Back to Journals » Journal of Inflammation Research » Volume 14
Acute Exposure to the Food-Borne Pathogen Listeria monocytogenes Does Not Induce α-Synuclein Pathology in the Colonic ENS of Nonhuman Primates
Authors Mancinelli AM, Vichich JM, Zinnen AD, Hugon AM, Bondarenko V, Metzger JM, Simmons HA, Golos TG, Emborg ME ![]()
Received 3 September 2021
Accepted for publication 1 December 2021
Published 22 December 2021 Volume 2021:14 Pages 7265—7279
DOI https://doi.org/10.2147/JIR.S337549
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Anthony M Mancinelli,1 Jonathan M Vichich,1 Alexandra D Zinnen,1 Anna Marie Hugon,2 Viktoriya Bondarenko,1 Jeanette M Metzger,1 Heather A Simmons,1 Thaddeus G Golos,1,3,4 Marina E Emborg1,5
1Wisconsin National Primate Research Center, University of Wisconsin-Madison, Madison, WI, USA; 2Department of Pathology and Laboratory Medicine, University of Wisconsin-Madison, Madison, WI, USA; 3Department of Comparative Biosciences, University of Wisconsin-Madison, Madison, WI, USA; 4Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, WI, USA; 5Department of Medical Physics, University of Wisconsin-Madison, Madison, WI, USA
Correspondence: Marina E Emborg Email [email protected]
Introduction: Gastrointestinal (GI) inflammation elicited by environmental factors is proposed to trigger Parkinson’s disease (PD) by stimulating accumulation of pathological α-synuclein (α-syn) in the enteric nervous system (ENS), which then propagates to the central nervous system via the vagus nerve. The goal of this study was to model, in nonhuman primates, an acute exposure to a common food-borne pathogen in order to assess whether the related acute GI inflammation could initiate persistent α-syn pathology in the ENS, ultimately leading to PD.
Methods: Adult female cynomolgus macaques were inoculated by oral gavage with 1× 108 colony-forming units (CFUs) Listeria monocytogenes (LM, n=10) or vehicle (mock, n=3) and euthanized 2 weeks later. Evaluations included clinical monitoring, blood and fecal shedding of LM, and postmortem pathological analysis of colonic and cecal tissues.
Results: LM inoculation of healthy adult cynomolgus macaques induced minimal to mild clinical signs of infection; LM shedding in feces was not seen in any of the animals nor was bacteremia detected. Colitis varied from none to moderate in LM-treated subjects and none to minimal in mock-treated subjects. Expression of inflammatory markers (HLA-DR, CD3, CD20), oxidative stress (8-OHDG), α-syn, and phosphorylated-α-syn in the enteric ganglia was not significantly different between treatment groups.
Discussion: Our results demonstrate that cynomolgus macaques orally inoculated with LM present with a clinical response that resembles human LM exposure. They also suggest that acute exposure to food-borne pathogens is not sufficient to induce significant and persistent α-syn changes in healthy adult female subjects. Based on the results of this limited experimental setting, we propose that, if LM has a role in PD pathology, other underlying factors or conditions, such as male sex, inflammatory bowel disease, exposure to toxins, dysbiosis, and/or aging, are needed to be present.
Keywords: Parkinson’s disease, alpha synuclein, nonhuman primate, listeria, inflammation, enteric nervous system
Introduction
Alpha-synuclein (α-syn) is a presynaptic protein ubiquitously expressed in neurons of the central, peripheral, and enteric nervous systems (ENS).1 Under normal conditions, soluble monomeric α-syn is found at nerve terminals where it binds SNARE proteins to assist with neurotransmitter vesicle release into the synaptic cleft.2 In pathological conditions like Parkinson’s disease (PD), α-syn becomes phosphorylated, misfolds, and forms aggregates termed Lewy bodies which affect proper protein function. Inflammation seems to facilitate α-syn phosphorylation and aggregation.3–5
PD is a progressive neurodegenerative disorder of unknown etiology that affects 1% of the population over 60 years old. It is diagnosed by the presence of typical motor symptoms associated with the severe loss of dopaminergic neurons in the substantia nigra pars compacta.6 Yet, current epidemiological evidence indicates that PD starts years earlier, heralded by symptoms such as constipation and loss of sense of smell.6
Based on PD prodromal clinical features and the patterns of distribution of pathological α-syn in the nervous system, Braak et al hypothesized that PD etiology could be due to ingestion of an environmental trigger.7–9 The exposure would lead to accumulation of pathological α-syn in neurons of the enteric nervous system (ENS), followed by propagation of the misfolded protein into the central nervous system (CNS) via the vagus nerve.7–9 Studies in northern European populations showed that truncal vagotomies, but not superselective vagotomies, lower the risk of PD, suggesting a relationship between the gastrointestinal (GI) system and PD onset.10–12 In rodents, inoculation of α-syn preformed fibrils (PFFs) through different routes of administration led to transneuronal propagation of misfolded α-syn across the neural axis.13–17 Furthermore, truncal vagotomy in mice injected into the duodenal and pyloric muscularis layer with PFFs prevented the spread of pathological α-syn from the ENS to the CNS.14 Taken as a whole, these reports confirmed the hypothesis that misfolded α-syn can propagate, yet they do not explain which naturally occurring event triggers α-syn changes in the ENS.
Colonic inflammation seems to play a role in PD pathophysiology.18 Patients with a history of inflammatory bowel disease (IBD) have an estimated 28–30% risk of developing PD. Furthermore, PD patients commonly have inflammation in the ascending colon, as well as elevated immune markers in feces compared to healthy controls.19,20 The appendix, an organ known to stimulate development of gut-associated lymphoid tissue (GALT), is rich in both lymphoid tissue and α-syn making it a potential “incubator” of α-syn pathology.21 Data compiled from the Swedish National Patient Registry and the Parkinson’s Progression Markers Initiative database related appendectomy to a decreased risk of PD in rural areas and delayed onset of PD, respectively.22 In this context, it is sensible to consider that exposure by ingestion to a common environmental pathogen may indirectly cause PD by inducing GI inflammation, thus facilitating α-syn misfolding and aggregation in the ENS.
Listeria monocytogenes (LM) is an intracellular Gram+ food-borne bacterium that has been reported in a wide variety of settings and food products and can replicate at a wide range of temperatures (3–42°C).23 In healthy individuals, LM infection (listeriosis) is self-limiting and results only in diarrhea and fever. However, in the elderly and immunocompromised individuals, LM can lead to encephalitis. LM in pregnant women can lead to spontaneous abortion, still birth, or neonatal infection.24 According to the US Centers for Disease Control, approximately 1600 patients present in clinic with listeriosis each year and 260 of these cases prove fatal.24
LM enters the host through the intestinal mucosa by direct invasion of enterocytes25 or by translocation across the microfold cells of the Peyer’s patches.26 LM has also evolved to survive in macrophages after phagocytic uptake, allowing a Trojan horse-like spread of LM that includes crossing of the blood–brain barrier.27,28 Listeriolysin-O is a hemolysin that allows LM to escape from macrophages, lyse host cell walls, and spread between neighboring epithelial cells.29–31 This cell-to-cell movement evades the humoral immune response by minimizing LM exposure to the extracellular milieu.
Inflammatory responses can be characterized as acute or chronic, with specific time-dependent immune cell infiltrates releasing different factors that impact the cellular environment.32,33 Acute inflammation is typically self-limiting and composed of neutrophils, followed by monocytic cells, which remove cellular debris and apoptotic neutrophils within ~24 to 48 hours after challenge.33,34 Chronic inflammation is a persistent immune response of mononuclear cells (macrophages, and lymphocytes) that can lead to local tissue damage.32 The innate immune response to LM is acute. It commences when pathogen-associated molecular patterns (PAMPs) on the surface of the bacterium engage pattern recognition receptors (PRRs) on intestinal epithelial cells.35 Stimulation of PRRs leads to increased IL-1R, associated kinases and downstream activation of transcription factors NF-κB and interferon regulatory factor 3 (IRF-3). IRF-3 and NF-κB induce proinflammatory cytokine release and type I interferon response, which bolsters a robust acute inflammatory response in the GI tract.36 Clearance of the LM relies on mononuclear CD4, CD8 and NK T-cells recruited in the adaptive immune response.37
Although the evidence linking chronic GI inflammation and increase risk of PD is increasing, whether PD etiology could be related to the ingestion of an environmental pathogen that causes an immune response, albeit acute, has not yet been evaluated. The wide spread and typically undetected exposure of the human population to LM led to our hypothesis that food-borne pathogens such as LM could be an environmental trigger of GI inflammation that initiates persistent α-syn pathology in the ENS and ultimately lead to PD. To test this hypothesis, we took advantage of an ongoing project at the Wisconsin National Primate Research Center (WNPRC) assessing the impact of listeriosis in the reproductive system (to be published at a later date) to model the typical acute LM exposure. Healthy non-pregnant adult female cynomolgus macaques were inoculated with LM, euthanized 2 weeks later, and their GI tracts assessed for colonic inflammation, α-syn, and phosphorylated α-syn (p- α-syn) expression in the ENS.
Materials and Methods
Ethics Statement
This experiment was performed in strict accordance with the recommendations of the National Research Council Guide for Care and Use of Laboratory Animals and at the AAALAC-accredited WNPRC of the University of Wisconsin–Madison. The colonic tissues used in this study were obtained from monkeys originally assigned to and euthanized under the University of Wisconsin–Madison Institutional Animal Care and Use Committee (IACUC) approved protocol G005061. Every effort was made to minimize the number of primates used and to limit animal distress.
The human brain tissues from a Parkinson’s disease patient used as positive controls for the immunostainings were obtained from the Wisconsin Alzheimer’s Disease Research Center Brain Bank following documented review and approval from the Institutional Review Board (IRB) and with written informed consent by the patient, as prescribed by the Declaration of Helsinki.
Subjects
Thirteen adult female cynomolgus macaques (Macaca fascicularis) were used in this study (Table 1). Animals were housed in group 3 or group 4 enclosures in accordance with the Animal Welfare Act and its regulations and the Guide for the Care and Use of Laboratory Animals. All animals were monitored twice daily by an animal researcher or veterinary technician for evidence of disease or injury. Body weight was monitored to ensure that all animals remained in properly sized cages. Animals were fed commercial nonhuman primate chow (2050 Teklad Global 20% Protein Primate Diet, Harlan Laboratories, Madison, WI) twice daily, supplemented with fruits or vegetables and a variety of environmental enrichment.
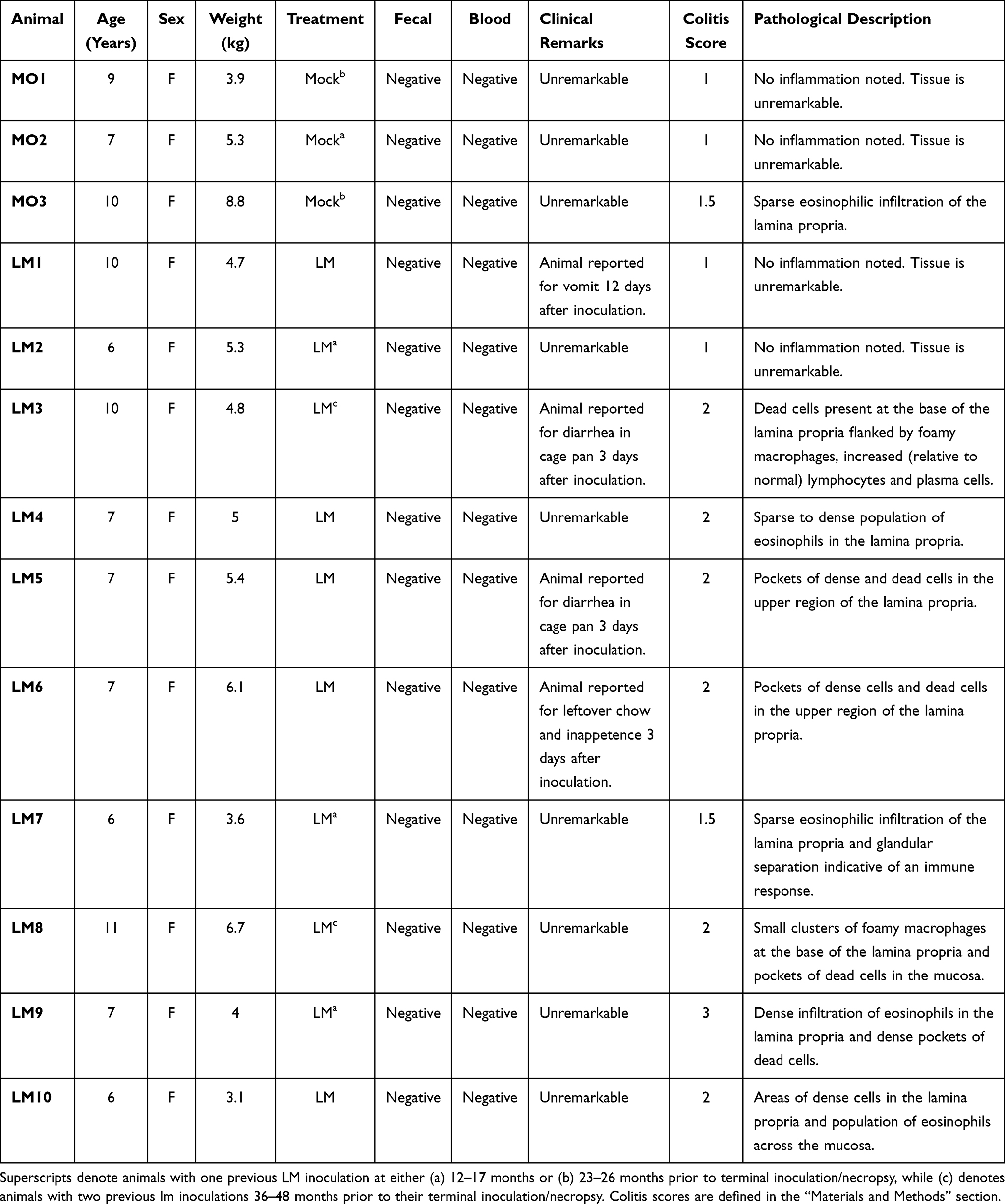 |
Table 1 Information per Study Subject on Treatment, Fecal and Blood Shedding, Colitis Score, Clinical and Pathological Remarks. |
Listeria Inoculation
Listeria monocytogenes (LM; log-phase cells of strain LM2203) were cultured at 37◦C in Tryptic Soy Broth (Becton Dickinson, Sparks, MD). The LM strain was isolated from a cluster of listeriosis in pregnant women that led to fetal demise.38 Each inoculum containing 1×108 colony forming units (CFUs) of LM was dissolved in 10mL whipping cream and delivered via oral gavage through a soft intragastric feeding tube under sedation (n=10), as previously described.38,39 Control inoculations (mock) consisted of 10mL whipping cream alone with no LM (n=3). Eight of the 13 animals were previously inoculated with LM following the same protocol. Those inoculations occurred 12–48 months prior to their terminal inoculation series. The timeline and number of previous inoculations per animal are detailed in Table 1.
To assure a live dose of LM was given to the subjects, 500 μL of the 10mL whipping cream inoculum was diluted in phosphate-buffered saline (PBS; Catalog #P5368, Sigma-Aldrich, St. Louis, MO), plated on Trypticase soy agar with 5% sheep blood (Becton Dickinson, Sparks, MD), and incubated at 37◦C. Subsequent growth on the agar plates was quantified to confirm the dose of live LM inoculum delivered.38
Fecal Shedding and Bacteremia
LM fecal shedding and bacteremia were evaluated during the 14-day period following LM inoculation, as previously described.38,39
Fecal samples were obtained daily, starting on the day of inoculation prior to the first dose of inoculum being given and ending on the day of necropsy. Samples were collected from cage pans. Serial fecal dilutions in PBS were plated on BBL blood agar plates and incubated at 37◦C for 48 hours. The number of colonies was quantified using NIH ImageJ colony-counting software at both 24 hours and 48 hours after being plated.
Whole peripheral blood samples were collected every 2–3 days40 and processed by the Clinical Pathology Laboratory within the School of Veterinary Medicine, at the University of Wisconsin–Madison. Bactec Peds Plus/F blood culture bottles were aseptically inoculated with 3 mL per bottle. The samples were then incubated at 35°C in a BD Bactec 9050 blood culture system (Becton Dickinson Diagnostic Systems, Sparks, MD) until a positive signal was observed or for a maximum of 5 days.
Tissue Collection and Processing
Fourteen days after LM inoculation the monkeys were anesthetized with ketamine hydrochloride (10–15 mg/kg, iv) and euthanized with pentobarbital sodium (minimum 25 mg/kg, iv).
Samples of proximal colon and cecum were collected, rinsed to remove fecal material from the mucosa, fixed in 4% paraformaldehyde for 24–72 hours, rinsed in PBS, and stored in 70% ethanol until processed and embedded in paraffin. Sections were cut at 5 μm on a rotary microtome and mounted on positively charged slides.
A representative section of colon and cecum was stained with hematoxylin and eosin (HE) to assess microanatomy and pathology in each animal.
Immunohistochemistry
Proximal colon sections were immunostained against human leukocyte antigen DR (HLA-DR), cluster of differentiation 3 (CD3), cluster of differentiation 20 (CD20), 8-hydroxy-2ʹ-deoxyguanosine (8OHdG), protein gene product 9.5 (PGP9.5), α-syn and α-syn phosphorylated at serine 129 (p-α-syn) (Table 2) with antibodies optimal for nonhuman primates (NHPs), following previously validated methods.41,42 Briefly, slides were deparaffinized in xylene for 30 minutes, rehydrated through ethanols, and treated for heat antigen retrieval in a microwave oven for 6 min at 100% power followed by 3 minutes at 60% power and a 1 hour cool down period. Tissue sections were treated with Super Block solution (ScyTek, Logan, UT, USA, AAA125) and then incubated overnight at room temperature with appropriate primary antibody. The sections were then treated with appropriate biotinylated secondary antibody (1:200) for 1 hour, followed by avidin–biotin complex peroxidase (VECTASTAIN Elite ABC HRP Kit, Vector Laboratories, Burlingame, CA, USA), and visualized with a commercial 3,3′- diaminobenzidine (DAB) kit. All sections were counterstained with hematoxylin (excluding those stained for 8-OHdG), dehydrated, and coverslipped (Cytoseal mounting medium, Thermo Scientific, Waltham, MA, USA).
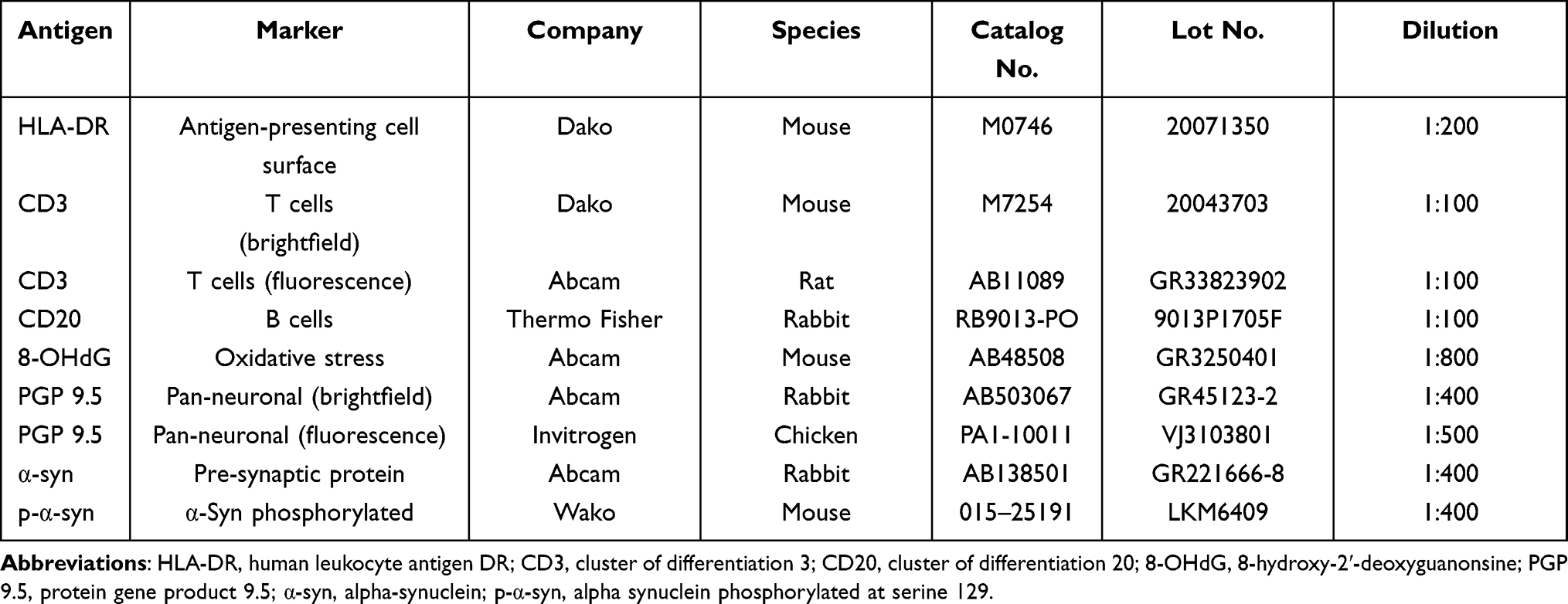 |
Table 2 Primary Antibodies Used for immunohistochemistry and Immunofluorescence |
Negative and positive controls were performed in parallel. Negative controls consisted of omission of primary antibody. Positive controls included healthy macaque colon for PGP9.5, tissue from the subject with highest inflammation score for HLA-DR, CD3, CD20, and 8-OHdG and PD patient brain tissue for α-syn and p-α-syn immunostainings.
Immunofluorescence
Double and triple labeling immunofluorescence stainings were performed to visualize colocalization of α-syn with neuronal PGP9.5 and to assess the spatial relationship between α-syn and the inflammatory cell markers HLA-DR and CD3. Slides were deparaffinized, rehydrated through ethanols, and treated for heat antigen retrieval in a microwave for 6 min at 100% power followed by 3 min at 60% and left to cool for 60 min at room temperature. Tissue sections were incubated with Super Block solution (ScyTek, Logan, UT, USA, AAA125) followed by incubation in primary antibodies overnight at 4°C. The sections were then incubated with Alexa Fluor-conjugated secondary antibody (1:1000) for 2 hours against the applicable species and coverlipped with Fluoro-Gel mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Negative controls were performed in parallel by omitting the primary antibodies. Confocal images were obtained using a Nikon A1 confocal microscope (Tokyo, Japan).
Pathological Evaluation
Representative HE-stained colon and cecum sections for each animal were evaluated by a WNPRC board-certified veterinary pathologist (HAS), familiar with histologic findings in the colony but blinded to the treatment groups. Criteria for tissue analysis included at least 12 visible myenteric ganglia and sufficient well-oriented mucosa and submucosa to evaluate and score for the presence and absence of colitis.
Colonic and cecal inflammation were scored using a previously validated scale.41 Inflammation (cellular infiltration of the lamina propria, submucosa, and muscular layers), glandular changes, and tissue morphology were evaluated and rated (1=no colitis, 2=mild, 3=moderate and, 4=severe) in increments of 0.5. A score of 1.5 corresponds to a qualitative assessment of minimal colitis. To assess intra-rater reliability, four sections were randomly selected and re-evaluated blindly; the sections received the same scores validating the results.
Quantification of Immunoreactivity
Expression of HLA-DR, CD3, CD20, 8-OHdG, PGP9.5, α-syn and p-α-syn in the myenteric ganglia was blindly quantified using the percent area above threshold (%AAT) and optical density (OD) functions of NIH ImageJ version 2.1.0 software with images obtained on a Zeiss Axio Imager M2 equipped with a Q Imaging camera following previously validated methods.41
Colon sections were divided into four equal quadrants with 3 myenteric ganglia randomly selected from each quadrant for a total of 12 ganglia per animal. Each immunostained myenteric ganglion was captured at 40x. The ganglia were outlined by a single blinded evaluator. In each photomicrograph, DAB color was separated from hematoxylin counterstain with the ImageJ Color Deconvolution filter. ImageJ was calibrated using a step tablet, and grayscale values were converted to OD units using the Rodbard function. The threshold is a minimum grayscale value of each pixel applied as a limit for inclusion in the %AAT analysis. The threshold was calculated by averaging the optimal individual threshold that best represents the immunoreactivity in the ganglia for four randomly selected photomicrographs of immunostained tissue sections from different animals. The following thresholds were utilized: HLA-DR=0.38, CD3=0.43, CD20=0.38, 8-OHdG=0.45, PGP9.5=0.30, α-syn=0.28 and p-α-syn=0.42.
Statistics
Data collection and analysis were performed by investigators blind to animals’ treatments and conditions. A p<0.05 was considered significant. Statistical analysis was performed using GraphPad Prism 9.0.
Datasets were compared between treatment groups of LM (n=10) vs mock (n=3) inoculation and between colitis conditions of no colitis (score 1; n=4) vs mild/moderate colitis (score 2–3; n=7) to further assess the impact of inflammation regardless of treatment. A Welch’s t-test was applied for comparisons between groups. Pearson correlation tests were utilized to assess the relationships between measures of α-syn-immunoreactivity (-ir) and colitis scores or HLA-DR-ir, and between α-syn-ir or p- α-syn-ir and number of LM inoculations per subject.
Results
LM Inoculation Induced Moderate to No Clinical Signs and Histologic Evidence of Colitis
Following oral gavage of LM or mock treatment, no emesis was reported in any monkey, ensuring full dose of inoculum. During the next 14 days, only 3 of the 10 LM inoculated animals presented with episodes of diarrhea or inappetance (Table 1). One additional monkey had one emetic episode 12 days post LM innoculation. LM fecal shedding was not observed in any of the 10 LM inoculated monkeys. LM bacteremia was not detected in the blood of any animal.
Analysis of HE-stained colon sections obtained 2 weeks post LM inoculation revealed some inflammation across subjects (Table 1 and Figure 1). The mock inoculated animals presented none (n=2) to minimal (n=1) colitis, although 12–26 months earlier they were inoculated with LM. The majority of LM treated monkeys had mild colitis (n=6), including the three animals with abnormal clinical observations. They presented lymphoplasmacytic and eosinophilic infiltration of the lamina propria, reactive macrophages, and regions of hyperdense cell death in the colonic mucosa. Of the remaining LM animals, two had no colitis and one had moderate colitis with hyperdense clusters of dead cells with inflammatory infiltration of the lamina propria and displacement of colonic glands. HE-stained cecal sections of all monkeys were also evaluated for a lingering or greater inflammatory reaction (not detected in the colon) after LM inoculation. Yet, the typhlitis scores in the cecum (Table 3) did not reveal any striking differences with the results obtained in the colon. The colitis and typhlitis scores of the five LM-treated subjects that were previously (12–48 months) inoculated with LM were similar to the animals with a single inoculation. Critically, the three mock-inoculated monkeys received one LM treatment 12–26 months earlier, and they displayed none to minimal inflammation, further confirming the acute inflammatory process triggered by LM.
 |
Table 3 Cecum Typhlitis Score per Study Subject |
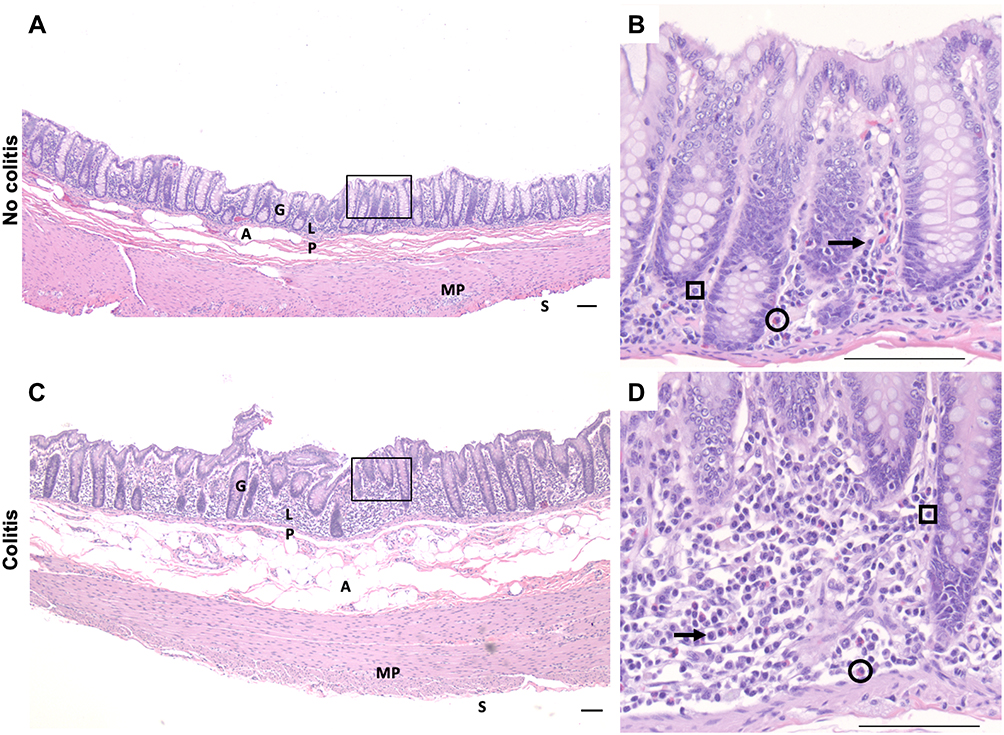 |
Figure 1 Representative HE-stained proximal colon sections images of cynomolgus macaques of no-colitis control (A, B); MO3) and moderate colitis (C, D; LM9) after oral LM inoculation. (A, C) at 4× magnification. (B, D) at 40× magnification. Note increased lymphoplasmacytic and eosinophilic inflammation in the lamina propria with glandular displacement in the colitis subject. Black arrow, plasma cell; circle, eosinophil; square, lymphocyte. Scale bar = 100µm for all panels. Abbreviations: A, adipocytes; G, gland; LP, lamina propria; MP, myenteric plexus; S, serosal surface. |
Immune Cell Markers Were Minimally Increased in the Colonic ENS 14 Days Post Inoculation
To further assess the immune response to LM, immune markers were evaluated in the colon and quantified in the ENS ganglia. Expression of HLA-DR (Figure 2A), a marker of antigen presenting cells, was minimally present and found on the surface of ovoid cells and foamy macrophage-like cells. CD3-ir (T cell marker; Figure 2B) and CD20-ir (B cell marker implicated in the adaptive immune response; Figure 2C) cells were primarily present in Peyer’s patches of both groups, and occasionally localized in the lamina propria. Expression of 8-OHdG (marker of oxidative stress) was minimal and localized in the nuclei of cells (Figure 2D). The %AAT and OD of immune marker expression in the ENS ganglia analyzed by the treatment group or by colitis condition did not identify significant differences between mock vs LM-inoculated.
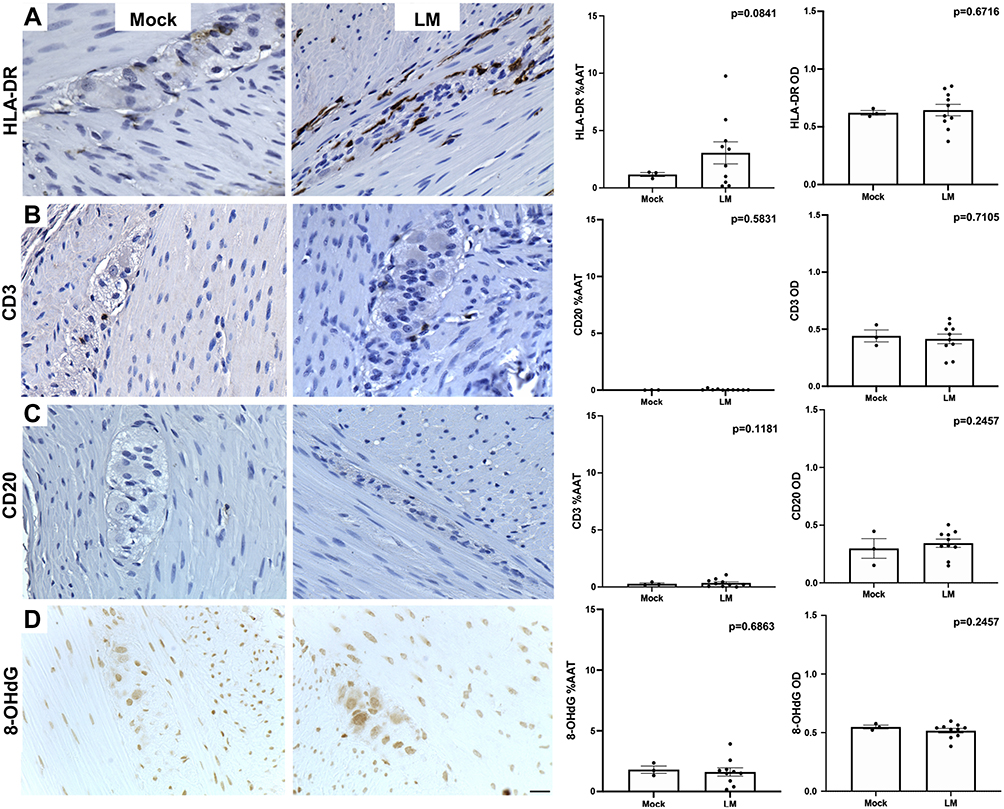 |
Figure 2 Images of proximal colon of cynomolgus macaques treated with mock (MO1) or LM (LM7) inoculations immunostained against markers of inflammation (A-C; HLA-DR, CD3, CD20) and oxidative stress (D; 8-OHdG) and their %AAT expression and OD expression (mean ± SEM). Scale bar = 25µm for all panels. |
α-Syn Expression Was Present in ENS Ganglia and Was Not Affected by LM Exposure
To have a measure of neuronal ENS integrity, the expression of the pan-neuronal marker PGP9.5 was evaluated in the colon of LM and mock inoculated animals. PGP9.5-ir was present throughout neuronal soma of myenteric ganglia and in nerve fibers running between muscle fibers and mucosa of all animals (Figure 3A). %AAT of PGP9.5-ir in myenteric ganglia was similar between treatment groups and colitis conditions, demonstrating that exposure to LM does not have a major effect on ENS neurons.
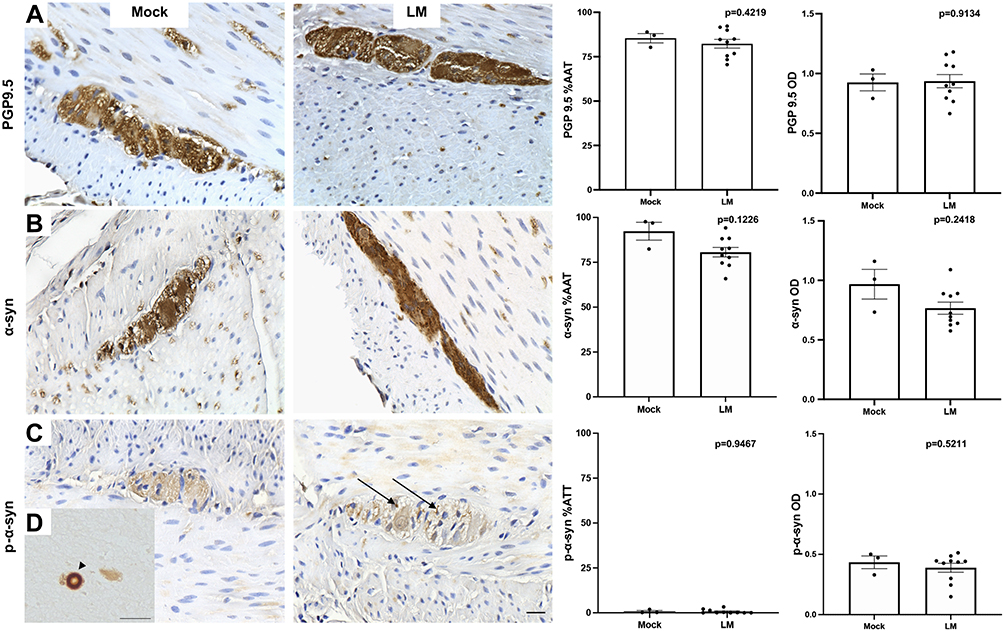 |
Figure 3 Images of proximal colon of cynomolgus macaques treated with mock (MO2) or LM (LM9) inoculations immunostained against pan-neuronal maker PGP9.5 (A), α-syn (B), p-α –syn (C, black arrows) and their %AAT expression and OD expression (mean ± SEM). Lewy body in human PD brain (D, black arrowhead). Scale bar = 25µm for all panels. |
When colon sections immunostained against α-syn were analyzed, the protein was ubiquitously found in the ENS myenteric plexus (Figure 3B). Double label immunofluorescence successfully colocalized α-syn-ir with PGP9.5-ir in myenteric ganglia, confirming α-syn location within ENS neurons (Figure 4). α-Syn-ir in the ENS ganglia measured by %AAT and OD was not significantly different when compared between treatment group or colitis condition. In addition, all animals presented minimal p-α-syn, limited to diffuse brown staining in ganglia (Figure 3C), compared to the PD brain tissue, where p-α-syn+ Lewy bodies were identified (Figure 3D). In the monkey with moderate colitis (LM9, received one previous inoculation 12 months earlier), some more intense p-α-syn-ir was observed in the ganglia (arrows in Figure 3C). Analysis of p-α-syn in the ENS ganglia measured by %AAT or OD was not significantly different when compared across treatment groups or colitis conditions. Lastly, α-syn-ir and p-α-syn-ir in the 5 LM-treated subjects that were previously inoculated with LM were similar to the expression in the animals that had a single inoculation.
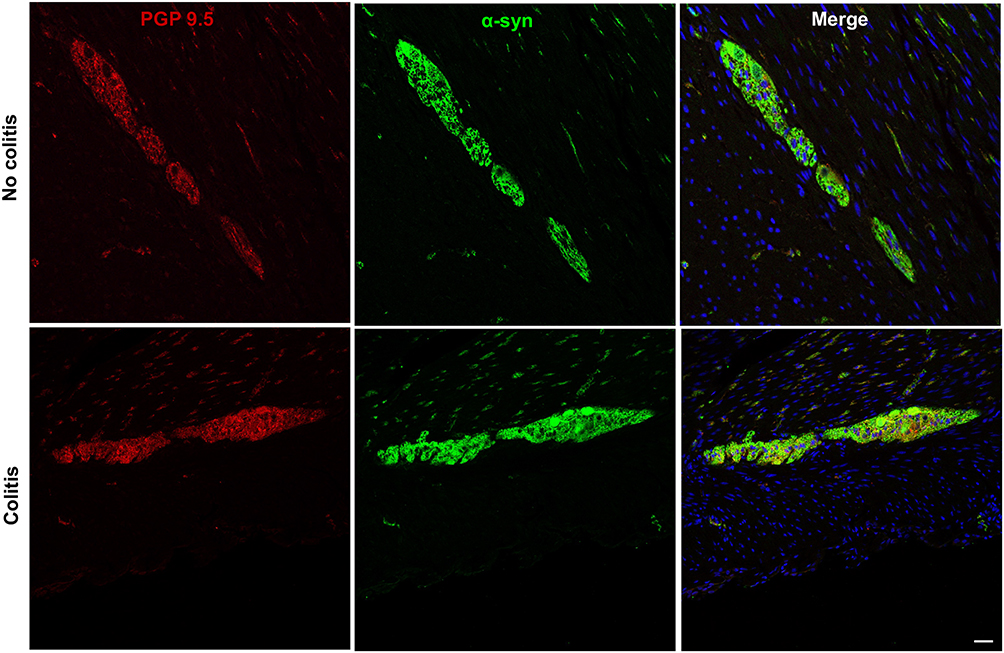 |
Figure 4 Double immunofluorescence images of proximal colon showing α-syn colocalization with pan-neuronal marker PGP9.5 in myenteric ganglia of no-colitis control (MO2) and colitis (LM9) cynomolgus macaques. Scale bar = 50µm for all panels. |
Pearson correlation tests were performed to assess if the individual differences observed in α-syn-ir were driven by colonic inflammation (Figure 5). Neither measures of α-syn-ir and the colitis score nor measures of α-syn-ir and HLA-DR-ir reach statistical significance. Pearson correlation tests to assess whether the number of LM inoculations per subject affected measures of α-syn-ir or p-α-syn-ir were also not significant.
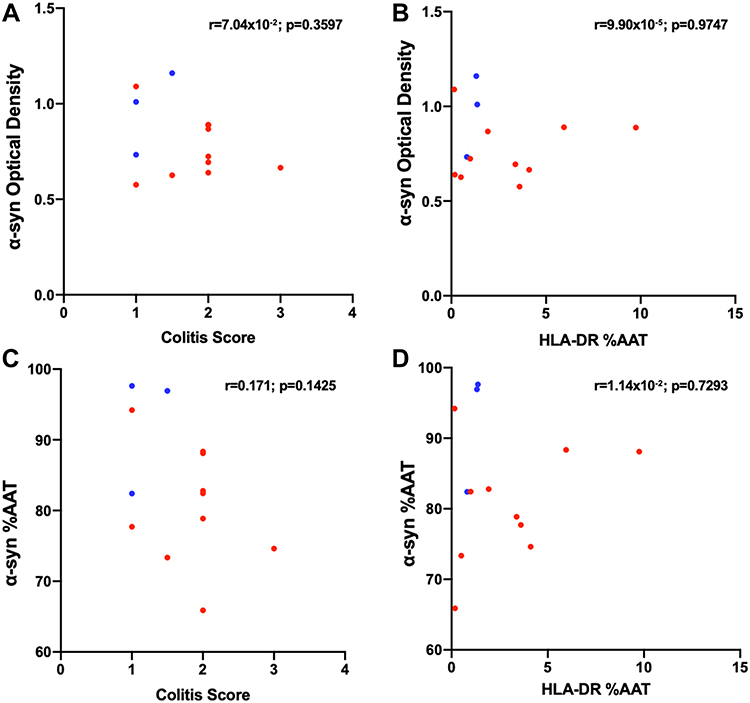 |
Figure 5 Pearson correlations graphs comparing colitis score vs α-syn optical density (A), HLA-DR %AAT vs α-syn OD (B), colitis score vs α-syn %AAT (C), and HLA-DR %AAT vs α-syn %AAT (D). Each point represents average per subject; the blue points correspond to Mock- and red points to LM- inoculated subjects. |
To further assess the spatial relationship between α-syn-ir and immune cells, triple labeling immunofluorescence for α -syn-ir, HLA-DR-ir and CD3-ir was performed (Figure 6). As expected, α-syn-ir was present in myenteric ganglia neurons. Some HLA-DR-ir was observed around the margins of ganglia, while CD3-ir was minimal, consistent with its minimal expression seen in brightfield.
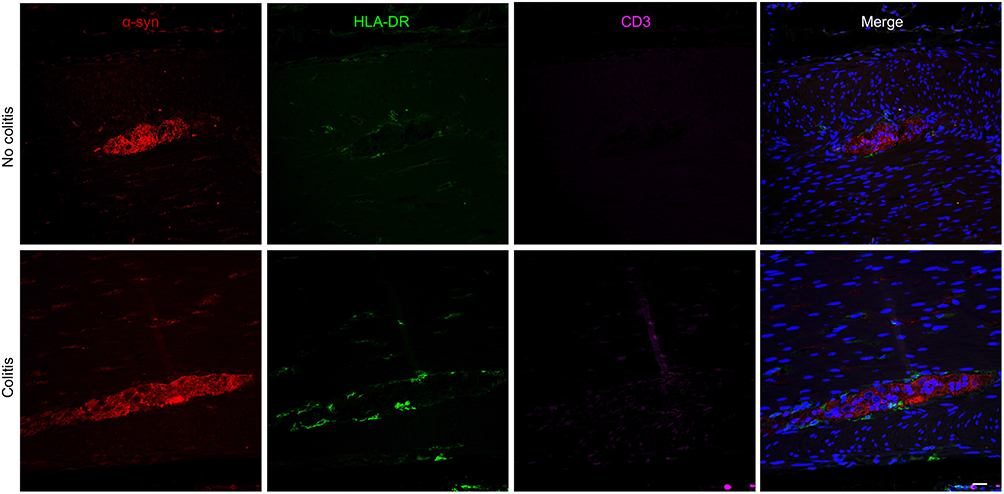 |
Figure 6 Triple immunofluorescence images of proximal colon sections showing α -syn-ir, HLA-DR-ir and CD3-ir in myenteric ganglia of no-colitis (MO2) and colitis (LM1) subjects. Scale bar = 50µm for all panels. |
Discussion
The present results demonstrate that 1) LM acute oral exposure can be successfully modeled in cynomolgus macaques, 2) the LM-related acute colitis resolves after 14 days, and 3) the acute immune response does not induce detectable lingering effects in α-syn expression in the ENS of healthy adult female subjects. These findings suggest that acute exposure to food-borne pathogens is not sufficient to trigger α-syn changes. The goal of this NHP project was to model the type of exposure to a common food-borne pathogen that would be experienced by the general population (a single inoculation to simulate a patient’s consumption of a contaminated product), in order to study whether the acute event could have long-term effects on α-syn in the ENS. To the best of our knowledge this is the first report to investigate this question in NHPs.
Cynomolgus macaques are an Old World NHP species frequently used in medical research, as their genetics, anatomy, physiology, and behavior provide clinically relevant outcomes. They were chosen for this experiment because when pregnant and orally inoculated with LM during the first trimester, they present with a variety of mild clinical signs in the dam, yet acute fetal demise is almost invariable. Thus, as with human pregnancy, listeriosis in macaque pregnancy is associated with adverse pregnancy outcomes.38,39 The mild clinical signs of listeriosis detected in the healthy adult monkeys after a prescribed LM oral inoculation also match the typical syndrome observed after LM exposure in healthy adult humans compared to pregnant, immunocompromised, or the elderly.24 To ensure the pathogenicity of the LM strain, we administered LM isolated from a cluster of human listeriosis cases that induced adverse pregnancy outcomes and verified the viability of the incoculum post-dosing by the growth of colonies in agar. The animals’ episodes of diarrhea or inappetance are a typical response to exposure to live LM. The lack of emesis post-dosing also ensured that a full inoculum was received. LM fecal shedding and bacteremia were not detected in any of the 10 LM inoculated monkeys, which is not uncommon in exposed humans and NHP subjects.38
LM is known to induce an acute immune response, recruiting neutrophils that play a role in primary pathogen defense, as well as dendritic cells and macrophages driven by IFNγ.43,44 Like most acute inflammatory responses, LM-induced inflammation typically resolves approximately 2 weeks post exposure. As such, the majority of the LM exposed animals presented mild inflammation scores, characterized by lymphoplasmacytic and eosinophilic infiltration of the lamina propria, reactive macrophages, and regions of hyperdense cell death in the colonic mucosa, reflecting the acute nature of the LM exposure and the effective self-limiting host immune response.45 The inflammation scores were confirmed by the minimal expression of the immune markers HLA-DR (macrophages), CD3 (T cells) and CD20 (B cells).
In humans, the appendix is rich in lymphoid tissue, has abundant α-syn expression and its surgical removal has been linked to a decreased risk of PD,22 suggesting that this organ may have a role in the onset of PD pathology. Although macaque monkeys lack an appendix, their cecum is considered an anatomical equivalent,46,47 thus we evaluated cecal sections for signs of more robust inflammation. The typhlitis scores matched the colonic findings of mild acute inflammation.
The macaque ENS, like the human ENS, is organized in a network of ganglia, each ganglion composed of PGP9.5+ neurons that also express α-syn.42 Our analysis confirmed the presence of PGP9.5-ir and α-syn-ir neurons in the cynologus macaque ENS ganglia. We also found minimal p-α-syn-ir across treatment groups, suggesting that LM did not induce permanent pre-pathological changes in α-syn. The immunostaining methods used to identify p-α-syn, including the choice of antibodies and positive control, are critical considerations for ensuring high quality, replicable data. Other reports have relied on different antibodies for identifying p-α-syn, yet those studies were performed in rodents.48 In this project, we took advantage of a p-α-syn antibody that was previously validated and optimized for use in NHP and human PD tissues.41,42 To minimize immunostaining variability, tissue sections from animals of different treatment groups were processed in parallel (and with positive and negative controls) and were incubated in solutions for the same periods of time. The human PD brain tissue used as a positive control for p-α-syn was processed with the same methods as the colonic tissue (ie, both tissues were paraffin embedded, cut at 5µm thickness, and stained in parallel with the cynomolgous tissue using all the same reagents). PD brain tissue is recommended as a positive control for this antibody and has been previously validated by our group as a positive control for p-α-syn immunostaining in the monkey colon.41,42 Lastly, proteinase-K treatment before p-α-syn immunstaining is a standard practice to confirm the presence of aggregated, insoluble p-α-syn.1 Although aggregated p-α-syn in the form of Lewy bodies was observed in the human PD brain, p-α-syn-ir was minimal and diffuse in the colon tissue sections and did not justify performing p-α-syn immunostaining with proteinase-K pre-treatment.
A limitation of this study was the use of only female cynomolgus macaques. As mentioned in the introduction, the reason for this experimental sex bias was our commitment to maximizing use of NHP resources while reducing the number of animals used in research. This study took advantage of an ongoing project assessing the impact of LM exposure in the reproductive organs of healthy female NHPs, thus reducing the need to infect additional animals with LM. PD is 1.5 times more likely to occur in men than women,49 whether this is the result of an X- or Y-linked factor or is related to environmental interactions leading to increased exposure by human males is not yet clear. Several publications have linked the increased risk of PD in IBD patients preferentially to men,50,51 while others found greater risk in women52 or no differences between sexes.53 Interestingly, a recent study showed that the brains of male mice with dextran-induced colitis had reduced tyrosine hydroxylase as well as sustained CD8+ T-cell infiltration and elevated expression of the lfng gene, which encodes for a cytokine produced by CD8+ T-cells,53 suggesting a sex-dependent response to colitis by the nervous system.
Compared to the lack of α-syn changes in the ENS colonic ganglia after LM-induced acute inflammation, our group previously reported that idiopathic chronic colitis in NHPs (mainly males) is associated with reduced α-syn expression and increased pathological phosphorylation of α-syn.41 Duration of lymphocyte stimulation in a chronic response could account for the differences. The acute self-limiting nature of inflammation induced in subjects by the LM challenge in our study may not be sufficient to generate changes in α-syn. In IBD, the immune cells recruited include neutrophils, dendritic cells, macrophages, and monocytes. Lymphocytes undergo long term stimulation resulting in chronic tissue damage.54 Notably, cytokines associated with an innate and/or chronically occurring immune response, such as TNF-α, IL-1β, IL-2, IL-4 and IL-6, are elevated in the striatum and cerebrospinal fluid of PD patients,55 further supporting the role of chronic inflammation in PD.
Current evidence suggests that the chronic GI inflammation that could lead to PD pathology is associated with GI dysbiosis and, that exposure to environmental toxins may start the pathological process. Chronic oral dosing of rotenone to mice is known to induce loss of nigrostriatal dopaminergic neurons and acumulation of misfolded p-α-syn. Interestingly, it can also induce inflammation in the gastrointestinal tract, alter the GI microbiome, and increase p-α-syn in the ENS.56–58 Whether inflammation (or the toxin itself) causes dysbiosis or vice versa is not clear. Moreover, PD is an age-related disorder and chronic inflammation and changes in the gut microbiome are both typical findings in older adults, further stimulating research to understand the role of the microbiome in PD.59–61
Inasmuch as inflammation seems to facilitate α-syn phosphorylation and aggregation,3,4 current evidence indicates that accumulation of pathological α-syn itself can initiate an immune response62 that may perpetuate a cycle of proteinopathy, neurodegeneration and inflammation. In PD brains, the amount of microglial activation measured by CD68 and HLA-DR expression is directly associated with α-syn load.63 In addition, the presence of neuromelanin inside activated microglia suggests that diseased dopaminergic neurons are being phagocytosed.64
It could be argued that multiple LM inoculations or tissue collection after a longer period of time post-exposure may have induced detectable α-syn pathology. As shown in Tables 1, 5 LM-treated subjects were previously (12–48 months) inoculated with LM, yet their colitis score, α-syn-ir, and p-α-syn-ir were similar to animals with a single LM treatment. Additionally, the three mock-inoculated monkeys received LM treatment 12–26 months earlier, and they displayed none to minimal inflammation and unremarkable α-syn and p-α-syn-ir expression. We would like to emphasize that exposure of the human population to listeria is an isolated event that typically occurs from an outbreak of infected pre-packaged dairy products or lunch meat products.65 Repeated exposure over multiple days/months is unlikely to be experienced by a patient as outbreaks are typically isolated and consumers are warned as soon as the infected product is detected in the market.
Although the interface between the immune system, α-syn, and PD remains to be fully elucidated, this study demonstrates that an environmental challenge that induces an acute inflammatory response is insufficient to trigger α-syn pathology and ultimately PD. Since the publication of Braak’s hypothesis7–9 the concept of an environmental trigger has been widely speculated. Given the reported results, we propose that, if an LM challenge has a role in PD pathology, other underlying factors or conditions, such as male sex, IBD, exposure to toxins, dysbiosis, and/or aging are needed to be present.
Conclusions
Our results in an NHP model of listeriosis demonstrate that acute oral exposure to a food-borne pathogen is not sufficient to trigger persistent α-syn changes in the ENS of healthy adult female subjects that could lead to PD.
Data Sharing Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
We gratefully acknowledge the Wisconsin Alzheimer’s Disease Research Center (P50-AG033514) for providing human PD brain tissue. LM isolate 2203 was a kind gift from Dr. Sophia Kathariou, North Carolina State University.
Funding
This research was supported by the Parkinson’s Foundation to AMM and MEE, NIH P51OD011106 to the Wisconsin National Primate Research Center, and R01 AI107157 to TGG.
Disclosure
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
1. Vermilyea SC, Emborg ME. α-Synuclein and nonhuman primate models of Parkinson’s disease. J Neurosci Methods. 2015;255:38–51. doi:10.1016/j.jneumeth.2015.07.025
2. Bendor JT, Logan TP, Edwards RH. The function of α-synuclein. Neuron. 2013;79(6):1044–1066. doi:10.1016/j.neuron.2013.09.004
3. Hald A, Lotharius J. Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? Exp Neurol. 2005;193(2):279–290. doi:10.1016/j.expneurol.2005.01.013
4. Matyszak MK. Inflammation in the CNS: balance between immunological privilege and immune responses. Prog Neurobiol. 1998;56(1):19–35. doi:10.1016/S0301-0082(98)00014-8
5. Rai SN, Singh P, Varshney R, et al. Promising drug targets and associated therapeutic interventions in Parkinson’s disease. Neural Regen Res. 2021;16(9):1730–1739. doi:10.4103/1673-5374.306066
6. Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–376. doi:10.1136/jnnp.2007.131045
7. Braak H, Rob U, Gai WP, et al. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna). 2003;110(5):517–536. doi:10.1007/s00702-002-0808-2
8. Braak H, Tredici KD, Rüb U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi:10.1016/S0197-4580(02)00065-9
9. Braak H, de Vos RAI, Bohl J, et al. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396(1):67–72. doi:10.1016/j.neulet.2005.11.012
10. Liu B, Fang F, Pedersen NL, et al. Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology. 2017;88(21):1996–2002. doi:10.1212/WNL.0000000000003961
11. Svensson E, Horváth-Puhó E, Thomsen RW, et al. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol. 2015;78(4):522–529. doi:10.1002/ana.24448
12. Tysnes OB, Kenborg L, Herlofson K, et al. Does vagotomy reduce the risk of Parkinson’s disease? Ann Neurol. 2015;78(6):1011–1012. doi:10.1002/ana.24531
13. Lohmann S, Bernis ME, Tachu BJ, et al. Oral and intravenous transmission of alpha-synuclein fibrils to mice. Acta Neuropathol. 2019;138(4):515–533. doi:10.1007/s00401-019-02037-5
14. Kim S, Kwon S-H, Kam T-I, et al. Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron. 2019;103(4):627–641e7. doi:10.1016/j.neuron.2019.05.035
15. Holmqvist S, Chutna O, Bousset L, et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128(6):805–820. doi:10.1007/s00401-014-1343-6
16. Van Den Berge N, Ferreira N, Mikkelsen TW, et al. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain. 2021;144(6):1853–1868. doi:10.1093/brain/awab061
17. Van Den Berge N, Ferreira N, Gram H, et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019;138(4):535–550. doi:10.1007/s00401-019-02040-w
18. Pajares M, I Rojo A, Manda G, Boscá L, Cuadrado A. et al. Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells. 2020;9(7):1687.
19. Devos D, Lebouvier T, Lardeux B, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50:42–48. doi:10.1016/j.nbd.2012.09.007
20. Houser MC, Chang J, Factor SA, et al. Stool Immune profiles evince gastrointestinal inflammation in Parkinson’s disease. Mov Disord. 2018;33(5):793–804. doi:10.1002/mds.27326
21. Kooij IA, Sahami S, Meijer SL, et al. The immunology of the vermiform appendix: a review of the literature. Clin Exp Immunol. 2016;186(1):1–9. doi:10.1111/cei.12821
22. Killinger BA, Madaj Z, Sikora JW, et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med. 2018;10::465.
23. Roberts BN, Chakravarty D, Gardner JC, et al. Response to anaerobic environments. Pathogens. 2020;9(3):210.
24. Charlier C, Perrodeau É, Leclercq A, et al. Clinical features and prognostic factors of listeriosis: the MONALISA national prospective cohort study. Lancet Infect Dis. 2017;17(5):510–519. doi:10.1016/S1473-3099(16)30521-7
25. Lecuit M. Understanding how Listeria monocytogenes targets and crosses host barriers. Clin Microbiol Infect. 2005;11(6):430–436. doi:10.1111/j.1469-0691.2005.01146.x
26. Corr S, Hill C, Gahan CG. An in vitro cell-culture model demonstrates internalin- and hemolysin-independent translocation of Listeria monocytogenes across M cells. Microb Pathog. 2006;41(6):241–250. doi:10.1016/j.micpath.2006.08.003
27. Greiffenberg L, Goebel W, Kim KS, et al. Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: inlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect Immun. 1998;66(11):5260–5267. doi:10.1128/IAI.66.11.5260-5267.1998
28. Drevets DA, Sawyer RT, Potter TA, et al. Listeria monocytogenes infects human endothelial cells by two distinct mechanisms. Infect Immun. 1995;63(11):4268–4276. doi:10.1128/iai.63.11.4268-4276.1995
29. Gaillard JL, Berche P, Sansonetti P. Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes. Infect Immun. 1986;52(1):50–55. doi:10.1128/iai.52.1.50-55.1986
30. Kathariou S, Metz P, Hof H, et al. Tn916-induced mutations in the hemolysin determinant affecting virulence of Listeria monocytogenes. J Bacteriol. 1987;169(3):1291–1297. doi:10.1128/jb.169.3.1291-1297.1987
31. Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol. 1989;109(4 Pt 1):1597–1608. doi:10.1083/jcb.109.4.1597
32. Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl 2):S3. doi:10.1186/ar1917
33. Ryan GB, Majno G. Acute inflammation. A review. Am J Pathol. 1977;86(1):183–276.
34. Freire MO, Van Dyke TE. Natural resolution of inflammation. Periodontol. 2000;63(1):149–164. doi:10.1111/prd.12034
35. Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27(8):352–357. doi:10.1016/j.it.2006.06.003
36. O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–364. doi:10.1038/nri2079
37. Ranson T, Bregenholt S, Lehuen A, et al. Invariant Vα14+NKT cells participate in the early response to enteric Listeria monocytogenes Infection. J Immunol. 2005;175(2):1137–1144. doi:10.4049/jimmunol.175.2.1137
38. Wolfe B, Kerr AR, Mejia A, et al. Sequelae of fetal infection in a non-human primate model of listeriosis. Front Microbiol. 2019;10:2021. doi:10.3389/fmicb.2019.02021
39. Wolfe B, Wiepz GJ, Schotzko M, et al. Acute fetal demise with first trimester maternal infection resulting from. mBio. 2017;8(1):e01938.
40. Lancaster DP, Friedman DF, Chiotos K, et al. Blood volume required for detection of low levels and ultralow levels of organisms responsible for neonatal bacteremia by use of bactec peds Plus/F, Plus Aerobic/F Medium, and the BD Bactec FX system: an in vitro study. J Clin Microbiol. 2015;53(11):3609–3613. doi:10.1128/JCM.01706-15
41. Resnikoff H, Metzger JM, Lopez M, et al. <p>Colonic inflammation affects myenteric alpha-synuclein in nonhuman primates. J Inflamm Res. 2019;12:113–126. doi:10.2147/JIR.S196552
42. Shultz JM, Resnikoff H, Bondarenko V, et al. Neurotoxin-induced catecholaminergic loss in the colonic myenteric plexus of rhesus monkeys. J Alzheimers Dis Parkinsonism. 2016;6:6.
43. Harty JT, Lenz LL, Bevan MJ. Primary and secondary immune responses to Listeria monocytogenes. Curr Opin Immunol. 1996;8(4):526–530. doi:10.1016/S0952-7915(96)80041-0
44. Czuprynski CJ, Brown JF, Maroushek N, et al. Administration of anti-granulocyte mAb RB6-8C5 impairs the resistance of mice to Listeria monocytogenes infection. J Immunol. 1994;152(4):1836–1846.
45. Vázquez-Boland JA, Kuhn M, Berche P, et al. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev. 2001;14(3):584–640.
46. Fisher RE. The primate appendix: a reassessment. Anat Rec. 2000;261(6):228–236. doi:10.1002/1097-0185(20001215)261:6<228::AID-AR1005>3.0.CO;2-O
47. Zinnen AD, Jeanette JV, Metzger M, et al. Alpha-synuclein and Tau are abundantly expressed in the appendix of humans and the ceca of common marmosets and rhesus macaques. Society for Neuroscience Abst. 2021;2021.
48. Delic V, Chandra S, Abdelmotilib H, et al. Sensitivity and specificity of phospho-Ser129 alpha-synuclein monoclonal antibodies. J Comp Neurol. 2018;526(12):1978–1990. doi:10.1002/cne.24468
49. Wooten GF, Currie LJ, Bovbjerg VE, Lee JK, Patrie J. Are men at greater risk for Parkinson’s disease than women? J Neurol Neurosurg Psychiatry. 2004;75(4):637–639. doi:10.1136/jnnp.2003.020982
50. Peter I, Dubinsky M, Bressman S, et al. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 2018;75(8):939–946. doi:10.1001/jamaneurol.2018.0605
51. Lin JC, Lin C-S, Hsu C-W, et al. Association between parkinson’s disease and inflammatory bowel disease: a Nationwide Taiwanese Retrospective Cohort Study. Inflamm Bowel Dis. 2016;22(5):1049–1055. doi:10.1097/MIB.0000000000000735
52. Weimers P, Halfvarson J, Sachs MC, et al. Inflammatory Bowel Disease and Parkinson’s Disease: a Nationwide Swedish Cohort Study. Inflamm Bowel Dis. 2019;25(1):111–123. doi:10.1093/ibd/izy190
53. Houser MC, Caudle WM, Chang J, et al. Experimental colitis promotes sustained, sex-dependent, T-cell-associated neuroinflammation and parkinsonian neuropathology. Acta Neuropathol Commun. 2021;9(1):139. doi:10.1186/s40478-021-01240-4
54. Lazaridis N, Germanidis G. Current insights into the innate immune system dysfunction in irritable bowel syndrome. Ann Gastroenterol. 2018;31(2):171–187. doi:10.20524/aog.2018.0229
55. Nagatsu T, Mogi M, Ichinose H, et al.Changes in cytokines and neurotrophins in Parkinson’s disease. J Neural Transm Suppl. 2000;(60):277–290. doi:10.1007/978-3-7091-6301-6_19
56. Yang X, Qian Y, Xu S, et al. Longitudinal analysis of fecal microbiome and pathologic processes in a rotenone induced mice model of Parkinson’s disease. Front Aging Neurosci. 2017;9:441. doi:10.3389/fnagi.2017.00441
57. Morais LH, Hara DB, Bicca MA, et al. Early signs of colonic inflammation, intestinal dysfunction, and olfactory impairments in the rotenone-induced mouse model of Parkinson’s disease. Behav Pharmacol. 2018;29(2 and 3–Spec Issue):199–210. doi:10.1097/FBP.0000000000000389
58. Pan-Montojo F, Anichtchik O, Dening Y, et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One. 2010;5(1):e8762. doi:10.1371/journal.pone.0008762
59. Romano S, Savva GM, Bedarf JR, et al. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis. 2021;7(1):27. doi:10.1038/s41531-021-00156-z
60. Keshavarzian A, Engen P, Bonvegna S, Cilia R. The gut microbiome in Parkinson’s disease: a culprit or a bystander? Prog Brain Res. 2020;252:357–450.
61. Buford TW. (Dis) Trust your gut: the gut microbiome in age-related inflammation, health, and disease. Microbiome. 2017;5(1):80. doi:10.1186/s40168-017-0296-0
62. Allen Reish HE, Standaert DG. Role of alpha-synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis. 2015;5(1):1–19. doi:10.3233/JPD-140491
63. Croisier E, Moran LB, Dexter DT, et al. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation. 2005;2:14. doi:10.1186/1742-2094-2-14
64. Depboylu C, Schäfer MK-H, Arias-Carrión O, et al. Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. J Neuropathol Exp Neurol. 2011;70(2):125–132. doi:10.1097/NEN.0b013e31820805b9
65. Bortolussi R. Listeriosis: a primer. CMAJ. 2008;179(8):795–797. doi:10.1503/cmaj.081377
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.