Back to Journals » Cancer Management and Research » Volume 14
Acute Mast Cell Leukemia Preceded by Malignant Mediastinal Germ Cell Tumor: A Case Report and Literature Review
Authors Wang H, Chen Y, Lin H, Ni W, Zhang Q ![]() , Lan J, Jin L
, Lan J, Jin L
Received 22 February 2022
Accepted for publication 12 May 2022
Published 23 May 2022 Volume 2022:14 Pages 1783—1794
DOI https://doi.org/10.2147/CMAR.S363508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Kattesh Katti
Huafang Wang,1 Yuan Chen,2 Huijun Lin,3 Wanmao Ni,4 Qiaolei Zhang,5 Jianping Lan,1 Lai Jin1
1Cancer Center, Department of Hematology, Zhejiang Provincial People’s Hospital (Affiliated People’s Hospital, Hangzhou Medical College), Hangzhou, Zhejiang, 310014, People’s Republic of China; 2Cancer Center, Department of Pathology, Zhejiang Provincial People’s Hospital (Affiliated People’s Hospital, Hangzhou Medical College), Hangzhou, Zhejiang, 310014, People’s Republic of China; 3Laboratory Medicine Center, Department of Clinical Laboratory, Zhejiang Provincial People’s Hospital (Affiliated People’s Hospital, Hangzhou Medical College), Hangzhou, Zhejiang, 310014, People’s Republic of China; 4Cancer Center, Key Laboratory of Tumor Molecular Diagnosis and Individualized Medicine of Zhejiang Province, Zhejiang Provincial People’s Hospital (Affiliated People’s Hospital, Hangzhou Medical College), Hangzhou, Zhejiang, 310014, People’s Republic of China; 5Department of Hematology, Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Hangzhou, Zhejiang, 310022, People’s Republic of China
Correspondence: Lai Jin; Jianping Lan, Cancer Center, Department of Hematology, Zhejiang Provincial People’s Hospital (Affiliated People’s Hospital, Hangzhou Medical College), No. 158 Shangtang Road, Hangzhou, Zhejiang, 310014, People’s Republic of China, Tel +86-571-85893497, Email [email protected]; [email protected]
Background: Mast cell leukemia (MCL) is a highly life-threatening and extremely rare subtype of systemic mastocytosis (SM). MCL often genetically contains one or more somatic mutations, particularly activating mutations of KIT. This study reported on an acute MCL patient who had a rare phenotype and genetic mutants with a history of primary malignant mediastinal germ cell tumor (GCT).
Case Presentation: A 30-year-old Asian male patient who underwent two rounds of surgery and chemotherapy with a history of primary mediastinal GCT (PM-GCTs) was admitted to our hospital due to persistent chest pain and severe fatigue. The diagnosis of acute MCL was confirmed via morphology analysis and chemical staining of marrow aspirate, as well as via marrow biopsy, with the addition of C-findings that included splenomegaly and cytopenia. The atypical MCs were phenotypically positive for CD117 and CD9 but weakly positive for CD2 and negative for CD25. Next-generation sequencing of the marrow aspirate identified heterozygous mutations in TP53 P301Qfs*44, FLT3 R973X, SETBP1 N272D, and JAK3 I688F, whereas mutations in KIT were not found. Although the initial therapy of corticosteroids, ruxolitinib, and dasatinib-based regimens was effective, he died of acute respiratory distress syndrome after the first cycle of chemotherapy with cladribine and cytarabine. The patient’s survival time was 2.4 months after the initial presentation of MCL.
Conclusion: In this case, MCL preceded by PM-GCTs had similar clinical symptoms and morphological manifestations but distinctly different genetic profiles than primary MCL. The characteristic morphology of MCL provides the most pivotal evidence that led our diagnosis in the correct direction. A competing hypothesis is that there is a common embryonal cancer stem cell between PM-GCTs and secondary MCL, and the latter is gradually developed in the context of additional “driver mutations”.
Keywords: case report, mast cell leukemia, germ cell tumor, primary mediastinal, KIT, TP53
Introduction
Mastocytosis is a malignant disease characterized by the clonal expansion and infiltration of mast cells (MCs) in the skin, marrow, and other organs. Mastocytosis is clinically divided into cutaneous mastocytosis, systemic mastocytosis (SM), and localized MC tumors. The first form usually appears in childhood and has a favorable prognosis, and the latter two forms frequently develop in adulthood.1 SM is a rare subtype characterized by multifocal infiltration of MCs in the bone marrow and other organs.
The diagnostic criteria of SM are classified into major and minor criteria. The major criteria indicate multifocal dense infiltrates of MCs (≥ 15 MCs in aggregates) in BM biopsies and/or in other extracutaneous organs. The minor criteria include a). > 25% of atypical MCs are detected on BM smears or are spindle-shaped in MC infiltrates that are detected on other organs; b). an activating point mutation at codon 816 of KIT in the marrow or another extracutaneous organ; c). MCs in the marrow, blood, or another extracutaneous organ exhibit CD2 and/or CD25; and d). the baseline serum tryptase level is > 20 ng/mL (unless there is an associated myeloid neoplasm, in which case item d is not valid). SM is confirmed with the major criteria and at least one minor criterion, or more than three minor criteria.1,2
MCL is an extremely rare subtype of SM, accounting for less than 0.5% of SM. It can be divided into de novo or secondary to earlier mastocytosis, and the ratio of the two subtypes is approximately 3:1. MCL is fatal because of its systemic nature and resistance to current therapeutic agents.3 The diagnostic criteria for MCL are as follows: 1) the establishment of SM diagnosis; 2) neoplastic infiltration by atypical MCs in BM biopsy; and 3) the presence of atypical MCs in marrow with or without other internal organs (more than 20% of BM nucleated cells).2 Traditionally, MCL includes an aleukemic variant (in most cases) when the percentage of atypical MCs < 10% of peripheral blood mononuclear cells (PBMCs) and a classical/leukemic variant when the percentage ≥ 10%.4 MCL can be further subdivided into chronic versus acute MCL types, and the latter type follows a more aggressive course, with the presence of ≥ 1 C-findings (including cytopenia, hepatomegaly, splenomegaly, and gastrointestinal or skeletal involvement).5 Neoplastic MCs usually express KIT (CD117), tryptase, and CD25, with or without coexpression of CD2. Genetically, they often contain one or more somatic mutations, represented by activating mutations of KIT.
Germ cell tumors (GCTs) are the most common cancer among men in adolescents and young adults. Most patients have a favorable prognosis with effective chemotherapy, whereas certain subtypes like primary mediastinal GCTs (PM-GCTs) have high incidences of secondary malignant neoplasms, wherein the hematologic malignancies particularly myeloid neoplasms are the most frequent concomitant neoplasms.6,7 This study reported an acute MCL case of a young male patient who had a rare phenotype and genetic mutants with a history of primary malignant mediastinal GCT.
Case Presentation
A 20-year-old Asian male patient, who had no family history of tumor and genetic diseases, presented with a painful anterior mediastinal mass in March 2010. The detection of tumor markers showed elevated serum alpha-fetoprotein (AFP) at 327.6 ng/mL (normal range, 0–10.0ng/mL) and human chorionic gonadotropin (β-hCG) at 116.1mIU/mL (normal range, 0–5.0 mIU/mL). He underwent a complete surgical tumorectomy, and the histopathology showed a malignant mixed germ cell tumor encompassing seminoma and immature teratoma tumor components (Figure 1A-F). After this resection, the patient recovered well, with significantly decreased AFP (85.4 ng/mL) and normal β-HCG (1.2 mIU/mL). He was then consolidated with 4 cycles of cisplatin-based chemotherapy concurrent with three-dimensional conformal radiotherapy. A total absorbed dose of 3060 centigrays (cGy) in 17 fractions of 180Gy each was administered to the primary mediastinal lesion, middle and superior mediastinum, and double supraclavicular area. However, at the end of 2014, he experienced the recurrence of GCT with a metastatic lesion located on the posterior basal segment of the lower lobe of the left lung. He then underwent pneumoresection, and the pathological result revealed a single immature teratoma (Figure 1G-I). He subsequently received 6 cycles of consolidated chemotherapy, including vindesine, ifosfamide, and cisplatin. Afterward, the patient remained in a stable remission condition.
 |
Figure 1 The morphology of hematoxylin-eosin staining mediastinal mass, including primary (A–F) and metastatic GCT (G–I). The primary mass contained seminoma (10%) and immature teratoma (90%) components. The seminoma cell was pleomorphic with abundant cytoplasm and distributed in clusters ((C–D), 200× and 400×). The components of immature teratoma mainly included immature neural tube ((A–B), 200× and 400×), cartilage ((E), 200×), and glands ((F), 400×). The metastatic mass was a mainly extensive deposition of osteoid connective tissue, surrounded by atypical epithelioid cells ((G), 200×, and (H–I), 400×). The neoplastic cells, featured with increased nuclear-to-cytoplasmic ratio and abnormal chromatin distribution, were arranged in irregular lacunar (H) and adenoid structures (I). The red or black triangles pointed to the corresponding structure. GCT, germ cell tumor. |
At the beginning of 2020, he was urgently admitted to our hospital with 1 day of persistent chest pain and severe fatigue. Clinical symptoms showed a low-grade fever, flushed skin, and splenomegaly. A complete blood cell count showed anemia (hemoglobin: 8.2 g/dL), leukocytosis (15.66*109/L), and thrombocytopenia (38*109/L). Coagulation tests demonstrated a higher D-dimer value (2340 µg/L) and a slightly prolonged activated partial thromboplastin time (37.4 s). The routine chemical analysis detected a high level of lactate dehydrogenase (LDH) (878 U/L) in the peripheral blood. The results of computed tomography scan and transabdominal ultrasound revealed hepatosplenomegaly and seroperitoneum. Positron emission tomography/computed tomography (PET/CT) detected a diffuse increase in systemic bone metabolism and hepatosplenomegaly with slightly increased FDG metabolism, which was consistent with the manifestation of blood system diseases. There was no evidence indicating the second recurrence of GCT.
A peripheral blood smear revealed the existence of 22% MC-like immature cells. BM aspiration showed substantial infiltration (67% of all nucleated cells) from morphologically heterogeneous atypical circles to oblong cells containing abundant cytoplasm with metachromatic coarse granules. Chemical staining results showed that atypical cells were negative for nonspecific esterase (NSE), peroxidase (POX), and naphthol AS-D chloroacetate esterase staining (NAS-DCE), except for toluidine blue staining (Figure 2). An analysis of the BM aspirate via multiparameter flow cytometry revealed that abnormal cells were positive for CD45, CD117, CD13, CD33, and CD9 but weakly positive for CD2 and CD22, and negative for CD16, CD7, κ, λ, CD19, CD38, HLA-DR, CD35, CD65, CD15, CD11b, CD123, CD79a, CD10, CD25, CD34, and MPO (Figure 3). The chromosomal G banding analysis showed a normal karyotype (46, XY). 16 myeloid leukemia-associated fusion genes, including MLL-AF6, CBFB-MYH11, AML1-ETO, PML-RARα, and BCR-ABL, were negative through reverse transcription-polymerase chain reaction (RT-PCR). In addition, fluorescence in situ hybridization detected that RARα rearrangement was negative. Next-generation sequencing (NGS) of the BM aspirate identified stable mutations in TP53 P301Qfs*44, FLT3 R973X, SETBP1 N272D, and JAK3 I688F, whereas KIT mutations were not detected. Their detailed nucleotide alterations and variant allele frequency (VAF) are shown in Table 1. Four mutated genes were sequenced again via Sanger sequencing (Figure 4). The PCR primers that were used are listed in Supplemental Table 1. The bone marrow biopsy revealed atypical cells that were mainly oval and short spindle cells distributed in clusters and eosinophilic by hematoxylin-eosin (HE) staining, accounting for 80% of the nucleated cells. Immunohistochemical staining confirmed positivity for CD117 and negativity for MPO, CD25, and CD34 (Figure 5).
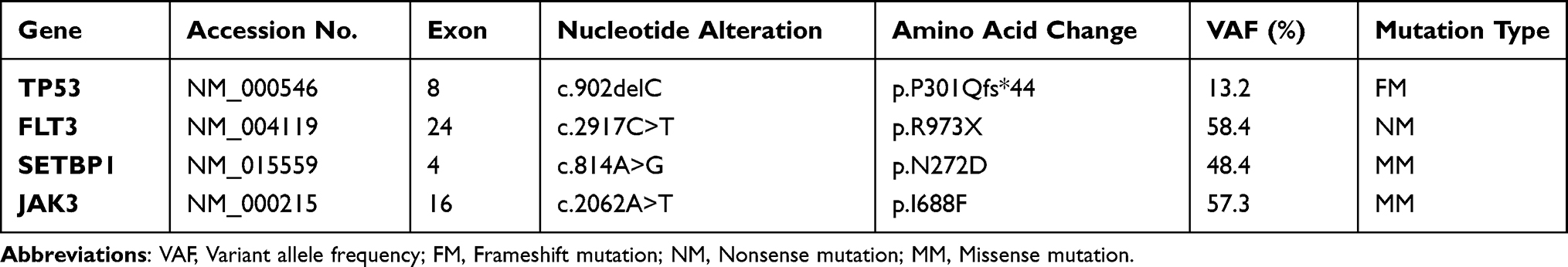 |
Table 1 Mutations Detected in Secondary MCL |
 |
Figure 2 The features of morphology and chemical staining in the case. The morphology of Wright-Giemsa-stained marrow smear (A–C) and peripheral blood smear (D) was shown, 1000×, and the red triangle pointed to atypical mast cells. The results of chemical staining were negative for POX (E), NAS-DCE (F), and NSE (G), and was positive for Toluidine Blue (H–I), 1000×. The red triangles pointed to the positive cells in the corresponding staining. |
 |
Figure 3 Multi-color flow cytometry analysis of cell surface markers of marrow mononuclear cells. Partial markers of the detected were presented. The cluster of cells colored in red indicated the leukemic cells. |
 |
Figure 4 Sanger sequencing of genomic PCR products of bone marrow mononuclear cells. The detailed information of 4 genetic mutations, including TP53, FLT3, SETBP1, and JAK3, was written below the corresponding peak diagram. The black arrow pointed to the mutant base. PCR, polymerase chain reaction. |
 |
Figure 5 Morphologic and immunohistochemical features of the case. Marrow biopsy showed the architecture was diffusely infiltration by clusters of oval and short spindle cells ((A and B), 200× and 400×). The neoplastic cells were positive for CD117 (C), but almost all negative for MPO (D), CD25 (E), and CD34 (F), 200×. |
The patient was first treated with 2 weeks of dexamethasone (10 mg/day for 1 week and then 5 mg/day for 1 week). Surprisingly, his anemia and thrombocytopenia exhibited significant improvements. When confirmed the diagnosis of MCL, the patient was transferred to another hospital for treatment. His following prognosis was further tracked. He was again diagnosed with MCL and treated with the combination therapy of dasatinib (100 mg/day), ruxolitinib (5 mg/bid for the first week and then 10 mg/bid as a maintenance treatment), and dexamethasone (20 mg/day for 1 week and then 10 mg/day for 4 days). After two weeks of treatment, his condition displayed evident remission, particularly a notable decrease in spleen size. Nevertheless, he subsequently died of acute respiratory distress syndrome after undergoing the first cycle of combination chemotherapy with cladribine (9.5 mg d1-5) and cytarabine (1930 mg d1-5). The patient’s survival time was 2.4 months after the initial presentation of MCL.
Discussion
In this case, a male patient was first reported as having acute MCL secondary to a previous PM-GCTs. As a group of neoplasms commonly occurring in the gonads, GCT is a model of curable cancer that often has a satisfactory outcome with cisplatin-based chemotherapy.8 However, primary mediastinal GCTs, which have a predilection for male patients and those with Klinefelter syndrome, tend to have a poor prognosis due to the evolution of neoplasms in other somatic types and resistance to cisplatin, especially when regarding secondary hematologic malignancies (S-HMs), with a median survival time of fewer than 6 months.9 The association of PM-GCTs and S-HMs is an uncommon but well-recognized entity (Table 2).6 The HMs preceded by or concurrent with PM-GCTs are mostly acute myeloid leukemia (AML) (the largest proportion is acute megakaryoblastic leukemia), which share more genetic similarities (characterized by isochromosome 12p [i12p] and/or TP53 mutations) with PM-GCTs rather than primary AML, thus supporting the hypothesis that a common cancer progenitor cell with the capacity to differentiate into germ cells and hematopoietic lineages may evolve into both tumors (Table 2).
 |
Table 2 Summary of Cytogenetic Features of PM-GCTs with Concomitant HMs. The Table Only Summarizes the Reported Patients (Published in PubMed Until April 2022) Whose Molecular Testing Was Performed in at Least One Sample Either Primary PM-GCT or Concomitant HM. Only the Most Relevant/Recurrent Molecular Abnormalities Reported are Shown |
In this case, the diagnosis of acute MCL was based on the implementation of SM with the major criteria (dense infiltration of over 15 aggregated MCs in the marrow) and one minor criterion (>25% atypical MCs in the marrow aspirate and biopsy), with the addition of up to 22% atypical MCs in PBMCs and C-findings that included splenomegaly and cytopenia. In addition, the typical morphology and positive results of chemical staining with toluidine blue played decisive roles in the differential diagnosis of MCs.
The patient’s leukemic cells were heterogeneous in morphology, varying widely in size and shape with a rare phenotype and genetic manifestations. The neoplasm cells were phenotypically positive for CD117 and CD9 but weakly positive for CD2 and negative for CD25. Previous reports of SM have revealed that the expression levels of CD2 and CD25 are gradually decreased, along with malignant progression.10,11 It has been reported that 38% of MCL cases have a double-negative CD2/CD25 immunophenotype, and the positive coexpression of CD2 and CD25 has a significantly higher proportion in MCL patients with KIT D816V than in those with no missense variant (66% vs 25%, respectively).3 Furthermore, the secondary MCL in the case had no representative genetic aberration of i(12p), which was identical with a previous report where two MCLs cases associated with PM-GCTs harbored the normal karyotype.9
MCL has typical genetic characteristics. The proportion of gain-of-function somatic mutations in KIT, including KIT D816V and other KIT mutants at exons 8, 9, 10, 11, 13, and 17, is approximately 90%.5 In addition, KIT D816V may be accompanied by additional genetic variants that jointly contribute to malignant expansion. Patients with additional aberrations have shorter overall survival (OS) than those with KIT D816V alone. A multivariable risk analysis of MCL patients indicates that mutations in SRSF2, ASXL1, or RUNX1 (S/A/Rpos), which have been revealed to have oncogenic functions in myeloid malignancies, are the only dependent risk factors. S/A/Rpos patients with MCL have a more aggressive phenotype, a lower response rate, more resistance to disparate treatment modalities, and poorer survival than S/A/Rneg MCL.12 However, secondary MCL exhibited a classical genetic aberrance of TP53 frameshift mutation as the patient developed in the setting of PM-GCTs, while also exhibiting three infrequent genetic mutations (Table 1).
In this case, the mutation locus of TP53 P301Qfs*44 is adjacent to the DNA binding domain (DBD, codons 94–297), a hot-spot mutation region. TP53 mutations in AML are considered independent high-risk factors with a poor prognosis.13 Consistently, TP53 mutation is regarded as one of the typical genetic characteristics in S-HM preceded by mediastinal dysgerminoma (Table 2). Besides, a previous study reported that an ASM patient had a history of ovarian dysgerminoma, and TP53 developed a somatic nonsense mutation in the dysgerminoma and bone marrow of the ASM period.14 FLT3 is an oncogene of the receptor tyrosine kinase family involved in the proliferation and differentiation of hematopoiesis. Activating mutations represented by FLT3-ITD are closely related to tumorigenesis, especially AML. As previously reported for the exclusive relationship of FLT3-ITD and TP53 mutations in AML,13 a rare nonsense mutation of FLT3 R973X occurred in our case, corresponding to the inactivating mutation of TP53.
SETBP1 mutations, first identified in Schinzel-Giedion syndrome, are considered biomarkers of myelodysplasia/myeloproliferative neoplasm overlap syndrome. Additionally, SETBP1 hotspot mutations within a conserved 11-nucleotide region (amino acids 868–871) are often detected in secondary AML and chronic myelomonocytic leukemia. It has further been reported that only hotspot mutations can induce the resistance and poor prognosis of myeloid neoplasms.15 The role of SETBP1 nonhotspot mutations is currently elusive, such as what occurred with SETBP1 N272D in our case. Along with SETBP1 mutations, a nonreceptor tyrosine kinase JAK3 mutation is identified as the secondary mutation in juvenile myelomonocytic leukemia (JMML), whereas the latter is involved not in the initiation but the progression of JMML, thus indicating a poor prognosis.16 JAK3 mutation is a driver mutation frequently reported in T lineage acute lymphoblastic leukemia. Recurrent JAK3 V722I is reported in malignant struma ovarii, which is a specific ovarian teratoma.17 Consistent with JAK3 V722I, the mutation site of JAK3 I688F is located in the same protein domain (protein kinase 1), thus potentially harboring a similar function.
When regarding the case, we propose the following hypothesis. First, the mutation burden caused by chemotherapies with platinum-based drugs and radiotherapy in the primary solid tumor influences the evolution of hematopoietic cells and further progression to secondary leukemia. Second, the leukemic conditions originating from a PM-GCTs progenitor cell are capable of undergoing hematopoietic differentiation into the subsequent hematological malignancy, which is consistent with an earlier study.18 Finally, a competing hypothesis is that there is a shared precursor in the germ cell lineage harboring the p53 pathway alteration for the primary malignant GCT and the secondary MCL, and the latter is gradually developed in the setting of additional “driver mutations”, which is supported by the latest evidence.7 Future studies to explore the potential germline genetic predisposition for the development of hematologic neoplasms in the setting of PM-GCTs are prospective.
There were some defects in this case. First, due to clinical laboratory limitations, the concentration of serum tryptase was not examined to investigate the correlation between tryptase expression and leukemic development. In addition, the patient’s relatives refused to perform gene sequencing, and specimens in the period of PM-GCTs were also not examined for genetic alterations. Thus, it is difficult to determine whether these gene mutations are germline or somatic.
Conclusion
In this case, acute MCL preceded by malignant mediastinal GCT had similar clinical symptoms and morphological manifestations but distinctly different genetic profiles than primary MCL. The characteristic morphology of MCL provides the most pivotal evidence that led our diagnosis in the correct direction. A competing hypothesis is that there is a common embryonal cancer stem cell between the PM-GCTs and the secondary MCL, and the latter is gradually developed in the setting of additional “driver mutations”. Future studies to explore the potential germline genetic predisposition for the development of hematologic neoplasms in the setting of PM-GCTs are prospective.
Abbreviations
MCL, Mast cell leukemia; MCs, Mast cells; SM, Systemic mastocytosis; GCT, Germ cell tumor; AML, Acute myeloid leukemia; PBMCs, Peripheral blood mononuclear cells; PCR, Polymerase chain reaction; LDH, Lactate dehydrogenase; PET/CT, Positron emission tomography/computed tomography; NSE, Nonspecific esterase; POX, Peroxidase; NAS-DCE, Naphthol AS-D chloroacetate esterase staining; PM-GCTs, Primary mediastinal GCTs; S-HMs, Secondary hematologic malignancies; AFP, Alpha-fetoprotein; β-hCG, Human chorionic gonadotropin; DBD, DNA binding domain; HE, Hematoxylin-eosin; OS, Overall survival.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Ethical Statement
The ethical approval and documentation for a case report were waived by the Ethical Committee of the Zhejiang Provincial People’s Hospital.
Consent for Publication
Written informed consent was obtained from the patient’s parent for publication of this Case report and any accompanying images.
Acknowledgments
Thanks to the technical staff of Tianjin Concorde Huamei Medical Diagnostic Technology Co., LTD for their professional support in the NGS and Sanger sequencing. We also thank Proofine English Studio and American Journal Experts for editing the manuscript. All authors have read the CARE Checklist (https://www.care-statement.org/checklist), and the manuscript was prepared and revised according to the CARE Checklist (2013 version).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to take responsibility and be accountable for the contents of the article.
Funding
This study was supported by the Zhejiang Provincial Natural Science Foundation of China (LY17H080008) and a Project of the Health Department of Zhejiang Province of China (2021KY517).
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Peter Valent CA, Dean D. Metcalfe. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420–1427.
2. Pardanani A. Systemic mastocytosis in adults: 2019 update on diagnosis, risk stratification and management. Am J Hematol. 2019;94(3):363–377.
3. Georgin-Lavialle S, Lhermitte L, Dubreuil P, Chandesris MO, Hermine O, Damaj G. Mast cell leukemia. Blood. 2013;121(8):1285–1295.
4. Preetesh Jain SW, Patel KP, Sarwari N, Cortes J, Kantarjian H, Verstovsek S. Mast cell leukemia (MCL): clinico-pathologic and molecular features and survival outcome. Leuk Res. 2017;59:105–109.
5. Leguit R, Hebeda K, Kremer M, et al. The Spectrum of Aggressive Mastocytosis: a Workshop Report and Literature Review. Pathobiology. 2020;87(1):2–19.
6. Urbini M, Schepisi G, Bleve S, et al. Primary Mediastinal and Testicular Germ Cell Tumors in Adolescents and Adults: a Comparison of Genomic Alterations and Clinical Implications. Cancers. 2021;13:20.
7. Taylor J, Donoghue M, Ho C, et al. Germ cell tumors and associated hematologic malignancies evolve from a common shared precursor. J Clin Invest. 2020;130(12):6668–6676.
8. Cheng L, Albers P, Berney D, et al. Testicular cancer. Nat Rev Dis Primers. 2018;4(1):29.
9. Hartmann J, Nichols C, Droz J, et al. Hematologic disorders associated with primary mediastinal nonseminomatous germ cell tumors. J Natl Cancer Inst. 2000;92(1):54–61.
10. Escribano L, Orfao A, Díaz-Agustin B, et al. Indolent systemic mast cell disease in adults: immunophenotypic characterization of bone marrow mast cells and its diagnostic implications. Blood. 1998;91(8):2731–2736.
11. Valent P, Sotlar K, Sperr WR, et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): a consensus proposal. Ann Oncol. 2014;25(9):1691–1700.
12. Jawhar M, Schwaab J, Meggendorfer M, et al. The clinical and molecular diversity of mast cell leukemia with or without associated hematologic neoplasm. Haematologica. 2017;102(6):1035–1043.
13. Terada K, Yamaguchi H, Ueki T, et al. Full-length mutation search of the TP53 gene in acute myeloid leukemia has increased significance as a prognostic factor. Ann Hematol. 2018;97(1):51–61.
14. Tsutsumi M, Miura H, Inagaki H, et al. An aggressive systemic mastocytosis preceded by ovarian dysgerminoma. BMC Cancer. 2020;20(1):1162.
15. Winkelmann N, Schäfer V, Rinke J, et al. Only SETBP1 hotspot mutations are associated with refractory disease in myeloid malignancies. J Cancer Res Clin Oncol. 2017;143(12):2511–2519.
16. Wakamatsu M, Okuno Y, Murakami N, et al. Detection of subclonal SETBP1 and JAK3 mutations in juvenile myelomonocytic leukemia using droplet digital PCR. Leukemia. 2021;35(1):259–263.
17. Poli R, Scatolini M, Grosso E, et al. Malignant struma ovarii: next-generation sequencing of six cases revealed Nras, Braf, and Jak3 mutations. Endocrine. 2021;71(1):216–224.
18. Lee KC. Hematopoietic precursor cells within the yolk sac tumor component are the source of secondary hematopoietic malignancies in patients with mediastinal germ cell tumors. Cancer. 1994;73(5):1535–1536.
19. Sowithayasakul P, Sinlapamongkolkul P, Treetipsatit J, et al. Hematologic Malignancies Associated With Mediastinal Germ Cell Tumors: 10 Years’ Experience at Thailand’s National Pediatric Tertiary Referral Center. J Pediatr Hematol Oncol. 2018;40(6):450–455.
20. Vlasveld LT, Splinter TA, Hagemeijer A, Van Lom K, Löwenberg B. Acute myeloid leukaemia with +i(12p) shortly after treatment of mediastinal germ cell tumour. Br J Haematol. 1994;88(1):196–198.
21. Ladanyi M, Samaniego F, Reuter VE, et al. Cytogenetic and immunohistochemical evidence for the germ cell origin of a subset of acute leukemias associated with mediastinal germ cell tumors. J Natl Cancer Inst. 1990;82(3):221–227.
22. Chaganti RS, Ladanyi M, Samaniego F, et al. Leukemic differentiation of a mediastinal germ cell tumor. Genes Chromosomes Cancer. 1989;1(1):83–87.
23. Nichols CR, Hoffman R, Einhorn LH, Williams SD, Wheeler LA, Garnick MB. Hematologic malignancies associated with primary mediastinal germ-cell tumors. Ann Intern Med. 1985;102(5):603–609.
24. Lu C, Riedell P, Miller CA, et al. A common founding clone with TP53 and PTEN mutations gives rise to a concurrent germ cell tumor and acute megakaryoblastic leukemia. Cold Spring Harbor Mol Case Studies. 2016;2(1):a000687.
25. Leonard JT, Raess PW, Dunlap J, Hayes-Lattin B, Tyner JW, Traer E. Functional and genetic screening of acute myeloid leukemia associated with mediastinal germ cell tumor identifies MEK inhibitor as an active clinical agent. J Hematol Oncol. 2016;9:31.
26. Oshrine BR, Olsen MN, Heneghan M, et al. Acquired isochromosome 12p, somatic TP53 and PTEN mutations, and a germline ATM variant in an adolescent male with concurrent acute megakaryoblastic leukemia and mediastinal germ cell tumor. Cancer Genet. 2014;207(4):153–159.
27. Akizuki K, Sekine M, Kogure Y, et al. TP53 and PTEN mutations were shared in concurrent germ cell tumor and acute megakaryoblastic leukemia. BMC Cancer. 2020;20(1):5.
28. Amra N, Zarate LV, Punia JN, et al. Mediastinal Germ Cell Tumor and Acute Megakaryoblastic Leukemia With Co-occurring KRAS Mutation and Complex Cytogenetics. Pediatric Dev Pathol. 2020;23(6):461–466.
29. Vaishampayan U, Dan ME, Hussain M. Unusual hematologic malignancies. Case 2. Mediastinal germ cell tumor, malignant histiocytosis, and acute leukemia. J Clin Oncol. 2002;20(17):3739–3742.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.