there were no severe TEAEs or deaths.
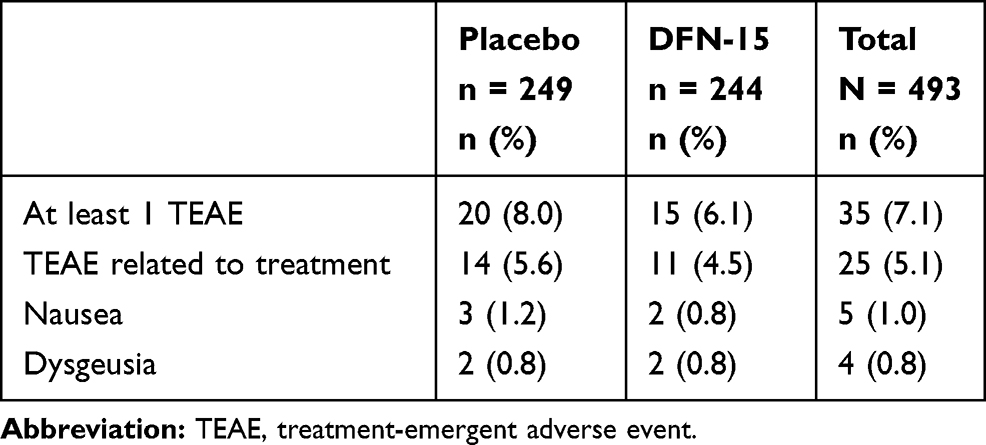 |
Table 4 Summary of Adverse Events Occurring in ≥2% of Patients Who Treated a Migraine Attack of Moderate or Severe Headache with DFN-15 or Placebo |
Discussion
This randomized, double-blind, placebo-controlled study was conducted to compare DFN-15, an oral solution of celecoxib, with placebo for the acute treatment of migraine at any baseline pain intensity. In this study, patients were re-randomized to treat a second attack of any pain intensity, having already treated a first attack. The results showed that DFN-15 was significantly more effective than placebo for the primary endpoints for the first treated attack, 2-hour pain freedom and 2-hour MBS freedom. Significant effects were also observed at other 2-hour endpoints, including pain relief; freedom from photophobia and phonophobia; change in functional disability; and treatment satisfaction.
The 2-hour results are directionally similar for the second attack and for the previously reported first attack.16 Although the 2-hour rates for freedom from pain and freedom from the MBS were higher for the second attack, there was a corresponding increase in the magnitude of the placebo rates. As a consequence, the placebo-subtracted rates were very similar. We hypothesized that treatment while pain is mild might increase the pain-free rates and the placebo-subtracted pain-free rates,19 but only 17.2% (85/567) of patients in this study treated during mild pain, which may partially obscure the benefit of early treatment. Based on this finding, the general advice to treat migraine with acute medications while pain is mild likely applies to DFN-15, although specifically designed studies in people with mild pain are needed.
In addition to the 2-hour endpoints, we explored the onset of benefits on a variety of measures. We found that pain relief and phonophobia freedom became statistically significant at 45 minutes and remained so for all subsequent time points. We also found that pain freedom and photophobia freedom achieved statistical significance at 2 hours post-dose and that this superiority was maintained for all subsequent time points. Functional disability significantly improved by 2 hours post-dose, which aligns with results from the first attack, when the 2-hour mean change from baseline in functional disability scores for DFN-15 and placebo were −0.9 and −0.7 at 2 hours (p = 0.004).16 The relatively rapid onset of treatment effects with DFN-15 is consistent with its relatively short Tmax of ≤1 hour.
We also examined sustained pain freedom and sustained pain relief at 24 hours and found highly significant differences favoring DFN-15. These enduring treatment effects were also demonstrated for the first attack and may reflect the relatively long half-life of DFN-15 (4.5 ± 1.6 hours).14
Finally, the safety and tolerability profile was favorable. TEAEs were predictable and generally mild. There were no clinically significant changes in vital signs or laboratory values.
This study has several limitations. Because we re-randomized participants to treat a second attack, the potential of carry-over effects must be considered. Although carry-over effects were demonstrated, they were equally distributed between the DFN-15 and placebo treatment arms; it is unlikely they materially influenced our findings. Nonetheless, the implications of this carry-over effect should be considered in the design of future studies of acute migraine treatments using re-randomization. Additionally, because the per protocol analysis designated freedom from pain and the MBS as the per protocol co-primary endpoints for the first attack only, analyses of results from the second attack must therefore be considered exploratory. The consistency of results from the first and second attacks on a range of clinically important endpoints (with the exception of nausea freedom) greatly reduces the possibility that these outcomes were achieved by chance, and the time–effect curves show that once statistical separation was achieved, it was maintained across all subsequent endpoints. Nevertheless, based on the lack of protocol-specified co-primary endpoints for the second attack, the analyses in this manuscript are effectively post-hoc.
Conclusions
DFN-15 was significantly superior to placebo on multiple efficacy endpoints, including 2-hour freedom from pain and the MBS, as well as headache relief and functional disability. Safety and tolerability results were predictable and similar to placebo.
Abbreviations
CI, confidence interval; COX, cyclo-oxygenase; MBS, most bothersome symptom; NSAID, nonsteroidal anti-inflammatory drug; PPMQ-R, Patient Perception of Migraine Questionnaire-Revised; SD, standard deviation; TEAE, treatment-emergent adverse event.
Data Sharing Statement
Data for this study are available to qualified researchers by reasonable request from Sagar Munjal, MD, MS.
Ethics Approval and Informed Consent
The conduct of this study adhered to the Declaration of Helsinki (WMA Brazil, 2013), the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines for current Good Clinical Practice, the US Food and Drug Administration’s Code of Federal Regulations, and other applicable regulatory requirements. The protocol and other relevant study documentation were reviewed and approved by the Copernicus Group IRB (Durham, NC, USA), and patients provided written informed consent before any study-related examinations or activities were performed.
Acknowledgments
Medical writing services were provided by Christopher Caiazza. DRL Publication #889.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study was supported and funded by the Dr. Reddy’s Laboratories group of companies, Princeton, NJ 08540, USA.
Disclosure
Richard B. Lipton has received research support from the NIH. He also receives support from the Migraine Research Foundation and the National Headache Foundation. He serves on the editorial board of Neurology, senior advisor to Headache, and associate editor to Cephalalgia. He has reviewed for the NIA and NINDS, holds stock options in eNeura Therapeutics and Biohaven Holdings; serves as consultant, advisory board member, or has received honoraria from: American Academy of Neurology, Allergan, American Headache Society, Amgen, Avanir, Biohaven, Biovision, Boston Scientific, Dr. Reddy’s (Promius), Electrocore, Eli Lilly, eNeura Therapeutics, Equinox, GlaxoSmithKline, Lundbeck (Alder), Merck, Pernix, Pfizer, Supernus, Teva, Trigemina, Vector, Vedanta. He receives royalties from Oxford University Press (Wolff’s Headache 7th and 8th Edition), Wiley, and Informa. Sagar Munjal is an employee of Dr. Reddy’s Laboratories and owns stock in the company. David W. Dodick reports the following conflicts within the past 12 months: Consulting: AEON, Amgen, Clexio, Cerecin, Ctrl M, Allergan, Alder, Biohaven, Linpharma, Lundbeck, Promius, Eli Lilly, eNeura, Novartis, Impel, Satsuma, Theranica, WL Gore, Nocira, XoC, Zosano, Upjohn (Division of Pfizer), Pieris, Revance, Equinox. Honoraria: CME Outfitters, Curry Rockefeller Group, DeepBench, Global Access Meetings, KLJ Associates, Academy for Continued Healthcare Learning, Majallin LLC, Medlogix Communications, MJH Lifesciences, Miller Medical Communications, Southern Headache Society (MAHEC), WebMD Health/Medscape, Wolters Kluwer, Oxford University Press, Cambridge University Press. Research Support: Department of Defense, National Institutes of Health, Henry Jackson Foundation, Sperling Foundation, American Migraine Foundation, Patient Centered Outcomes Research Institute (PCORI). Stock Options/Shareholder/Patents/Board of Directors: Ctrl M (options), Aural analytics (options), ExSano (options), Palion (options), Healint (Options), Theranica (Options), Second Opinion/Mobile Health (Options), Epien (Options/Board), Nocira (options), Matterhorn (Shares/Board), Ontologics (Shares/Board), King-Devick Technologies (Options/Board), Precon Health (Options/Board). Patent 17189376.1-1466:vTitle: Botulinum Toxin Dosage Regimen for Chronic Migraine Prophylaxis. Stewart Tepper reports the following conflicts: Grants for research (no personal compensation): Allergan, Amgen, ElectroCore, Eli Lilly, Lundbeck, Neurolief, Novartis, Satsuma, Zosano. Consultant and/or Advisory Boards (honoraria): Aeon, Align Strategies, Allergan/Abbvie, AlphaSights, Amgen, Aperture Venture Partners, Aralez Pharmaceuticals Canada, Axsome Therapeutics, Becker Pharmaceutical Consulting, BioDelivery Sciences International, Biohaven, ClearView Healthcare Partners, CoolTech, CRG, Currax, Decision Resources, DeepBench, DRG, Eli Lilly, Equinox, ExpertConnect, GLG, Guidepoint Global, Healthcare Consultancy Group, Health Science Communications, HMP Communications, Impel, InteractiveForums, Krog and Partners, Lundbeck, M3 Global Research, Magellan Rx Management, Medicxi, Navigant Consulting, Neurolief, Nordic BioTech, Novartis, Pulmatrix, Reckner Healthcare, Relevale, SAI MedPartners, Satsuma, Slingshot Insights, Spherix Global Insights, Sudler and Hennessey, Synapse Medical Communications, System Analytic, Teva, Theranica, Thought Leader Select, Trinity Partners, XOC, Zosano.
Salary: Dartmouth-Hitchcock Medical Center, American Headache Society, Thomas Jefferson University.
CME honoraria: American Academy of Neurology, American Headache Society, Cleveland Clinic Foundation, Diamond Headache Clinic, Elsevier, Forefront Collaborative, Hamilton General Hospital, Ontario, Canada, Headache Cooperative of New England, Henry Ford Hospital, Detroit, Inova, Medical Learning Institute Peerview, Medical Education Speakers Network, Miller Medical Communications, North American Center for CME, Physicians’ Education Resource, Rockpointe, ScientiaCME, WebMD/Medscape.
Charlie Iaconangelo was a paid consultant of Dr. Reddy’s Laboratories.
Daniel Serrano was a paid consultant of Dr. Reddy’s Laboratories.
The authors report no other conflicts of interest related to this work.
References
1. Silberstein SD. Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55(6):754–762. doi:10.1212/WNL.55.6.754
2. Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: the American Headache Society evidence assessment of migraine pharmacotherapies. Headache. 2015;55(1):3–20. doi:10.1111/head.12499
3. Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. A meta-analysis. Ann Intern Med. 1991;115(10):787–796. doi:10.7326/0003-4819-115-10-787
4. Henry D, McGettigan P. Epidemiology overview of gastrointestinal and renal toxicity of NSAIDs. Int J Clin Pract Suppl. 2003;135:43–49.
5. Hernandez-Diaz S, Rodriguez LA. Association between nonsteroidal anti-inflammatory drugs and upper gastrointestinal tract bleeding/perforation: an overview of epidemiologic studies published in the 1990s. Arch Intern Med. 2000;160(14):2093–2099. doi:10.1001/archinte.160.14.2093
6. Moore RA, Derry S, Makinson GT, McQuay HJ. Tolerability and adverse events in clinical trials of celecoxib in osteoarthritis and rheumatoid arthritis: systematic review and meta-analysis of information from company clinical trial reports. Arthritis Res Ther. 2005;7(3):R644–665. doi:10.1186/ar1704
7. Loo CY, Tan HJ, Teh HS, Raymond AA. Randomised, open label, controlled trial of celecoxib in the treatment of acute migraine. Singapore Med J. 2007;48(9):834–839.
8. Walker C. Are all oral COX-2 selective inhibitors the same? A consideration of celecoxib, etoricoxib, and diclofenac. Int J Rheumatol. 2018;2018:1302835. doi:10.1155/2018/1302835
9. Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N Engl J Med. 2016;375(26):2519–2529. doi:10.1056/NEJMoa1611593
10. Celebrex (celecoxib capsule) prescribing information. Available from: http://labeling.pfizer.com/showlabeling.aspx?id=793.
11. Lipton RB, Hamelsky SW, Dayno JM. What do patients with migraine want from acute migraine treatment? Headache. 2002;42(Suppl 1):3–9. doi:10.1046/j.1526-4610.2002.0420s1003.x
12. Davies GM, Santanello N, Lipton R. Determinants of patient satisfaction with migraine therapy. Cephalalgia. 2000;20(6):554–560. doi:10.1046/j.1468-2982.2000.00082.x
13. Smelt AF, Louter MA, Kies DA, et al. What do patients consider to be the most important outcomes for effectiveness studies on migraine treatment? Results of a Delphi study. PLoS One. 2014;9(6):e98933. doi:10.1371/journal.pone.0098933
14. Pal A, Shenoy S, Gautam A, et al. Pharmacokinetics of DFN-15, a novel oral solution of celecoxib, versus celecoxib 400-mg capsules: a randomized crossover study in fasting healthy volunteers. Clin Drug Investig. 2017;37(10):937–946. doi:10.1007/s40261-017-0548-6
15. Munjal S, Bennett A. Efficacy and safety of DFN-15, an oral liquid formulation of celecoxib, in adults with migraine: a multicenter, randomized, placebo-controlled, double-blind, crossover study. Neuropsychiatr Dis Treat. 2017;13:2797–2802. doi:10.2147/NDT.S151834
16. Lipton RB, Munjal S, Brand-Schieber E, Tepper SJ, Dodick DW. Efficacy, tolerability, and safety of DFN-15 (celecoxib oral solution, 25 mg/mL) in the acute treatment of episodic migraine: a randomized, double-blind, placebo-controlled study. Headache. 2020;60(1):58–70. doi:10.1111/head.13663
17. Headache Classification Committee of the International Headache Society. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33(9):629–808. doi:10.1177/0333102413485658
18. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2018. Available at https://www.R-project.org/.
19. Goadsby PJ, Zanchin G, Geraud G, et al. Early vs. non-early intervention in acute migraine-’Act when Mild (AwM)’. A double-blind, placebo-controlled trial of almotriptan. Cephalalgia. 2008;28(4):383–391. doi:10.1111/j.1468-2982.2008.01546.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.