Back to Journals » OncoTargets and Therapy » Volume 14
ACTL6A Promotes the Proliferation of Esophageal Squamous Cell Carcinoma Cells and Correlates with Poor Clinical Outcomes
Authors Li R, Li Y, Qin H, Li S
Received 26 October 2020
Accepted for publication 21 December 2020
Published 13 January 2021 Volume 2021:14 Pages 199—211
DOI https://doi.org/10.2147/OTT.S288807
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Rui-zhe Li,1 Yun-yun Li,1,2 Hui Qin,1 Shan-shan Li1
1Department of Pathology, School of Basic Medical Sciences, Zhengzhou University and First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450000, People’s Republic of China; 2Department of Stomatology, First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450000, People’s Republic of China
Correspondence: Shan-shan Li
Department of Pathology, School of Basic Medical Sciences, Zhengzhou University and First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450000, People’s Republic of China
Tel +86 138 3815 6395
Fax +86 371 6796 6155
Email [email protected]
Background: ACTL6A, a regulatory subunit of ATP-dependent chromatin-remodeling complexes SWI/SNF, has been identified as a central oncogenic driver in many tumor types.
Materials and Methods: We used immunohistochemistry (IHC) to detect ACTL6A expression in esophageal squamous cell carcinoma (ESCC) tissues. Then, the effect of ACTL6A on proliferation and DNA synthesis was explored by using cell counting kit 8 (CCK8) and EdU retention assays. The potential oncogenic mechanism of ACTL6A in ESCC cells was also analyzed by flow cytometry and Western blotting. We further established an ESCC xenograft mouse model to validate the in vitro results.
Results: ACTL6A expression, localized in cancer cell nuclei, was markedly higher in ESCC tissues than in the corresponding noncancerous tissues (P< 0.001) and was positively associated with tumor size, histological differentiation, T stage and tumor-node-metastasis (TNM) stage. Kaplan–Meier analysis revealed that high ACTL6A expression was significantly associated with poor overall survival (OS) (P = 0.008, HR= 2.562, 95% CI: 1.241– 5.289), and decision curve analysis (DCA) demonstrated that ACTL6A could increase the clinical prognostic efficiency of the original clinical prediction model. Further in vitro experiments showed that ACTL6A knockdown led to inhibition of cell proliferation and DNA synthesis in ESCC cell lines, while overexpression of ACTL6A had the opposite effects. ACTL6A knockdown resulted in G1 phase arrest, with downregulation of cyclin D1, CDK2 and S6K1/pS6 pathway proteins and upregulation of p21 and p27, while overexpression of ACTL6A facilitated the entry of more cells into S phase with upregulated cyclin D1, CDK2 and S6K1/pS6 pathway proteins and downregulated p21 and p27. Finally, a xenograft mouse model of ESCC cells validated the results in vitro.
Conclusion: ACTL6A expression may affect the proliferation and DNA synthesis of ESCC cells by facilitating ESCC cell cycle redistribution via the S6K1/pS6 pathway. Therefore, ACTL6A may potentially become an alternative therapeutic target for ESCC.
Keywords: ACTL6A, esophageal squamous cell carcinoma, cell cycle, proliferation, pS6
Introduction
Esophageal squamous cell carcinoma (ESCC) remains the most prominent subtype of esophageal cancer, accounting for 335,080 new cases and 300,878 deaths in East Asia in 2018.1,2 ESCC is thought to have obvious exposure-driven causality, driven by factors such as tobacco consumption, hot beverage drinking and poor nutrition.3 Exposure of the squamous epithelium of esophageal tract to these carcinogenic compounds is likely to change the genetic and epigenetic makeup of squamous epithelium cells, thereby facilitating neoplastic transformation.3 The prognosis of ESCC is poor because patients often present with distant metastasis to the liver, lung and lymph nodes.4 Despite advances in treatment, including tumor resection, chemotherapy, radiotherapy and immunotherapy, the 5-year survival rate of ESCC patients is still quite low, ranging from 15% to 25%5 underscoring the strong demand for deeper insight into the molecular mechanism behind the tumorigenicity and development of ESCC, identification of downstream targets of related signaling pathways as new therapeutic targets and the establishment of a clinical prediction model with high efficiency.
Actin-like protein 6A (ACTL6A), a regulatory subunit of the ATP-dependent SWI/SNF chromatin-remodeling complexes, is known as a central oncogenic driver and is commonly subject to genomic amplification and overexpression in a variety of tumors.6–13 In general, the SWI/SNF complex acts as a tumor suppressor; however, protein subunits of this complex are frequently mutated or lost in tumors, creating circumstances that are permissive for cancer development.14,15 ACTL6A was initially found to cooperate with β-actin to maintain maximal Brg1 ATPase activity.16,17 Deficiency of Brg1 was previously shown to induce cell cycle arrest through an RB-dependent mechanism, and Brg1 is preferentially recruited by p53 to a subset of p53-dependent promoters, including the p21 promoter, in melanoma.18 Thus, ACTL6A may function to regulate proliferation and the cell cycle indirectly by affecting the stability of Brg1 and SWI/SNF chromatin-remodeling complexes. Nevertheless, ACTL6A can also act independently of the SWI/SNF complex to promote cancer progression.19–22 ACTL6A expression leads to cell cycle arrest in head and neck squamous cell carcinoma (HNSCC) and epidermal squamous cell carcinoma (SCC) by interacting with the p21 promoter directly.7,10 Additionally, ACTL6A can cooperate with c-myc to maintain cancer stem cell self-renewal capacity and is associated with cancer cell motility.8,11,23–25 Moreover, ACTL6A has significant prognostic value in various tumor types.7,8,11 However, the exact oncogenic function of ACTL6A is largely unknown in ESCC.
A recent bioinformatics study performed KEGG analysis and gene set enrichment analysis (GSEA) via Gene Expression Profiling Interactive Analysis (GEPIA) based on The Cancer Genome Atlas (TCGA) datasets and demonstrated that ACTL6A significantly affects ribosomal pathways.26 Since S6 kinase (S6K1) is a key member of the pathway, that participates in translational control, and the S6 kinase (S6K1)-S6 signaling pathway is critical for regulating proliferation and survival in response to growth factors, we focused on S6K1 as a downstream target of ACTL6A.27–29 S6K1 always phosphorylates S6 protein at Ser235, Ser236, Ser240, Ser244, and Ser247.29 Aberrant upregulation of S6 phosphorylation and increased S6K activity are frequently present in various tumors including ESCC.30–32 Although phosphorylated-ribosomal protein S6 (p-S6) is considered to be a potential target in ESCC in vitro and S6 phosphorylation has prognostic significance in ESCC, the relationship between S6 and ACTL6A remains unclear.30
Here, we assessed the clinical relevance of ACTL6A and its prognostic impact on the survival of patients with ESCC by measuring the levels of ACTL6A in ESCC tissues and evaluating the relationship of these parameters with clinical outcome. Additionally, we also performed decision curve analysis (DCA) to investigate the practical clinical prognostic value of ACTL6A. We further examined the functional role of ACTL6A in ESCC cell proliferation in vitro and in vivo.
Materials and Methods
Patients and Tissue Samples
Formalin-fixed, paraffin-embedded ESCC tissue samples and corresponding adjacent normal mucosa epithelium of 128 ESCC patients, with histopathologically confirmed ESCC according to the World Health Organization criteria, were obtained from the Department of Pathology of the First Affiliated Hospital of Zhengzhou University (Zhengzhou, China) between January 2013 and December 2013. None of the enrolled patients received chemotherapy or radiotherapy prior to esophagectomy. The clinicopathological characteristics of the 128 ESCC patients are shown in Table 1. This retrospective study passed the review of the ethics committee of the First Affiliated Hospital of Zhengzhou University, and informed consent for the use of surgical specimens for research purposes was obtained from all the patients. The patients were followed up from the date of surgical resection, with an average follow-up of 41.0±2.1 months. Overall survival (OS) was defined as the period between the date of initial surgery and death or the last follow-up. The work has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki).
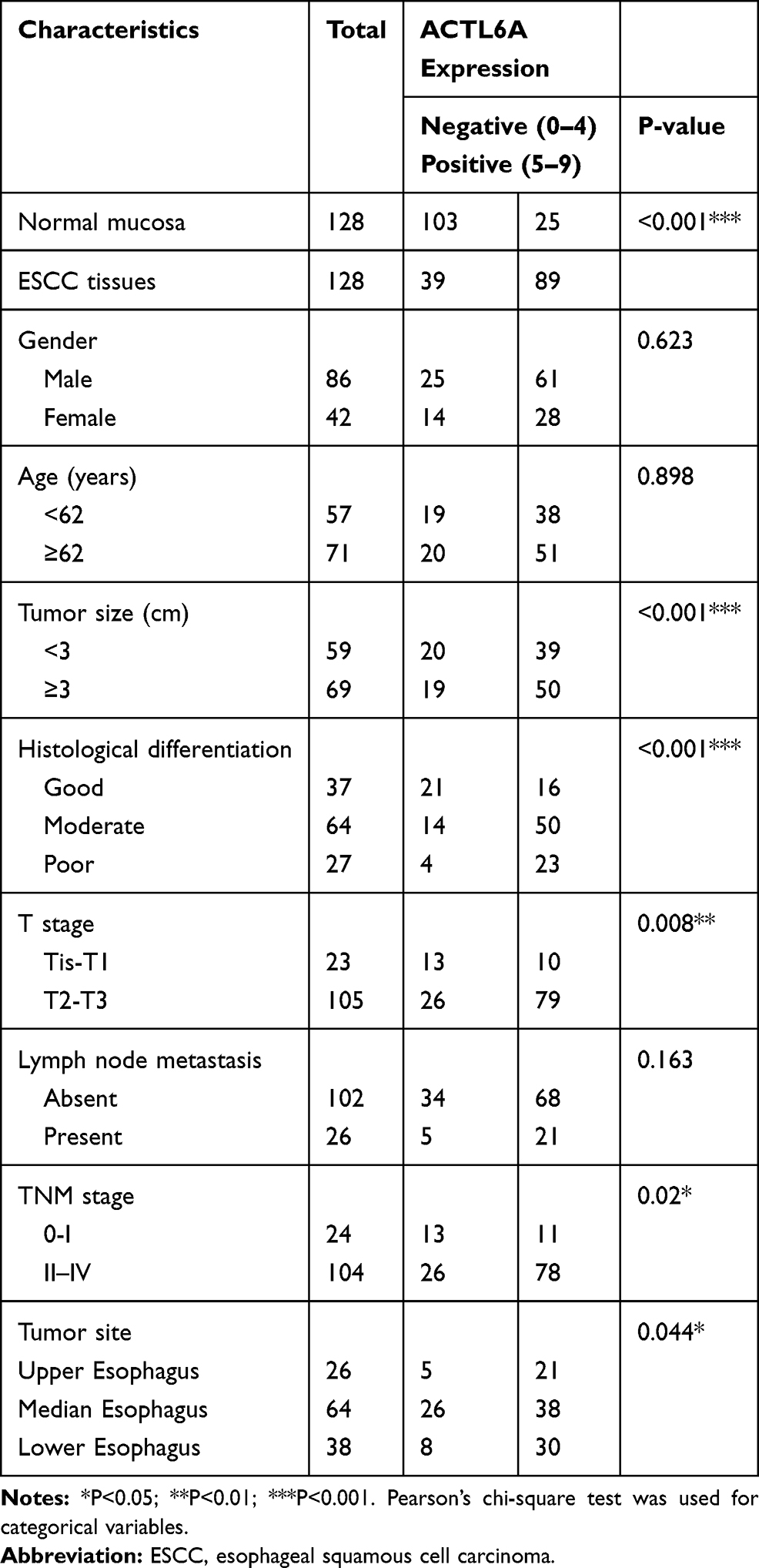 |
Table 1 Correlations of ACTL6A Expression and Clinicopathological Parameters in ESCC Patients |
Immunohistochemistry and the Evaluation of the Results
Immunohistochemistry was performed using rabbit anti-human ACTL6A primary antibody (1:200, Novus Biologicals, USA). A commercial biotin-streptavidin system (ZSGB-BIO, SP-9000) was used to detect the expression of ACTL6A. Two pathologists independently scored each of the sections. The staining was scored in terms of positive cell proportion (0, 0–10%; 1, 11%≤30%; 2, 30–70%; 3, >70%) and intensity (0, none; 1, weak; 2, moderate; 3, strong). The proportion and intensity scores were then multiplied to obtain a total score, with a range of 0 to 9. Patients with a final score of ≤1 were classified as the negative group and those with scores >1 were classified as the positive group. The pathologists discussed any cases showing a discrepancy in scores until a consensus was reached.
Construction of Lentivirus
shRNA-1 (5′-TCCAAGTATGCGGTTGAAA-3′) and shRNA-2 (5′-GTACTTCAAGTGTCAG ATT-3′) constructed to target ACTL6A were inserted into a super silencing vector (pHU6/EGFP + puromycin) provided by GenePharma (Shanghai, China). A nonsilencing control shRNA (5′-TTCTCCGAACGTGTCACGT-3′) was also used. Full-length ACTL6A cDNA (GenBank accession number NM_178042) was cloned into the LV5-expression vector (GenePharma). A specific sequence was used as a control. The above five kinds of plasmids were added with PSPAX2 and PMD2G plasmids into the supernatant of revived 293T cell culture. Growth medium was removed after 24 hours of incubation, and fresh DMEM-high glucose (Sigma, USA) with 10% FBS (Dakewe, New Zealand) was added. Then, 293T cells were cultured for another 48 hours. The corresponding lentiviruses (Lv-shACTL6A-1, Lv-shACTL6A-2, Lv-shNC, LV-ACTL6A and LV-CON) were obtained after ultracentrifugation of the collected supernatant for 4 hours.
Cell Culture, Reagents and Treatment
Human esophageal squamous carcinoma EC1, EC109, EC9706, TE7 and 293T cell lines were purchased from the Tumor Research Institute of the Chinese Academy of Sciences and KYSE450, KYSE510 and KYSE150 cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). All the cell lines were routinely cultured.
EC1 cells were transfected with virus cloned with stable ACTL6A knockdown sequence and TE7 cells were transfected with virus cloned with full-length ACTL6A sequence. The above cells were then purified with puromycin (Solarbio) for 2 weeks. The concentration of puromycin for EC1 and TE7 was 1.5 ug/mL and 3 ug/mL respectively. Real-time quantitative PCR TRIzol (Solarbio) was used to extract total RNA from each cell line according to a traditional protocol. Genomic DNA was removed and cDNA was synthesized by using the PrimescriptTM RT Reagent Kit with gDNA Eraser (perfect real time) (Takara, Japan). Real-time quantitative PCR was then conducted by using the following specific primers: 5ʹ-GCTGAACGGGAAGCTCACTG and 5ʹ-GTGCTCAGTGTAGCCCAGGA-3ʹ for GAPDH and 5ʹ-TCAGAGGCACCGTGGAATACTAG-3ʹ and 5ʹ-AGGACATAGCCATCGTGGACTG-3ʹ for ACTL6A. Fluorescent quantitation was accomplished by using the SYBR-Green I method (CWBio, China).
Western Blot Analysis
Cells were collected in RIPA buffer (Solarbio, China) with phenylmethylsulfonylfluoride (PMSF) and phosphatase cocktails (Thermo Scientific). The concentration of the total protein of the above lysates was detected using the BCA Protein Assay Kit (Solabio, China). Total protein (20–30μg) was added to each mini pool of SDS-PAGE gels, separated at 80 V for 2 hours and transferred to a polyvinylidene difluoride membrane (Millipore, USA) at 200 mA for 2 hours.
Primary antibodies used in the whole experiment are listed below: ACTL6A (NB100-61628) from Novus Biologicals (USA); p21 (#2946), CDK2 (#18048), S6 (#2217) and pS6 (#5364) from Cell Signaling Technology (USA); S6K1 (sc-8418) from Santa Cruz; GAPDH (10494-1-AP), cyclinD1 (26939-1-AP) and p27 (25614-1-AP) from Proteintech (Wuhan, China).
Cell Viability Assay
Cells were seeded in triplicate into 96-well plates at a density of 4×103 cells per well. Cell Counting Kit 8 (Dojindo, Japan) was used to detect the proliferation speed of cells in the following 4 days according to the manufacturer’s instructions. The absorbance values at specific time points were used to plot cell viability curves.
Edu Assay
Cells (2–4×104) were seeded into each well of a 12-well plate and cultured for 24 hours in an incubator at 37°C and 5% CO2. The kFluor647 Click-iT EdU imaging detection kit (Keygentec, China) was used to evaluate the proliferation speed of cells of different treatments. Cells were incubated with 50 µM EdU for 3 hours, and Triton X-100 was used to increase the permeability of the cells. Cells reacted with an Apollo reaction cocktail, containing kFluor647-azide for 30 minutes and then with Hoechst 33342 for another 30 minutes.
Cell Cycle Analysis
ESCC Cells (2×105) were collected into clean Eppendorf (EP) tubes after digestion with 0.25% trypsin digestive solution without EDTA and washed with PBS buffer 3 times. Then, 70% ethanol was used to fix the cells at 4°C for 2 hours. After washing 3 times with PBS buffer, the cells were incubated with working solution of propidium iodide (PI)/RNase A (Keygentec, China) at a rate of 9:1 and shielded from light for 30 minutes. Fluorescence-activated cells were sorted to determine the distribution in different cell cycle stages by using a NovoCyte Advanteon (ACEA Biosciences, USA), and the result was analyzed by NovoExpress software (ACEA Biosciences, USA).
Cell Apoptosis Analysis
ESCC cells were digested using the same methods for cell cycle detection. The experiment was performed according to the manufacturer’s instructions for the Annexin V-PE/7-AAD Apoptosis Detection Kit (Keygentec, China). Cells were incubated with 50 μL binding buffer, containing 5 μL 7-AAD dye, shielded from light for 15 minutes and then with 450 μL binding buffer, containing 1 μL annexin V-PE, shielded from light for another 15 minutes. Apoptosis was analyzed by NovoCyte Advanteon (ACEA Biosciences, USA), and the result was processed by FlowJo vX.0.7.
In vivo Xenograft Study
ESCC cells (5×106) from each group, stably transfected with Lv-shNC, Lv-shACTL6A-1, Lv-CON and Lv-ACTL6A, were injected with the flanks of male BALB/C nude mice, which were purchased from Charles River (Beijing, China). Subsequently, the volume of each tumor was measured every 3 days. After 22–25 days in total, mice were euthanized and tumors were collected and analyzed by Western blot and immunohistochemistry. Tumors were weighed and tissues were processed for H&E staining and immunohistochemistry. All animal experiments were conducted according to the requirements of the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978) and were approved by the Laboratory Animal Management Committee of the First Affiliated Hospital of Zhengzhou University.
Statistical Analysis
Student’s t test was used to analyze continuous variables and Pearson’s chi-square test was used to analyze categorical variables. The cumulative OS rates were calculated using the Kaplan-Meier method, and differences were compared using a Log-rank test. Cox univariate and multivariate analyses were also conducted to determine independent prognostic factors. All statistical analyses were performed by using IBM SPSS statistics 21. Kaplan-Meier curves were plotted by using PRISM 7 software. R studio software was also used to conduct DCA. P<0.05 was considered to indicate a statistically significant result.
Results
ACTL6A Expression in Human ESCC Tissues
We examined the expression of ACTL6A in ESCC tissues and corresponding adjacent noncancerous tissues (≥5 cm from the tumor lesion boundary) from 128 ESCC patients. The results demonstrated that ACTL6A localized in the cancer cell nuclei of ESCC tissues and was either not expressed or was weakly expressed in the basal cells of adjacent noncancerous tissues. In immunohistochemical staining of ESCC tissues, ACTL6A immunoreactivity was negative in 39 ESCC tissues (30%), with scores of 0 and 1 in 5 (4%) and 34 (26%), respectively, whereas positive expression (scores 2–9) of ACTL6A in the primary tumor was present in 89 of the 128 samples; this rate was significantly higher than that observed in the corresponding noncancerous tissues (24 of 128 positivity, 19%; P<0.001; Table 1, Figure 1A–F).
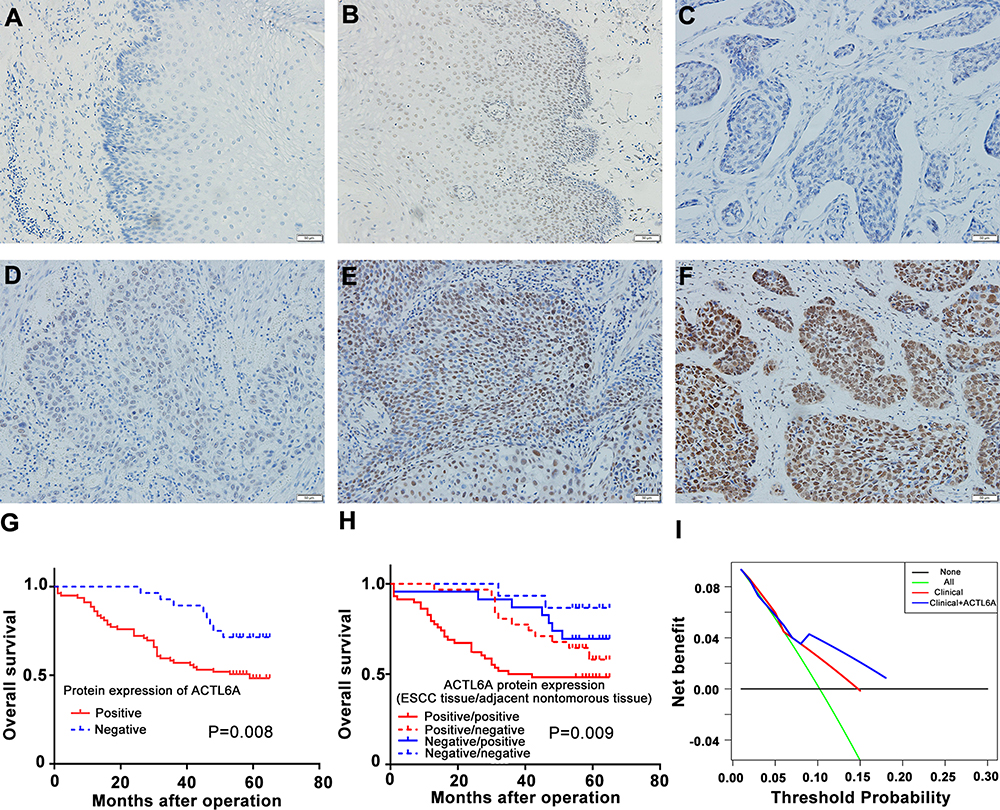 |
Figure 1 IHC analysis of ACTL6A in human ESCC and the clinical significance of its expression level. (A–F) ACTL6A staining in ESCC: (A) Negative noncancerous esophageal tissue; (B) Positive noncancerous esophageal tissue and (C–F) ESCC (C) non-staining (score: 0), (D) weak (score: 1), (E) moderate (score: 6), and (F) strong staining (score: 8) patterns. Scale bars, 50 μm. (G) Kaplan-Meier survival curve of ESCC patients based on ACTL6A expression level in ESCC tissue. (H) Kaplan-Meier survival curve of ESCC patients based on ACTL6A expression level in ESCC tissue and corresponding noncancerous esophageal tissue. (I) DCA to show the comparison of performance. The horizontal black solid line represents the assumption that no patient should take the necessary measures, while the green solid line represents the assumption that all patients should take the necessary measures. The y-axis represents the net benefit, which was calculated by adding points associated with benefits and subtracting those associated with harms. Based on the threshold probabilities obtained, our findings indicated that the clinicopathological model involving the ACTL6A expression level provided a greater net benefit than that without the ACTL6A expression level. Based on the threshold probabilities obtained, our findings indicated that the clinicopathological model involving the ACTL6A expression level (blue solid line) provided a greater net benefit than that without the ACTL6A expression level. (red solid line). |
To assess the significance of ACTL6A expression in ESCC, the associations between ACTL6A expression and the clinicopathological features of 128 ESCC patients were analyzed. As shown in Table 1, positive ACTL6A expression showed no association with patient sex (P=0.623), age (P=0.898), lymph node status (P=0.163) or tumor site (P=0.044). However, positive ACTL6A expression had a positive relationship with tumor size (P<0.001), histological differentiation (P<0.001), tumor status (P=0.008) and TNM stage (P=0.02). Overall, the results above indicated that high ACTL6A expression endowed ESCC cells with aggressive behaviors.
High ACTL6A Expression Has Clinical Prognostic Value and Indicates Poor Outcomes of ESCC Patients
To accomplish a prognostic evaluation, we performed Kaplan–Meier survival analysis of the 128 ESCC patients according to ACTL6A expression level and clinical follow-up time. Our results revealed that positive ACTL6A expression was significantly associated with worse OS (P =0.008) than negative ACTL6A expression (P = 0.008, HR= 2.540, 95% CI: 1.235–5.223; Figure 1G). We also divided ESCC patients into four groups based on the expression of ACTL6A in primary ESCC tissues and corresponding noncancerous tissues. The OS prognosis of the group with positive ACTL6A expression in both ESCC tissues and corresponding noncancerous tissues was the worst among all four groups (P=0.009, Figure 1H).
In addition, univariate and multivariate Cox regression analysis showed that positive ACTL6A expression was a significant and independent prognostic marker of the OS of ESCC patients (Table 2). To conduct further clinical prognostic evaluation, DCA was also performed to depict the net clinical benefit of the original clinical record with and without the data of ACTL6A expression level at certain threshold probabilities (Figure 1I). The results demonstrated that the ACTL6A expression level is negatively associated with the OS of ESCC patients. That is, ACTL6A is a tumor promoter and prognostic biomarker of ESCC.
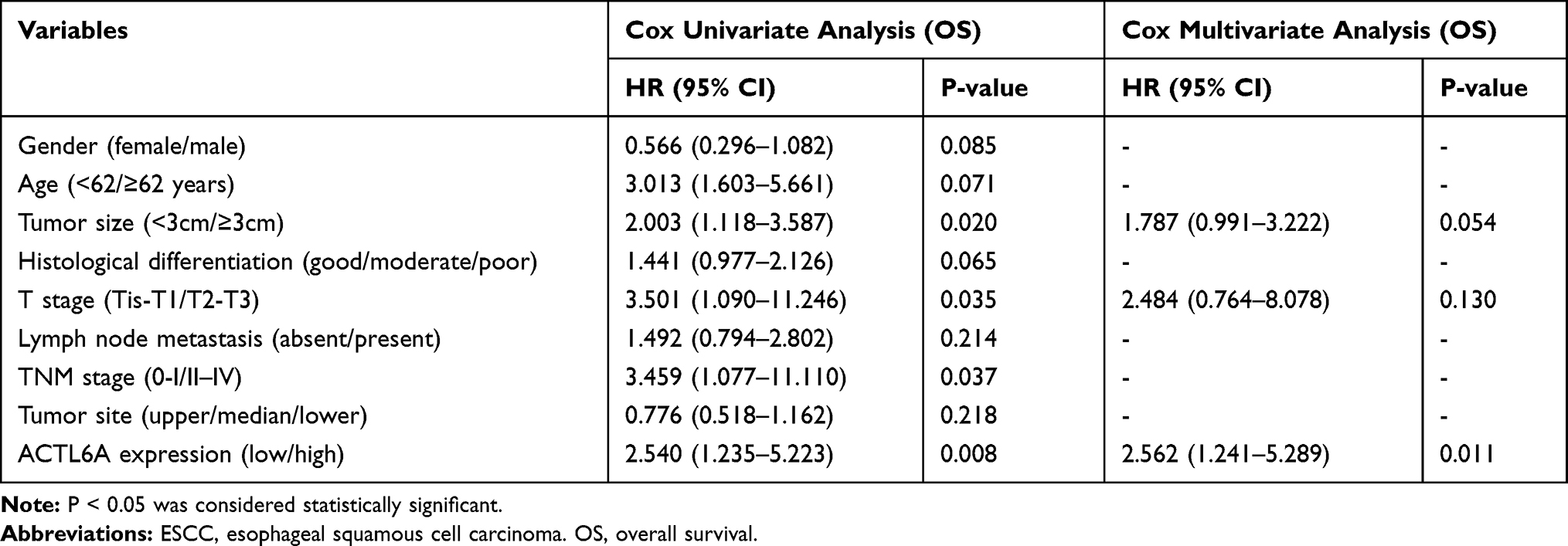 |
Table 2 Cox Univariate and Multivariate Regression Analysis of Prognostic Factors for Overall Survival in 128 ESCC Patients |
ACTL6A Promotes ESCC-Cell Proliferation in vitro
To analyze the proliferation stimulation effect of ACTL6A on ESCC, we used seven ESCC cell lines: TE7, KYSE510, KYSE150, KYSE450, EC1, EC9706 and EC109. Under normal circumstances, we detected the transcription and expression levels of ACTL6A in the above seven cell lines by using RT-PCR and Western blotting. The trend of the mRNA level in the cell lines was consistent with that of the protein level (Figure 2A). According to transcription and expression level of ACTL6A, TE7 cell line with the lowest ACTL6A expression was used to knockdown and EC1 with the highest ACTL6A expression was used to overexpress. After validating gain or loss-of-function of ACTL6A in TE7 and EC1 cell lines, we assayed the growth stimulating activity of ACTL6A using TE7 cells transduced with Lv-ACTL6A to overexpress ACTL6A and with an empty vector as control (Figure 2B). To detect the effects of knockdown of ACTL6A, EC1 cells were transduced with Lv-shACTL6A-1 and Lv-shACTL6A-2 that targeted ACTL6A mRNA for constant degradation. The effect of changed ACTL6A expression on proliferation was tested by CCK8 assay. We found that cell growth was inhibited in cells transduced with Lv-shACTL6A-1 and Lv-shACTL6A-2 in comparison to mock-transduced cells (P<0.05; Figure 2C). However, overexpression of ACTL6A accelerated cell growth in TE7 cells (P<0.05; Figure 2C). The EdU retention assay also validated the proliferation stimulation of ACTL6A in ESCC cells. The percentage of EdU-positive TE7 cells overexpressing ACTL6A was significantly increased compared with that of the control cells (P<0.05, Figure 2D and 2E), while compared with mock-transduced cells, EC1 cells with downregulated ACTL6A displayed a decreased percentage of EdU-positive cells (P<0.05, Figure 2D and E). Thus, ACTL6A functions in the proliferation of human ESCC cells in vitro.
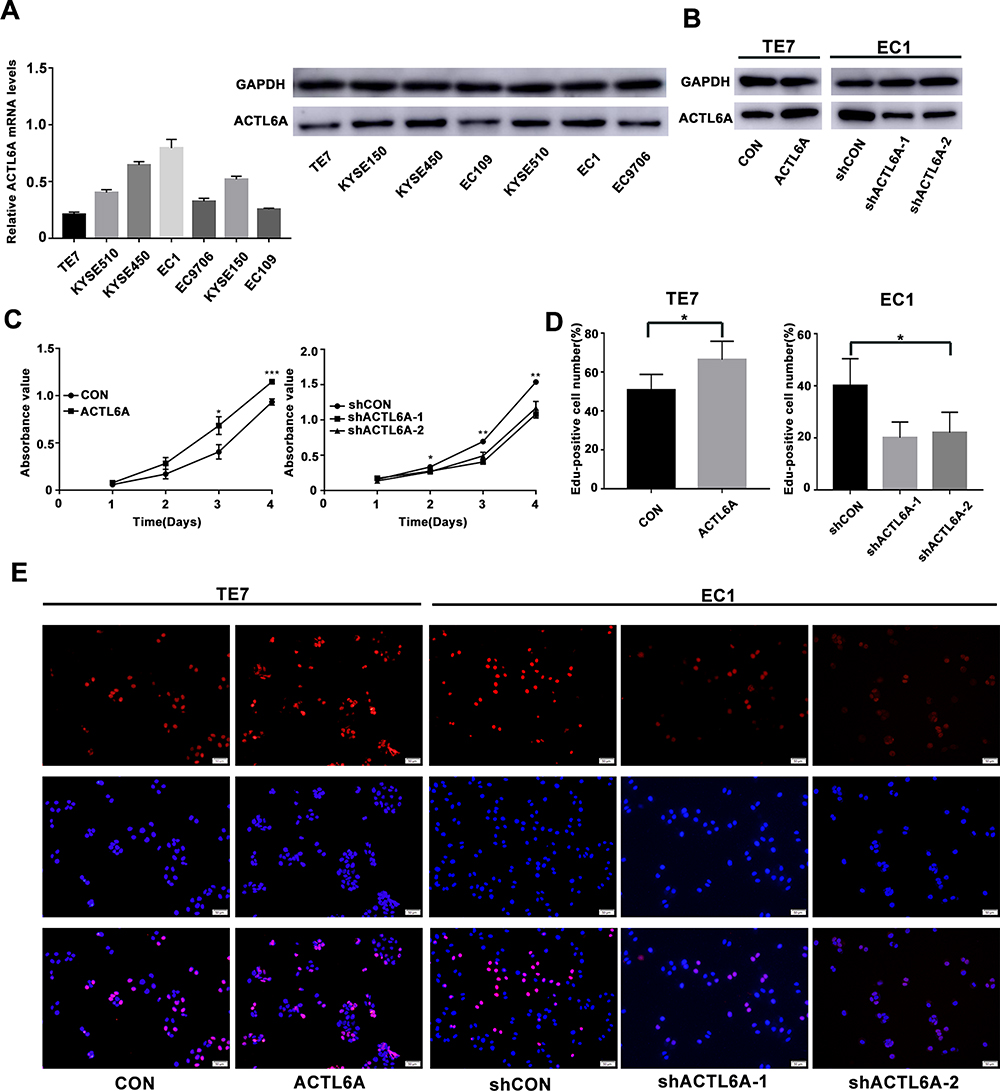 |
Figure 2 ACTL6A expression affects the proliferation of ESCC cell lines. (A) ACTL6A mRNA and protein levels in seven ESCC cell lines were detected by using real-time quantitative PCR and Western blotting. (B) Gain or loss-of-function of ACTL6A in TE7 and EC1 cell lines. (C) Effect of ACTL6A on the cell proliferation rate in TE7 and EC1 cells, which was determined by CCK8 assay at the indicated time points (*P<0.05, **P<0.01, ***P<0.001). Values are the mean±SD (n=3). (D and E) EdU retention assay of ACTL6A overexpression TE7 cells and ACTL6A knockdown EC1 cells. Number of EdU-positive cells is shown in the bar chart (*P<0.05). Values are the mean±SD. Scale bars, 50 μm. |
Underlying Mechanisms of ACTL6A Growth Stimulating Activity in ESCC Cell Lines
To determine the mechanism by which ACTL6A stimulates the growth of ESCC cells, we detected the effect of ACTL6A on cell division and apoptosis. Flow cytometry was performed to assess apoptosis and cell cycle distribution with annexin V-PE/7-AAD and PI, respectively. These results demonstrated that the proportion of cells in G1 phase significantly increased in EC1 cells transduced with Lv-shACTL6A compared with mock-transduced EC1 cells, while the proportion of cells in S phase significantly increased in TE7 cells transduced with Lv-ACTL6A compared with TE7 cells transduced with empty vector (Figure 3A). Given that cell cycle proteins are essential to regulate cell viability, we then determined whether ACTL6A has an effect on the expression of cell cycle regulators in the G1/S phase transition. Western blotting was used to detect the expression levels of not only cyclin D1 and CDK2, but also p21 and p27, cyclin-dependent kinase 4/6 inhibitors, in ESCC cells. As shown in Figure 3B, suppression of ACTL6A in EC1 cells caused G1 arrest with downregulation of cyclin D1 and CDK2 and upregulation of the negative cell cycle regulators p27 and p21, while overexpression of ACTL6A in TE7 led to upregulation of cyclin D1 and CDK2 and downregulation of p27 and p21. However, the level of ACTL6A expression had no effect on the apoptosis of ESCC cells (Figure 3C).
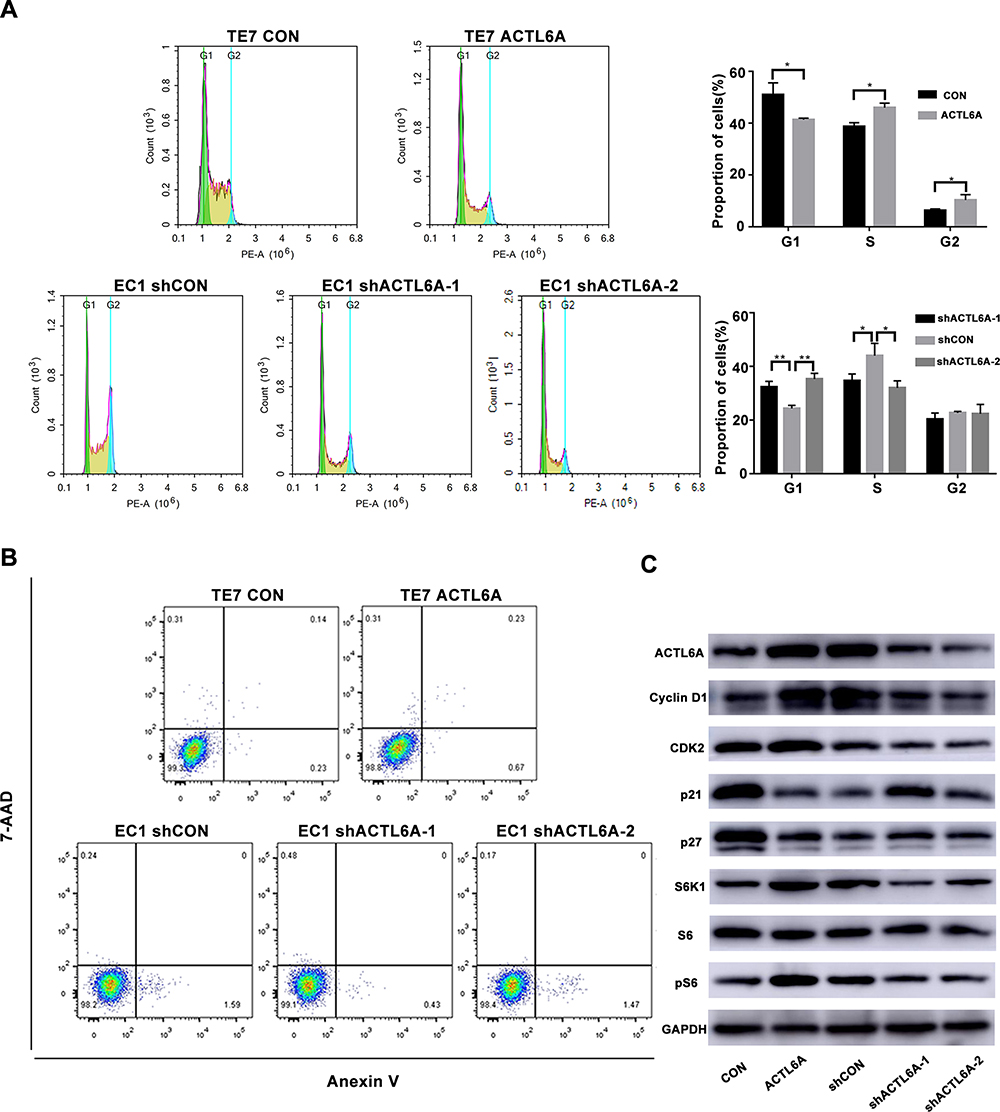 |
Figure 3 Effect of ACTL6A expression on the cell cycle and apoptosis in ESCC cells. (A) Knockdown of ACTL6A resulted in cell cycle arrest in G1 phase of TE7 cells and overexpression of ACTL6A caused more EC1 cells to enter S phase (*P<0.05, **P<0.01). Values are the mean±SD (n=3). (B) ACTL6A expression level barely affected the apoptosis of ESCC cells. The statistical analysis showed that these changes were not significant (not shown, n=3). (C) Cell lysates were analyzed by Western blotting using the indicated antibodies. |
The S6K1/pS6 pathway is critical in regulating cell viability, and pS6 expression is significantly associated with cell cycle protein expressions.30 We also determined whether ACTL6A affects the S6K1/pS6 pathway. ACTL6A expression resulted in upregulated S6K1 and pS6, while ACTL6A knockdown downregulated these proteins; the expression level of S6 remained the same, indicating that ACTL6A regulates the S6K1/pS6 pathway by affecting pS6 phosphorylation in TE7 and EC1 cells (Figure 3C).
Effect of ACTL6A Expression on ESCC Cell Growth in Xenograft Mice
To validate the effect of ACTL6A on ESCC cells in vivo, we subcutaneously injected TE7 cells transduced with Lv-ACTL6A or Lv-CON and EC1 cells transduced with Lv-shACTL6A-1 or Lv-shCON into the flank region of 5-week-old nude mice to establish a mouse xenograft model. As shown in Figure 4A–C, stable shRNA-mediated downregulation of ACTL6A markedly decreased tumor growth in the xenograft mouse model of EC1 cells compared with the control model, whereas stable upregulation of ACTL6A significantly accelerated tumor growth in the xenograft mouse model of TE7 cells compared with the control group. We found that ACTL6A affected the expression of cyclinD1, CDK2 and pS6 by using Western blotting in vivo, which is consistent with the results in vitro (Figure 4D). Immunohistochemical analysis also showed that Ki67, S6K1 and pS6 expression was significantly higher in xenograft mouse model tissues with ACTL6A overexpression than in control tissues and was markedly lower in xenograft mouse model tissues with suppressed ACTL6A than in mock-transduced vector tissues, which indicated that the expression of ACTL6A played a vital role in the proliferation of ESCC cells in vivo (Figure 4E).
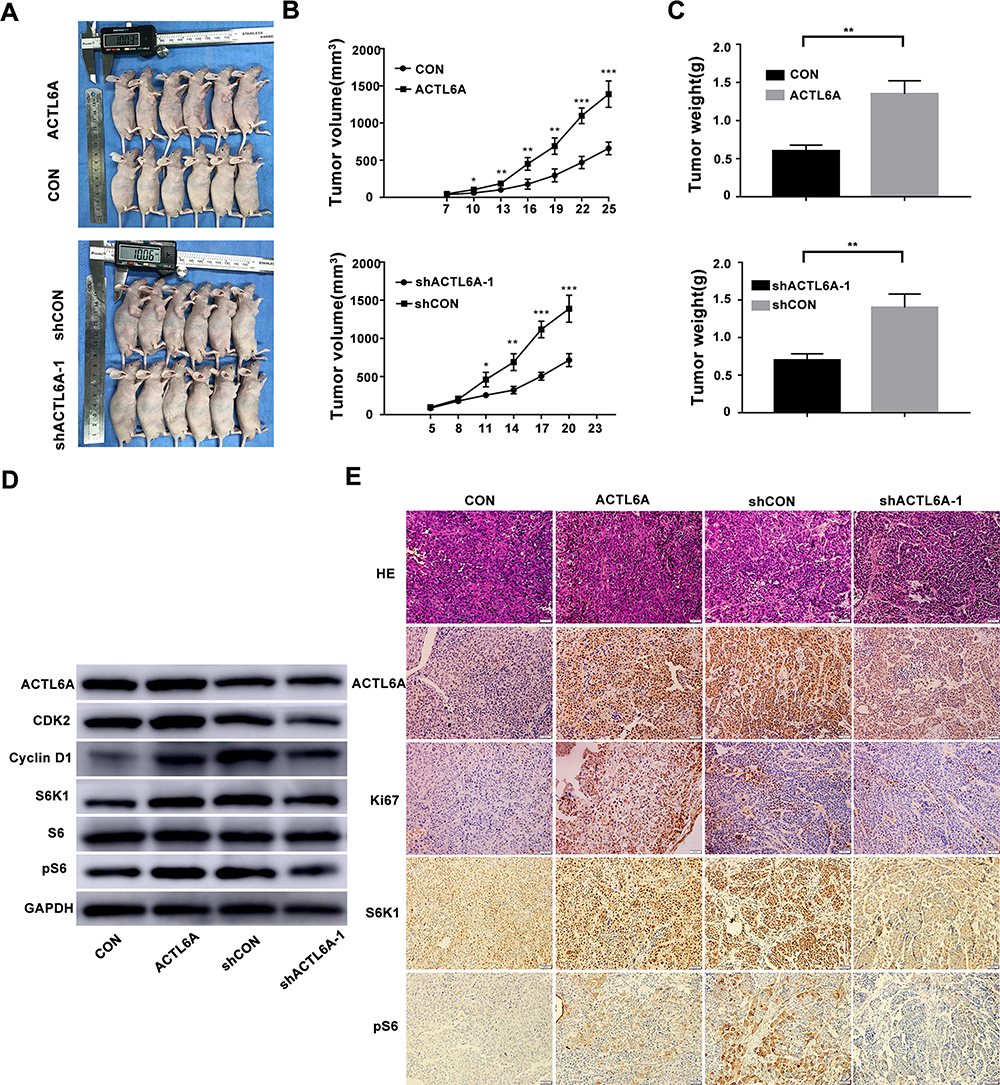 |
Figure 4 ACTL6A expression level plays a role in ESCC tumor growth in vivo. (A–C) Images of nude mice implanted with either ACTL6A-knockdown or ACTL6A overexpressing ESCC cells. Graphs show tumor growth and average tumor weights. *P<0.05, **P<0.01, ***P<0.001 based on Student’s t-test. (D) Western blotting of samples from xenograft mice transplanted with ESCC cells with differential ACTL6A expression levels probed with the indicated antibodies. (E) Representative H&E and IHC staining for Ki67, ACTL6A, S6K1 and pS6 in sections of xenograft mouse masses. Scale bars, 50 μm. |
Discussion
Despite advances achieved in therapeutic approaches, the 5-year survival rate of ESCC is still poor, indicating the demand for identifying new biomarkers for the early screening of high-risk ESCC patients.4 In this research, we demonstrated that patients with positive ACTL6A expression had obviously shortened OS compared with those with negative ACTL6A expression. Additionally, we found that ACTL6A was an independent prognostic factor of ESCC patients. Additionally, our study showed that ACTL6A was overexpressed in ESCC tissues compared to that in corresponding noncancerous mucosal tissue and was significantly associated with tumor differentiation grade, TNM stage, tumor size and T stage. DCA depicted that the clinical model involving ACTL6A provided a greater net benefit than the clinical model without ACTL6A, underscoring that the former possessed better clinical prognostic efficacy. These results are in line with a previous study showing that ACTL6A is upregulated in a variety of cancer types and has a relationship with poor outcome in osteosarcoma and glioma, which indicates that ACTL6A plays an essential role in ESCC progression.8,9,11,22,25,33
Our subsequent mechanistic research demonstrated the tumor promoting effects of ACTL6A upregulation and the antitumor effects of ACTL6A knockdown in ESCC cells in vitro and in vivo. Overexpression of ACTL6A significantly stimulated cell proliferation, while ACTL6A downregulation dramatically inhibited it. Further study indicated that the expression of ACTL6A has a significant effect on the distribution of the cell cycle in ESCC cells, which is in accord with previous research showing that cell cycle dysregulation is associated with sustained proliferative signaling.34 Our flow cytometry results showed that ACTL6A knockdown led to G1 phase arrest, whereas ACTL6A upregulation caused cells to accumulate in S phase in ESCC cells upon PI labeling. To reveal the underlying molecular mechanisms, Western blotting was conducted to assess the expression of key proteins shown to regulate the G1 checkpoint in many previous studies.30,35 We found that suppression of ACTL6A inhibited the expression of cyclin D1 and CDK2 and induced the expression of p27 and p21, which are negative cell cycle regulators, while overexpression of ACTL6A promoted the expression of cyclinD1 and CDK2 and downregulated the expression of p27 and p21. These results are in line with the discovery for laryngeal squamous cell carcinoma cells that ACTL6A cooperates with p63 to inhibit p21, a cell-cycle-regulatory gene, and epidermal growth factor (EGF) family ligand NRG1, and the loss of endogenous ACTL6A results in the accumulation of cells in G1/G07. However, our results demonstrated that upregulation of ACTL6A also led to an increase in the number of cells in G2/M phase. It is probable that the increase in S phase cells was large and resulted in more cells entering G2/M phase. Since the proliferation of ACTL6A overexpressing ESCC cells was enhanced, the possibility that G2/M arrest led to more cells in G2/M phase can almost be excluded. However, to elucidate the exact mechanism, more research related to cell cycle progression should be conducted. We also detected the effect of the ACTL6A expression level on the apoptosis of ESCC cells, and no relationship was found, which further demonstrated that ACTL6A affects the proliferation of ESCC cells mainly by the redistribution of the cell cycle rather than the regulation of apoptosis, in line with a previous study.7
The results of EdU assays demonstrated that overexpression of ACTL6A accelerated DNA synthesis in ESCC cells, which is in line with the detection of cell cycle distribution that upregulated ACTL6A led to more cells entering S phase. Several previous studies may explain this phenomenon: Naoki et al found that the ACTL6A protein itself may play a crucial role in Brg1 complex assembly and maintenance of the complex stability that affects transcription by RNA polymerase I, II and III and regulates cell growth of HeLa cells by regulating a series of genes as transcription factors.17 Bao et al discovered that during differentiation, ACTL6A downregulation facilitates activation of KLF4 and SWI/SNF chromatin modeling complexes to bind to promotors of differentiation genes.23 Ji et al revealed that ACTL6A overexpression promotes cellular proliferation in vitro and in vivo possibly through the direct interaction and stabilization of the transcriptional regulator YAP/TAZ.33 Additionally, our immunohistochemistry detection of human cancer xenografts in nude mice showed that the expression level of Ki67 was consistent with that of ACTL6A. Overall, the above results indicate that ACTL6A possibly contributes to the progression of ESCC cells in vitro and in vivo by increasing DNA synthesis which is completed in S phase.
We also detected the regulatory relationship between ACTL6A and the S6K1/pS6 pathway in vitro and in vivo. Our results showed that knockdown of ACTL6A led to suppression of cell cycle progression by inhibiting the expression of cyclin D1 and CDK2, while changes in the expression of S6K1 and pS6 had the same tendency. Kim et al demonstrated that knockdown of S6K1 and pS6 hindered cell cycle progression by inhibiting the expression of cyclin D and CDK2 in ESCC.30 Thus, it is reasonable to speculate that deletion of ACTL6A leads to suppression of the S6K1/pS6 pathway, resulting in a delay in entry into S phase in ESCC. Additionally, S6K1/pS6 is downstream of the AKT/mTOR pathway, which contributes to the proliferation of esophageal cancer cells, supporting the role of S6K1 in cancer cell proliferation.36 In addition, liver-specific S6 knockout in mice led to severe cell cycle arrest, indicating the critical role of S6 in cell cycle progression.37 At present, the direct regulatory mechanism by which ACTL6A regulates the phosphorylation of S6 to control cell cycle regulators remains unclear.
In conclusion, the present study indicates that a clinical model including the ACTL6A expression level may predict patient outcome more efficiently than a model that does not include ACTL6A expression, and mechanistic studies demonstrated that ACTL6A may function to promote oncogenesis and growth in vitro and in vivo via phosphorylation of S6. Our study, therefore, provides new insight into the mechanisms governing the redistribution of ESCC cells in the cell cycle and the level of DNA synthesis in S phase and possibly indicates an alternative therapeutic target for ESCC.
Acknowledgments
We thank Dr. Dao Xin, Ping Ma and Lulu Guan, who provided help with data collection for this study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Secrier M. Erratum: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2020;70(4):313. doi:10.3322/caac.21609
2. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Talukdar FR, Di Pietro M, Secrier M. di Pietro M, Secrier M, et al. Molecular landscape of esophageal cancer: implications for early detection and personalized therapy. Ann N Y Acad Sci. 2018;1434(1):342–359. doi:10.1111/nyas.13876
4. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349(23):2241–2252. doi:10.1056/NEJMra035010
5. Pan D, Su M, Zhang T, et al. A Distinct Epidemiologic Pattern of Precancerous Lesions of Esophageal Squamous Cell Carcinoma in a High-risk Area of Huai’an, Jiangsu Province, China. Cancer Prev Res. 2019;12:449–462. doi:10.1158/1940-6207.CAPR-18-0462
6. Bao X, Rubin AJ, Qu K, et al. A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 2015;16(1):284. doi:10.1186/s13059-015-0840-9
7. Saladi SV, Ross K, Karaayvaz M, et al. ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell. 2017;31(1):35–49. doi:10.1016/j.ccell.2016.12.001
8. Sun W, Wang W, Lei J, et al. Actin-like protein 6A is a novel prognostic indicator promoting invasion and metastasis in osteosarcoma. Oncol Rep. 2017;37(4):2405–2417. doi:10.3892/or.2017.5473
9. Zeng Z, Yang H, Xiao S. ACTL6A expression promotes invasion, metastasis and epithelial mesenchymal transition of colon cancer. BMC Cancer. 2018;18(1):1020. doi:10.1186/s12885-018-4931-3
10. Shrestha S, Adhikary G, Xu W, et al. ACTL6A suppresses p21(Cip1) expression to enhance the epidermal squamous cell carcinoma phenotype. Oncogene. 2020;39(36):5855–5866. doi:10.1038/s41388-020-1371-8
11. Meng L, Wang X, Liao W, et al. BAF53a is a potential prognostic biomarker and promotes invasion and epithelial-mesenchymal transition of glioma cells. Oncol Rep. 2017;38:3327–3334.
12. Jin ML, Kim YW, Jeong KW. BAF53A regulates androgen receptor-mediated gene expression and proliferation in LNCaP cells. Biochem Biophys Res Commun. 2018;505(2):618–623. doi:10.1016/j.bbrc.2018.09.149
13. Panwalkar P, Pratt D, Chung C, et al. SWI/SNF complex heterogeneity is related to polyphenotypic differentiation, prognosis, and immune response in rhabdoid tumors. Neuro Oncol. 2020;22(6):785–796. doi:10.1093/neuonc/noaa004
14. Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. 2015;1(5):e1500447. doi:10.1126/sciadv.1500447
15. St Pierre R, Kadoch C. Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr Opin Genet Dev. 2017;42:56–67. doi:10.1016/j.gde.2017.02.004
16. Zhao K, Wang W, Rando OJ, et al. Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell. 1998;95(5):625–636. doi:10.1016/S0092-8674(00)81633-5
17. Nishimoto N, Watanabe M, Watanabe S, et al. Heterocomplex formation by Arp4 and beta-actin is involved in the integrity of the Brg1 chromatin remodeling complex. J Cell Sci. 2012;125:3870–3882. doi:10.1242/jcs.104349
18. Keenen B, Qi H, Saladi SV, et al. Heterogeneous SWI/SNF chromatin remodeling complexes promote expression of microphthalmia-associated transcription factor target genes in melanoma. Oncogene. 2010;29(1):81–92. doi:10.1038/onc.2009.304
19. Lu W, Fang L, Ouyang B, et al. Actl6a protects embryonic stem cells from differentiating into primitive endoderm. Stem Cells. 2015;33(6):1782–1793. doi:10.1002/stem.2000
20. Krasteva V, Buscarlet M, Diaz-Tellez A, et al. The BAF53a subunit of SWI/SNF-like BAF complexes is essential for hemopoietic stem cell function. Blood. 2012;120(24):4720–4732. doi:10.1182/blood-2012-04-427047
21. Zhu B, Ueda A, Song X, et al. Baf53a is involved in survival of mouse ES cells, which can be compensated by Baf53b. Sci Rep. 2017;7(1):14059. doi:10.1038/s41598-017-14362-4
22. Taulli R, Foglizzo V, Morena D, et al. Failure to downregulate the BAF53a subunit of the SWI/SNF chromatin remodeling complex contributes to the differentiation block in rhabdomyosarcoma. Oncogene. 2013;33:2354–2362. doi:10.1038/onc.2013.188
23. Bao X, Tang J, Lopez-Pajares V, et al. ACTL6a enforces the epidermal progenitor state by suppressing SWI/SNF-dependent induction of KLF4. Cell Stem Cell. 2013;12(2):193–203. doi:10.1016/j.stem.2012.12.014
24. Park J, Wood MA, Cole MD. BAF53 forms distinct nuclear complexes and functions as a critical c-Myc-interacting nuclear cofactor for oncogenic transformation. Mol Cell Biol. 2002;22(5):1307–1316. doi:10.1128/MCB.22.5.1307-1316.2002
25. Xiao S, Chang RM, Yang MY, et al. Actin-like 6A predicts poor prognosis of hepatocellular carcinoma and promotes metastasis and epithelial-mesenchymal transition. Hepatology. 2016;63:1256–1271. doi:10.1002/hep.28417
26. Sima X, He J, Peng J, et al. The genetic alteration spectrum of the SWI/SNF complex: the oncogenic roles of BRD9 and ACTL6A. PLoS One. 2019;14(9):e0222305. doi:10.1371/journal.pone.0222305
27. Kim K, Pyo S, Um SH. S6 kinase 2 deficiency enhances ketone body production and increases peroxisome proliferator-activated receptor alpha activity in the liver. Hepatology. 2012;55(6):1727–1737. doi:10.1002/hep.25537
28. Um SH, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431(7005):200–205. doi:10.1038/nature02866
29. Meyuhas O. Ribosomal Protein S6 Phosphorylation: four Decades of Research. Int Rev Cell Mol Biol. 2015;320:41–73.
30. Kim S-H, Jang YH, Chau GC, et al. Prognostic significance and function of phosphorylated ribosomal protein S6 in esophageal squamous cell carcinoma. Modern Pathology. 2012;26(3):327–335. doi:10.1038/modpathol.2012.161
31. Karbowniczek M, Yu J, Henske EP. Renal Angiomyolipomas from Patients with Sporadic Lymphangiomyomatosis Contain Both Neoplastic and Non-Neoplastic Vascular Structures. Am J Pathol. 2003;162(2):491–500. doi:10.1016/S0002-9440(10)63843-6
32. Plas DR, Thomas G. Tubers and tumors: rapamycin therapy for benign and malignant tumors. Curr Opin Cell Biol. 2009;21:230–236. doi:10.1016/j.ceb.2008.12.013
33. Ji J, Xu R, Zhang X, et al. Actin like-6A promotes glioma progression through stabilization of transcriptional regulators YAP/TAZ. Cell Death Dis. 2018;9:517. doi:10.1038/s41419-018-0548-3
34. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
35. Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33(39):4724–4734. doi:10.1038/onc.2013.418
36. Liu XJ, Song MQ, Wang PL, et al. Targeted therapy of the AKT kinase inhibits esophageal squamous cell carcinoma growth in vitro and in vivo. International Journal of Cancer. 2019;145:1007–1019. doi:10.1002/ijc.32285
37. Wang Y, Ding Q, Yen C, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 2012;21(3):374–387. doi:10.1016/j.ccr.2011.12.028
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.