Back to Journals » International Journal of Nanomedicine » Volume 16
Active Tumoral/Tumor Environmental Dual-Targeting by Non-Covalently Arming with Trispecific Antibodies or Dual-Bispecific Antibodies on Docetaxel-Loaded mPEGylated Nanocarriers to Enhance Chemotherapeutic Efficacy and Minimize Systemic Toxicity
Authors Cheng WJ, Lin SY, Chen M, Chen LC, Ho HO ![]() , Chuang KH, Sheu MT
, Chuang KH, Sheu MT ![]()
Received 13 January 2021
Accepted for publication 7 May 2021
Published 10 June 2021 Volume 2021:16 Pages 4017—4030
DOI https://doi.org/10.2147/IJN.S301237
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Phong A Tran
Wei-Jie Cheng,1,* Shyr-Yi Lin,2,3,* Michael Chen,4 Ling-Chun Chen,5 Hsiu-O Ho,1 Kuo-Hsiang Chuang,4,6 Ming-Thau Sheu1
1School of Pharmacy, College of Pharmacy, Taipei Medical University, Taipei, Taiwan; 2Division of Gastroenterology, Department of Internal Medicine, Wan Fang Hospital, Taipei Medical University, Taipei, Taiwan; 3Department of General Medicine, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan; 4PhD Program in Clinical Drug Development of Chinese Herbal Medicine, Taipei Medical University, Taipei, Taiwan; 5Department of Biotechnology and Pharmaceutical Technology, Yuanpei University of Medical Technology, Hsinchu, Taiwan; 6Graduate Institute of Pharmacognosy, Taipei Medical University, Taipei, Taiwan
*These authors contributed equally to this work
Correspondence: Kuo-Hsiang Chuang; Ming-Thau Sheu Email [email protected]; [email protected]
Purpose: This study was aimed at developing the trispecific antibodies (anti-EGFR/anti-FAP/anti-mPEG, TsAb) or dual bispecific antibodies (anti-EGFR/anti-mPEG and anti-FAP/anti-mPEG) docetaxel (DTX)-loaded mPEGylated lecithin-stabilized micelles (mPEG-lsbPMs) for improving the targeting efficiency and therapeutic efficacy.
Methods: mPEG-lsbPMs were simply prepared via thin film method. The trispecific antibodies or bispecific antibodies bound the mPEG-lsbPMs by anti-mPEG Fab fragment. The formulations were characterized by DLS and TEM; in vitro and in vivo studies were also conducted to evaluate the cellular uptake, cell cytotoxicity and therapeutic efficacy.
Results: The particle sizes of mPEG-lsbPMs with or without the antibodies were around 100 nm; the formulations showed high encapsulation efficiencies of 97.12%. The TsAb and dual bispecific antibodies were fabricated and demonstrated their targeting ability. Two EGFR-overexpressed cell lines (HT-29 and MIA PaCa-2) were co-cultured with FAP-overexpressed WS1 cells (HT-29/WS1; MIA PaCa-2/WS1) to mimic a tumor coexisting in the tumor microenvironment. Cellular binding study revealed that the binding of anti-FAP micelles to three co-culture ratios (4:1, 1:1, and 1:4) of HT-29/EGFR to WS1/FAP was significantly higher than that for TsAb micelles and dual (1:1) micelles, and the binding of those targeting antibodies to WS1/FAP and MIA PaCa-2/EGFR was equally efficacious resulting in a similar binding amount of the TsAb and dual BsAbs (1:1) with the co-culture of MIA PaCa-2/EGFR and WS1/FAP at a 1:1 ratio. Antitumor efficacy study showed that treatment with DTX-loaded mPEG-lsbPMs modified with or without BsAbs, dual BsAbs (1:1), and TsAbs was enhanced in inhibiting tumor growth compared with that for Tynen® while showing fewer signs of adverse effects.
Conclusion: Active targeting of both tumors and TAF-specific antigens was able to increase the affinity of DTX-loaded mPEG-lsbPMs toward tumor cells and TAFs leading to successive uptake by tumor cells or TAFs which enhanced their chemotherapeutic efficacy against antigen-positive cancer cells.
Keywords: lecithin-stabilized mPEGylated mixed polymeric micelle, bispecific antibody, trispecific antibody, active targeting, tumor-associated fibroblast, tumor antigen
Introduction
Chemotherapeutic drug-loaded nanocarriers bring various benefits to traditional chemo-therapeutic agents for treating solid tumors. Methoxy polyethylene glycol (mPEG)ylated modification of the surface of nanocarriers achieves a sheltering effect that precludes non-specific interactions with the reticuloendothelial system (RES)1 and serum proteins2 leading to an extension of the in vivo half-life,3 a reduction in side effects,4 and enhancement of therapeutic efficacies.5 With the presence of a mPEGylated modification, e.g., 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000), Kao et al. offered a simple one-step method of non-covalent binding to mPEGylated nanocarriers (NCs) with anti-mPEG/antitumor antigen bispecific antibodies (BsAbs) to achieve active targeting of tumors, resulting in increased tumor accumulation and enhanced therapeutic efficacies of chemotherapeutic drugs.6,7 The BsAbs used for this purpose were constituted of antigen-binding fragments (Fabs) or single-chain variable fragments (scFvs) of an anti-mPEG Ab (that binds to the terminal methoxy portion of mPEG) and an antitumor Ab (that binds to specific tumor antigens). When mPEGylated NCs were modified with the anti-mPEG/antitumor antigen BsAbs, the BsAbs were attached onto their surface and offered anti-tumor binding abilities and enhanced cellular uptake or therapeutic efficacy to the chemodrugs loaded in the NCs. Such a BsAb-based modification, compared with the random chemical coupling utilized by the formation of Ab-drug conjugates (ADCs), offers an achievable and efficacious procedure to generate Ab-modified NCs or other nanomedicines, and ensures an outward orientation of functional antitumor Abs on NCs.6,8,9 By employing a suitable antitumor moiety to BsAbs, this approach can be adjusted to various cancers in which tumor-specific (-associated) antigens (TSAs or TAAs) are overexpressed on cell surfaces, such as human epidermal growth factor receptor (EGFR) 2 (HER2), EGFR, prostate-specific membrane antigen (PSMA), etc.10,11
However, another barrier encountered with solid tumor treatment with such BsAb non-covalently bound mPEGylated NCs is that tumor-associated fibroblasts (TAFs) in tumor environments block the entry of active targeting NCs and reduce their therapeutic efficacy. TAFs are commonly considered to be derived from stromal fibroblasts under stimulation of transforming growth factor (TGF)- β secreted by cancer cells.12 In vitro research showed that resident fibroblasts upregulate gene expressions of TAF surface markers, including smooth muscle actin (SMA) and fibroblast activation protein (FAP), and increase collagen production.13 TAFs also contribute to tumor promotion, tissue remodeling, and metastasis by secreting TGF-β, vascular endothelial growth factor (VEGF), and matrix metalloproteinases (MMPs).14 More importantly, an abnormal collagen morphology and altered composition of the extracellular matrix (ECM) regulated by TAFs form physical barriers and increase interstitial fluid pressure, leading to the inefficient entry of therapeutic agents into tumor cell areas.15,16 High levels of TAFs in various types of solid tumors were shown to be correlated with poor prognosis of such tumors.17 Several studies further confirmed that FAP could be a valuable and safe target, and TAF-targeting NCs could be combined with other more effective anticancer agents to achieve synergistic effects.18–21
To simultaneously target tumor cells and TAFs to enhance therapeutic efficacy and minimize systemic toxicity for chemodrug-loaded mPEGylated NCs, a trispecific Ab (TsAb), viz., anti-mPEG/anti-FAP/antitumor antigen TsAb, or dual BsAbs (anti-mPEG/anti-FAP BsAb and anti-mPEG/antitumor antigen BsAbs) that enable targeting of both cancer cells and TAFs (FAP+) were developed, as shown in Figure 1A. In this study, the TsAb was genetically engineered to contain an anti-mPEG Fab in which light and heavy chains were linked with an anti-FAP scFv and an anti-EGFR scFv, respectively (with the resulting TsAb being designated anti-EGFR-FAP-TsAb). Dual BsAbs were genetically engineered to contain an anti-mPEG Fab, in which light and heavy chains were respectively linked with an anti-EGFR scFv (designated anti-EGFR/anti-mPEG BsAb or anti-EGFR-BsAb) and an anti-FAP scFv (designated anti-FAP/anti-mPEG BsAb or anti-FAP-BsAb). Previously developed docetaxel (DTX)-loaded mPEGylated lecithin-stabilized polymeric micelles (DTX-loaded mPEG-lsbPMs)8 were non-covalently modified with either the anti-EGFR-FAP-TsAb, individual BsAbs (anti-EGFR-BsAbs or anti-FAP-BsAbs), or dual BsAbs (anti-EGFR-BsAbs and anti-FAP-BsAbs) via the anti-mPEG terminus to offer binding affinity to the EGFR and FAP. Selective targeting and cellular uptake by EGFR+ cancer cells (HT-29/MIA-PaCa-2) and FAP+ fibroblasts (WS1/FAP) of anti-EGFR-FAP-TsAb-DTX-loaded mPEG-lsbPMs was then examined by a cell-based enzyme-linked immunosorbent assay (ELISA) and flow cytometry, and compared with that of DTX-loaded mPEG-lsbPMs modified with each individual or dual BsAbs (anti-EGFR/FAP-BsAbs-DTX-loaded mPEG-lsbPMs) and non-targeting DTX-loaded mPEG-lsbPMs. We also investigated the cytotoxicity of drugs against individual cell lines (HT-29/MIA PaCa2) or a co-culture of each of those two cell lines with WS1/FAP cells. Last, in vivo treatment of a xenograft mice model co-implanted with MIA PaCa-2 cells and WS1/FAP cells was performed to examine the enhanced therapeutic effect and reduced systemic toxicity of anti-EGFR-FAP-TsAb-DTX-loaded mPEG-lsbPMs and anti-EGFR/FAP-BsAbs-DTX-loaded mPEG-lsbPMs in tumors surrounded by TAFs.
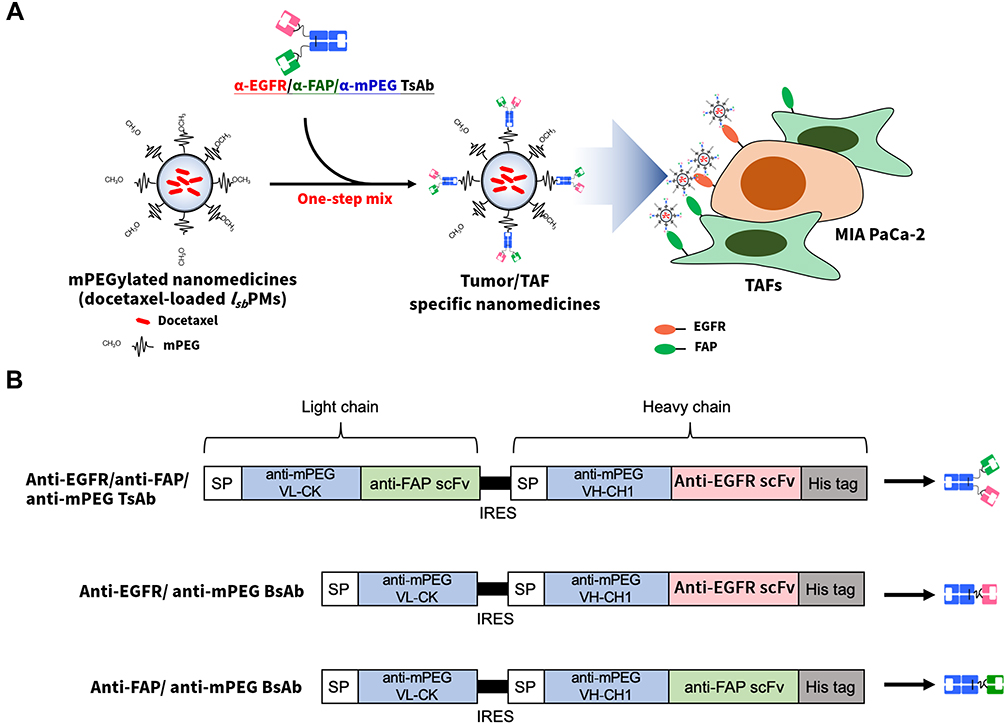 |
Figure 1 Overview of the trispecific antibody (TsAb)-modified nanomedicine strategy for simultaneously eliminating tumors containing tumor-associated fibroblasts (TAFs). (A) The one-step arming strategy relied on the non-covalent modification of mPEGylated nanomedicines (liposomal doxorubicin, Lipo-Dox) with an anti-human epidermal growth factor receptor 2 (HER2)/anti-fibronectin activation protein (FAP)/anti-mPEG TsAb via the anti-mPEG Fab part. Armed mPEGylated nanomedicines can actively target HER2+ breast cancer cells and FAP+ TAFs via the anti-HER2 single-chain variable fragment (scFv) and anti-FAP scFv parts, respectively. (B) Genetic structures of the TsAb and bispecific antibodies (BsAbs), which were composed of a murine immunoglobulin kappa chain leader sequence (IgK SP), a humanized anti-mPEG light chain (VL-CK), an internal ribosome entry site (IRES) sequence, a humanized anti-mPEG heavy chain fragment (VH-CH1), a glycine-serine peptide linker, scFv fragments of anti-HER2 or anti-FAP, and a polyhistidine-tag (6x His). |
Materials and Methods
Materials and Cell Lines
Docetaxel was purchased from ScinoPharm Taiwan (Tainan, Taiwan). Soybean lecithin (S 100) was provided by Lipoid (Ludwigshafen, Germany). 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) and 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG5000) were purchased from NOF (Tokyo, Japan). Kolliphor® TPGS was obtained from BASF (Florham Park, NJ, USA). 3.30-Dioctadecyloxacarbocyanine perchlorate (DIO) was obtained from Sigma-Aldrich (St. Louis, MO, USA). Tynen® (docetaxel), a generic product, was provided by TYY Pharmaceutical (Taipei, Taiwan). Fetal bovine serum, non-essential amino acids and sodium pyruvate were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Dulbecco’s Modified Eagle Medium (DMEM), Minimum Essential Medium (MEM) and penicillin/streptomycin/amphotericin B solution were purchased from Corning Incorporated (Corning, NY, USA). Other reagents and solvents were of analytical grade. WS1, MIA PaCa-2 and HT-29 were supplied by American Type Culture Collection (Manassas, VA, USA). WS1/FAP cells were human WS1 fibroblasts engineered to overexpress human FAP by lentiviral infection.
Preparation of DTX-Loaded mPEG-lsbPMs and LsbMDDs Bound with TsAbs or BsAbs
The DTX-loaded mPEG-lsbPMs were prepared via thin film hydration method as in the previous report8 with minor modifications. Briefly, 30 mg of DTX and 150 mg of DSPE-2000 were dissolved in 10 mL of methanol with addition of an adequate amount of TPGS, which acted as antioxidants. The methanol solution was evaporated in a rotary evaporator (R-114; Buchi, Flawil, Switzerland) to form the thin film. 200 mg of lecithin and 81.5 mg of DSPE-PEG5000 were dispersed in 10 mL of ddH2O and subjected to ultrasonication (VCX 750; Sonics & Materials, Newtown, CT, USA) to form the nanosuspension. Then, the 10 mL of nanosuspension were used to hydrate the thin film and sonicated at 100% amplitude to form self-assembled DTX-loaded mPEG-lsbPMs. Any unencapsulated drug was discarded by filtering with the 0.22 µm membrane (Millipore, Billerica, MA, USA). An appropriate amount of the trehalose was added into the solution. The solution was lyophilized and stored at 4°C. For cellular study, the DIO-loaded mPEG-lsbPMs was prepared with the same procedure, instead of using the DTX. To form the TsAbs LsbPMs, the lsbPMs were simply mixed with TsAbs at the TsAbs/mPEG molar ratio of 1:100; then, the mixed solution was incubated at room temperature for 30 mins. Preparation of BsAbs lsbPMs followed the same procedure.
Preparation and Binding Affinity and of BsAbs and TsAbs
Gene structure of BsAbs and TsAbs are shown in Figure 1B. Preparation of antibodies was similar with the previous report6 with slight modification. In short, the genes containing the BsAbs or TsAbs were incorporated into the pLNCX vector. The pLNCX vector was transfected into the Expi293 cells for scale production. After a few days, the BsAbs or TsAbs were harvested via affinity purification by HisTrap HP columns (GE Healthcare, Little Chalfont, UK). Concentrations of BsAbs and TsAbs were evaluated by bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Waltham, MA). For binding affinity, WS1/FAP, HT-29 and MIA PaCa-2 cells were used to assess the binding specificity of BsAbs and TsAbs to FAP and EGFR. The cells were incubated with either BsAbs or TsAbs; then goat anti-Human IgG Fab FITC was added to label the BsAbs and TsAbs. The antibody binding was quantitatively evaluated by flow cytometry. The binding affinity to mPEG was evaluated by enzyme-linked immunosorbent assay (ELISA), which coated the mPEG onto the 96-well plate and added the gradient concentration of BsAbs or TsAbs. Then, goat anti-Human IgG Fab HRP was added to detect the BsAbs or TsAbs. The plates were washed with PBS and ABTS substrate was added for detection. The absorbance was measured at 405 nm with the ELISA reader (Cytation 3; BioTek Instruments, Winooski, VT, USA).
Physical Characterization of DTX-Loaded mPEG- lsbPMs and TsAbs LsbPMs
The particle size and zeta potential were measured using Zetasizer ZSP (Malvern Instruments, Malvern, UK) by adding 1 mL of the DTX-loaded mPEG-LsbPMs solution to a desired concentration of particle numbers into a folded capillary zeta cell. The concentration of drugs was analyzed by HPLC. The encapsulation efficacy percentage and drug loading percentage were calculated by the following formulas:
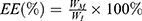
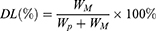
where WM is the amount of drug in the nanoparticles, WI is the amount of the initial feeding drug, and WP is the amount of the total polymers.
Cell Culture and Co-Culture
For the HT-29 and WS1/FAP co-culture experiment, the HT-29 and WS-1 were separately maintained in MEM, supplemented with 10% fetal bovine serum, 1% non-essential amino acids solution, 1% sodium pyruvate, 1% L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate, 0.3% N-acetyl-L-cysteine, and 1% penicillin/streptomycin/amphotericin B solution. For the MIA PaCa-2 and WS1/FAP co-culture experiment, the MIA PaCa-2 and WS1/FAP were separately cultured in half DMEM and half MEM, supplemented with 10% fetal bovine serum, 1.25% horse serum, 0.5% non-essential amino acids solution, 0.5% sodium pyruvate, 0.5% L-Ascorbic acid 2-phosphate sesquimagnesium salt hydrate, 0.15% N-acetyl-L-cysteine, and 1% penicillin/streptomycin/amphotericin B solution.
In vitro Drug Release Studies
To evaluate the drug release profile of docetaxel, each sample solution containing 0.25 mg/mL of docetaxel was transferred into the dialysis bag (MWCO 6k-8k, Cellu-Sep® T1; Orange Scientific, Seguin, TX, USA). The bag was soaked in the 25 mL of pH 7.4 PBS with 0.5% Tween 80 at 37°C with gentle shaking of 100 rpm. 1 mL of medium was collected at the predetermined time intervals (1, 2, 4, 6, 8, 10, 24, 48, and 72 h) and replaced with 1 mL of fresh buffer. The concentration of released docetaxel was measured by HPLC as mentioned previously.
In vitro Cellular Binding of DIO-Loaded mPEG-lsbPMs
Co-cultures of HT-29/EGFR:WS1/FAP (1:4, 1:1, and 4:1) and MIA PaCa-2/EGFR:WS1/FAP (1:4, 1:1, and 4:1) were seeded at a density of 5⋅104 cells/well on 12-well microplates. The non-targeting DIO-loaded mPEG-lsbPMs (labeled Micelle), anti-EGFR-BsAbs-DIO-loaded mPEG-lsbPMs (labeled Anti-EGFR Micelle), anti-FAP-BsAbs-DIO-loaded mPEG-lsbPMs (labeled Anti-FAP Micelle), anti-EGFR-/anti-FAP-DIO-loaded mPEG-lsbPMs (labeled Dual (1:1) Micelle), or anti-EGFR-FAP-TsAbs-DIO-loaded mPEG-lsbPMs (labeled TsAbs Micelle) with a molar ratio of BsAbs or TsAbs to mPEG of 0.01:1) were added to separate wells and incubated for 2 h. Cells were collected, and analyzed by flow cytometry to quantitatively evaluate cellular binding to HT-29/EGFR:WS1/FAP and MIA PaCa-2/EGFR:WS1/FAP co-cultures at three different ratios (1:4, 1:1, and 4:1).
Cell Viability
Co-cultures of HT-29/EGFR:WS1/FAP and MIA PaCa-2/EGFR:WS1/FAP different ratios (1:4, 1:1, and 4:1) were incubated to mimic the tumor microenvironment (TME). The in vitro cytotoxicity of different formulations was evaluated. Co-culture cells were seeded in triplicate into 24-well plates at a density of 5⋅104 cells/well. After incubation overnight, the medium was replaced with fresh medium containing either Docetaxel, Micelle, Anti-FAP Micelle, Dual (1:1) Micelle, Anti-EGFR Micelle or TsAbs Micelle. After 48 h of incubation, 5.5 mg/mL of MTT was added into each well and the cells were incubated for 3 h. The medium was dried out, and 200 µL of DMSO was added to dissolve the formazan crystals. The absorbance was measured at 550 nm with an ELISA reader (Cytation 3; BioTek Instruments, Winooski, VT, USA).
In vivo Pharmacokinetic (PK) Studies of Intravenous Administration
Eight-week-old male Sprague-Dawley rats were administered a single dose of 10 mg/kg of Docetaxel, DTX-loaded mPEG-lsbPMs (labeled Micelle), anti-EGFR-BsAbs-DTX-loaded mPEG-lsbPMs (labeled Anti-EGFR Micelle), anti-FAP-BsAbs-DTX-loaded mPEG-lsbPMs (labeled Anti-FAP Micelle), anti-EGFR-/anti-FAP-DTX-loaded mPEG-lsbPMs (labeled Dual (1:1) Micelle), or anti-EGFR-FAP-TsAbs-DTX-loaded mPEG-lsbPMs (labeled TsAbs Micelle) via a jugular vein injection (with three rats per group). Blood was collected from the jugular vein in heparinized tubes at 5, 15, and 30 min and 1, 2, 4, 6, 8, 10, 24, 48, and 72 h after administration. Blood samples were centrifuged at 3000 rpm for 10 min to obtain plasma and analyzed by TQ-XS (Waters Corp., Manchester, UK).
Tumor-Inhibition Studies
All male nu/nu mice received a subcutaneous injection of 100 µL (containing 5⋅105 cells) of the MIA PaCa-2:WS1/FAP (1:1) cell suspension in Matrigel into their right thigh. These tumor-bearing mice with around 150 mm3 tumor volumes were randomized into seven groups (5 mice per group): one control group (which received phosphate-buffered saline (PBS)) and six groups including Docetaxel, DTX-loaded mPEG-lsbPMs (labeled Micelle), anti-EGFR-BsAbs-DTX-loaded mPEG-lsbPMs (labeled Anti-EGFR Micelle), anti-FAP-BsAbs-DTX-loaded mPEG-lsbPMs (labeled Anti-FAP Micelle), anti-EGFR-/anti-FAP-DTX-loaded mPEG-lsbPMs (labeled Dual (1:1) Micelle), or anti-EGFR-FAP-TsAbs-DTX-loaded mPEG-lsbPMs (labeled TsAbs Micelle) (5 mg DTX/kg, n = 5). The mice were treated through the tail vein every 3 days 4 times. The length and width of the tumor as well as the weight of mice were recorded every 3 days. The tumor volume was calculated by using the formula, 1/2 length x width2.
In vivo Biodistribution Studies
Biodistribution studies were performed in MIA PaCa-2/EGFR:WS1/FAP-bearing BALB/c nude mice. After the tumor volume reached 150 mm3, the mice were randomly assigned to six groups, Docetaxel, DTX-loaded mPEG-lsbPMs (labeled Micelle), anti-EGFR-BsAbs-DTX-loaded mPEG-lsbPMs (labeled Anti-EGFR Micelle), anti-FAP-BsAbs-DTX-loaded mPEG-lsbPMs (labeled Anti-FAP Micelle), anti-EGFR-/anti-FAP-DTX-loaded mPEG-lsbPMs (labeled Dual (1:1) Micelle), or anti-EGFR-FAP-TsAbs-DTX-loaded mPEG-lsbPMs (labeled TsAbs Micelle). The mice were intravenously administered with 5 mg DTX through the tail vein. At the various time points (2 and 8 h), the mice were anesthetized and sacrificed by CO2. The mice were transcardially perfused with PBS buffer containing 10 IU heparin until the major organ is cleared of blood. The major organs including heart, liver, spleen, lungs, kidneys, and tumor were harvested and weighed. Organs were homogenized with 400 μL of PBS/10 IU heparin solution and extracted. The DTX extraction was analyzed by TQ-XS.
Statistical Analysis
Data are presented as the mean ± standard deviation (SD) of each group. The significance among samples was determined with a one-way analysis of variance (ANOVA). Significant differences between groups were indicated by * p<0.05, ** p<0.01, and *** p<0.001.
Results
Physical Characterization of DTX-Loaded mPEG-lsbPMs
In order to implement mPEGylation on the previously developed robust and promising delivery systems recognized as lecithin-stabilized polymeric micelles (mPEG-lsbPMs),22,23 the thin film of self-assembling micelles was first formed and then hydrated with a lecithin/DSPE-PEG5K nanosuspension in this study as shown in Figure 2A. The micellar core of the so-obtained DTX-loaded mPEG-lsbPMs was composed of DTX and DSPE-PEG2K, while the lipid shell consisted of lecithin and DSPE-PEG5K at a ratio of 40:15 (w/w). As depicted in Figure 2C, the average particle size of DTX-loaded mPEG-lsbPMs was found to be 109.7±1.3 nm, and the polydispersity index (PDI) was determined to be 0.176±0.018 with a zeta potential of −33.8±1.60 mV. The encapsulation efficiency (EE) was 97.1% and drug loading (DL) was 6.3%. The structures of DTX-loaded mPEG-lsbPMs as observed in TEM images (Figure 2B) exhibited a spherical morphology and were well dispersed and separated. With non-covalent modification of BsAbs or TsAbs, the particle size of BsAbs LsbPMs or TsAbs LsbPMs was respectively 92.90±0.73 and 94.48±0.63 nm; the zeta potential increased to −12.9±0.96 and −10.4±0.30 mV, respectively. The larger particle size of TsAbs LsbPMs may result from the higher molecular weight (MW = 104 KD) compared with the BsAbs (MW = 75 KD). Results indicated that BsAbs and TsAbs can successfully bind the mPEG-lsbPMs and altered the physical property of mPEG-lsbPMs.
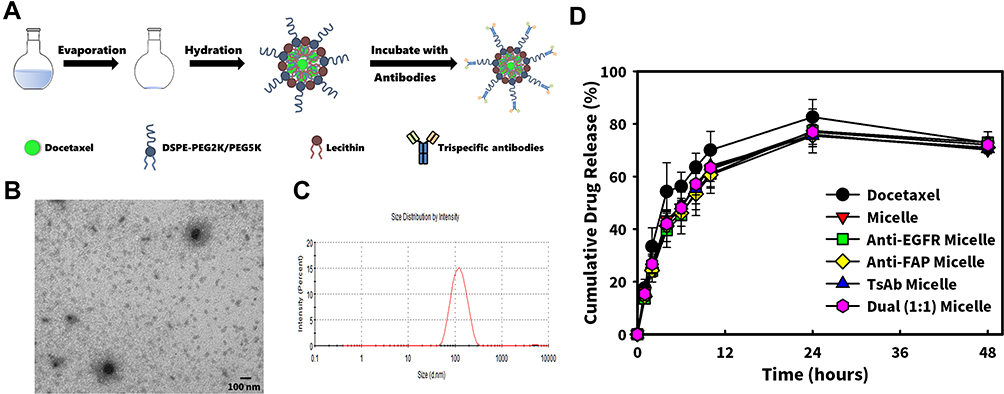 |
Figure 2 (A) Schematic illustration of the preparation of docetaxel (DTX)-loaded mPEGylated lecithin-stabilized polymeric micelles (mPEG-lsbPMs); (B) TEM images and (C) particle size analysis of mPEG-lsbPMs; (D) drug release profile of docetaxel. |
Assessment of Binding Affinity of BsAbs and TsAbs
The mPEG and tumor antigen binding affinities of antibodies were evaluated by flow cytometry and ELISA, respectively. As shown in Figure 3A, the fluorescence intensity of both the Anti-FAP-BsAbs and Anti-EGFR-FAP-TsAbs showed the significantly highest amount compared with the Anti-EGFR-BsAbs and control groups. Besides, the fluorescence intensity of Anti-EGFR-BsAbs was comparable with Anti-EGFR-FAP-TsAbs, which was far greater than that of Anti-FAP-BsAbs and control groups, shown in Figure 3B. In MIA PaCa-2 cells, the fluorescence intensity showed a similar result to that of HT-29 cells, as depicted in Figure 3C. As for mPEG binding affinity in Figure 3D, the absorbance of three groups, Anti-FAP-BsAbs, Anti-EGFR-BsAbs, and Anti-EGFR-FAP-TsAbs, increased with the increase of the antibody’s concentration, which indicated that the ability of BsAbs and TsAbs bound to the mPEG. Results demonstrated mPEG, FAP and EGFR binding affinity, which enabled the DTX-loaded mPEG-LsbPMs to target the tumors.
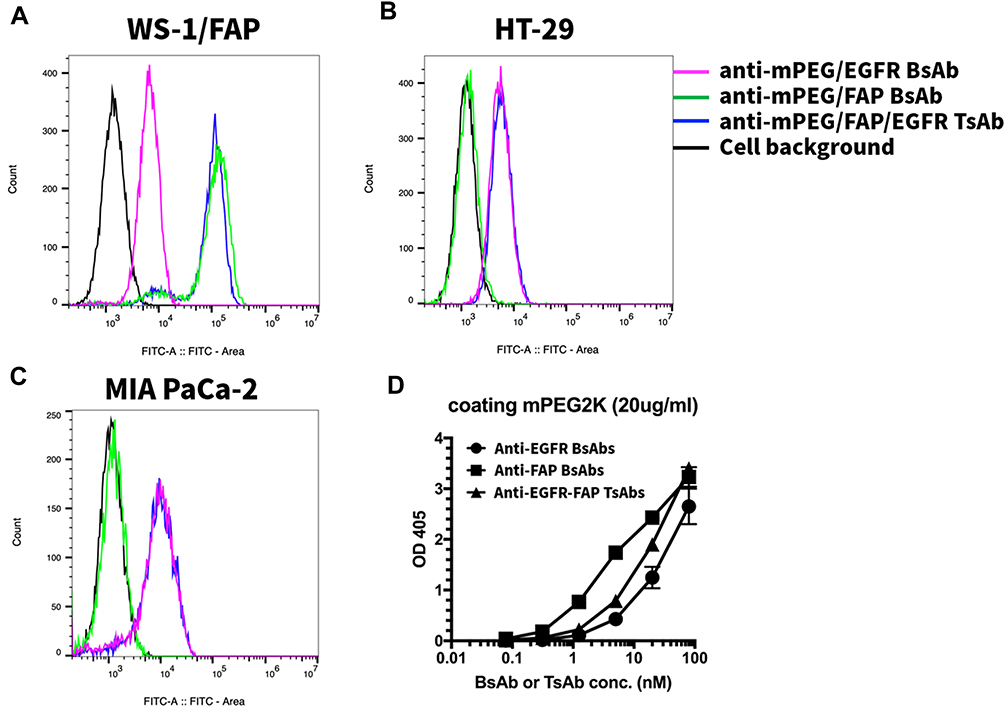 |
Figure 3 Assessment of binding affinity to FAP and EGFR on (A) WS-1/FAP cells, (B) HT-29 cells and (C) MIA PaCa-2 cells and (D) mPEG. |
In vitro Drug Release Studies
The drug release profile of docetaxel was evaluated at pH 7.4 PBS. As depicted in Figure 2D, the cumulative release of docetaxel reached a plateau after 24 h. Within 24 h, the release rate of the formulation groups was slightly slower than that of free drug, which may result from the controlled release ability of micelles. Besides, the release rate of Micelle was similar to that of other micelles with BsAbs or TsAbs, which indicated that modification of BsAbs or TsAbs did not affect the drug release.
In vitro Cellular Uptake
Specific targeting and cellular binding of DIO-loaded mPEG-lsbPMs modified without or with TsAb or BsAbs were examined using co-cultures of HT-29/EGFR and WS1/FAP or MIA PaCa-2/EGFR and WS1/FAP at various ratios (1:4, 1:1, and 4:1). In cell-based ELISAs, the amount of DIO-loaded mPEG-lsbPMs binding to cells was detected by an anti-PEG Ab. Results are shown in Figure 4 and demonstrated that unmodified DIO-loaded mPEG-lsbPMs (labeled Micelle) presented the least amount of binding with individual cells or co-culture of two cell lines (HT-29/EGFR and MIA PaCa-2/EGFR) with WS1/FAP at three different ratios (1:4, 1:1, and 4:1). As depicted in Figure 4A, the uptake of BsAb/TsAb-micelles was consistent with the further binding affinity of BsAb and TsAb, and the uptake of micelles was increased significantly with the BsAb and TsAb, which demonstrated that the non-covalent modification of BsAb or TsAb endowed lsbPMs with an active targeting ability and enhanced the amount of cellular uptake. For co-cultures of HT-29/EGFR and WS1/FAP at these three ratios (1:4, 1:1, and 4:1), binding of the Anti-FAP Micelle, TsAbs Micelle, and Dual (1:1) Micelle to the co-culture of HT-29/EGFR with WS1/FAP was observed to significantly increase with an increasing ratio of WS1/FAP to HT-29/EGFR, whereas that for Anti-EGFR Micelle to all three different ratios of HT-29/EGFR to WS1/FAP was demonstrated to be minimal as shown in Figure 4B, compared with the Micelle group. This indicated that the binding of those targeting antibodies to FAP which is overexpressed on WS1 cells was more efficacious, whereas that to EGFR overexpressed on HT-29 cells was less efficacious. It further revealed that the binding of the Anti-FAP Micelle to three co-culture ratios of HT-29/EGFR to WS1/FAP was significantly (p < 0.001) higher than that for the TsAb Micelle and Dual (1:1) Micelle. For co-culture of MIA PaCa-2/EGFR and WS1/FAP at various ratios (1:4, 1:1, and 4:1), binding of the Anti-FAP Micelle and Anti-EGFR Micelle to the co-culture of MIA PaCa-2/EGFR with WS1/FAP was observed to respectively increase and decrease, with respect to an increase in the ratio of WS1/FAP to MIA PaCa-2/EGFR, whereas that for the TsAbs Micelle and Dual (1:1) Micelle to the co-culture of MIA PaCa-2/EGFR with WS1/FAP was found to decrease with an increasing ratio of MIA PaCa-2/EGFR to WS1/FAP, as shown in Figure 4C. Compared with the Micelle group, uptake of BsAbs and TsAbs Micelles significantly increases at various ratios. These results illustrate that the binding of those targeting antibodies to FAP overexpressed by WS1 cells and EGFR overexpressed by MIA PaCa-2 cells was equally efficacious, resulting in similar binding amounts of TsAbs and Dual (1:1) on the co-culture of MIA PaCa-2/EGFR and WS1/FAP at a 1:1 ratio.
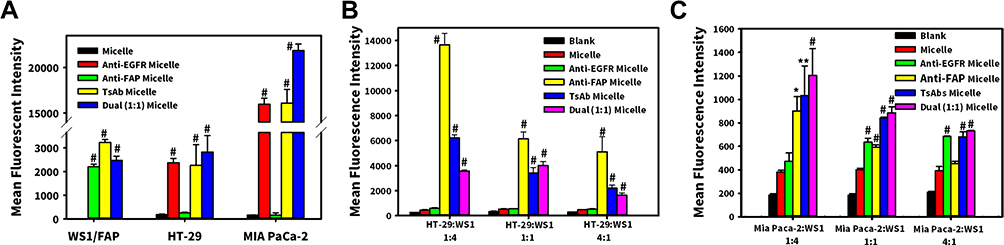 |
Figure 4 Cell uptake study of (A) individual cells (HT-29, MIA PaCa-2 and WS1/FAP) and (B) co-culture of HT-29:WS1/fibronectin activation protein (FAP) (at ratios of 1:4, 1:1, and 4:1); (C) co-culture of MIA PaCa-2:WS1/FAP (at ratios of 1:4, 1:1, and 4:1). * p < 0.05, ** p < 0.01, # p < 0.001 compared with Micelle. |
Cell Viability
Two cell lines of HT-29/EGFR and MIA PaCa-2/EGFR were co-cultured with WS1/FAP at various ratios of 1:4, 1:1, and 4:1 to mimic the tumor surrounded by the TME. To investigate the enhancement of cell-specific cytotoxicity, those individual cell lines (HT-29, MIA PaCa-2, and WS1/FAP) and three co-culture ratios of the two cell lines were treated with Micelle, Anti-EGFR Micelle, Anti-FAP Micelle, or TsAbs Micelle and were then evaluated by an MTT assay. Results are shown in Figure 5 which illustrates that the cytotoxicity of individual cells and three co-culture ratios of the two cell lines with WS1/FAP treated with DTX-loaded mPEGylated micelles armed with individual BsAbs or TsAbs was higher than that for micelles that were not armed with targeting antibodies. For individual cell line studies, the modification with BsAb or TsAb obviously increased the cytotoxicity in WS1 cells. The cytotoxicity in HT-29 cells was similar to that in MIA PaCa-2 cells, whose both IC50 values were around 30 ng/mL. Particularly, for co-culture studies, the cytotoxicity in MIA PaCa-2/WS1 was higher than that in HT-29/WS1. Therefore, with the correct specificity to the overexpressed antigen on targeted cells, the TsAb and BsAbs improved cellular binding, drug accumulation, and cytotoxicity of modified DTX-loaded mPEG-lsbPMs. It was concluded that the TsAb and BsAbs confirmed the tumor specificity and enhanced the cytotoxicity of DTX-loaded mPEG-lsbPMs toward antigen-positive cancer cells. The results also illustrated that Anti-EGFR BsAbs seemed to show a lower binding efficiency to HT-29 cells, which could have been due to lower expression of the EGFR. Therefore, MIA PaCa-2/WS1 was selected as tumor model for subsequent studies.
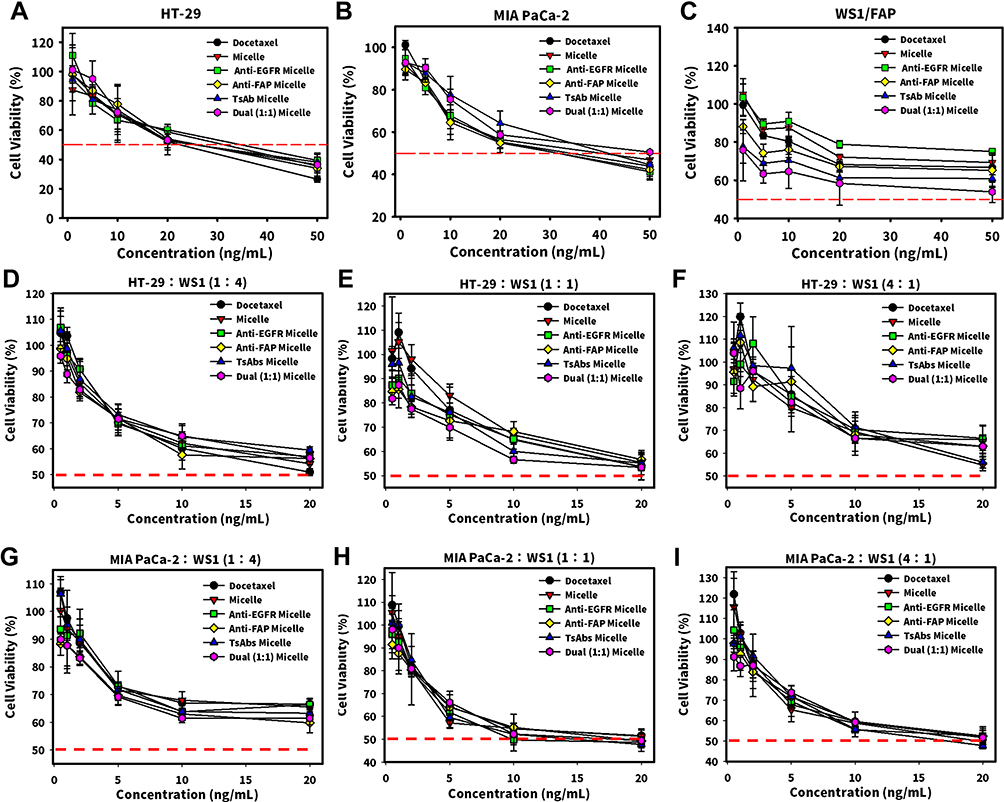 |
Figure 5 Cytotoxicity study of individual (A) HT-29, (B) MIA PaCa-2, (C)WS1/FAP cells and co-culture of HT-29/epidermal growth factor receptor (EGFR):WS1/fibronectin activation protein (FAP) at ratios of 1:4 (D), 1:1 (E), and 4:1 (F); and MIA PaCa-2/EGFR:WS1/FAP at ratios of 1:4 (G), 1:1 (H), and 4:1 (I). |
In vivo Pharmacokinetic (PK) Studies
The mean plasma concentrations of DTX after administration with a single dose of 10 mg/kg of Docetaxel, Micelle, Anti-EGFR Micelle, Anti-FAP Micelle, Dual (1:1) Micelle, or TsAbs Micelle via a jugular vein injection (with three rats per group) are shown in Figure 6A for a time period of 72 h and Figure 6B for a time period of 10 h. The PK profiles of Micelle alone, Anti-EGFR Micelle, Anti-FAP Micelle, Dual (1:1) Micelle, or TsAbs Micelle were analyzed and compared with Docetaxel. All of the PK profiles plotted in Figure 6A and B show a high initial DTX concentration after the injection, followed by a rapid decline to the terminal phase, which gradually reached a steady-state concentration. All of the declining rates for Micelle alone or those micelles armed with BsAbs, Dual BsAbs, or TsAbs (Anti-EGFR Micelle, Anti-FAP Micelle, Dual (1:1) Micelle, or TsAbs Micelle) were observed to be faster than that for Docetaxel.
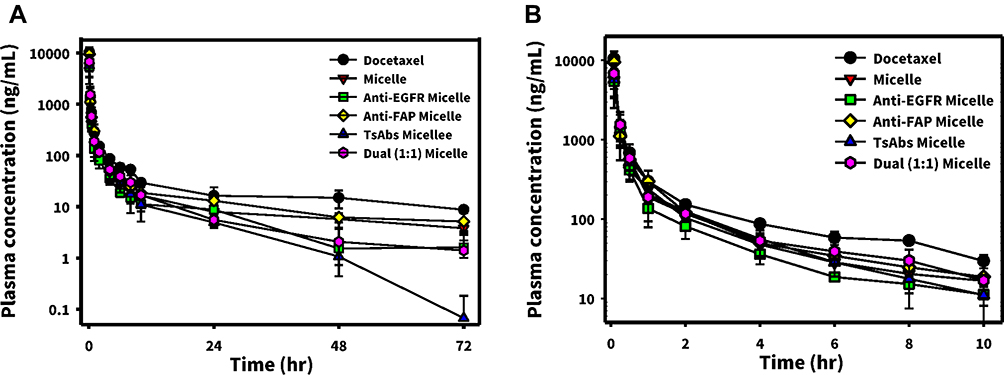 |
Figure 6 Plasma concentration-time curves of docetaxel ((A) 0~72 h; (B) 0~10 h) after intravenous administration of docetaxel, micelle, anti-epidermal growth factor receptor (EGFR) micelle, anti-fibronectin activation protein (FAP) micelle, dual (1:1), and trispecific antibody (TsAb) micelles (10 mg/kg) to Sprague-Dawley rats. |
Related PK parameters estimated by WinNonlin are listed in Table 1, which illustrates that a higher initial concentration (C0) of Docetaxel was observed compared with that for Micelle alone or those micelles armed with BsAbs, Dual BsAbs, or TsAbs (Anti-EGFR Micelle, Anti-FAP Micelle, Dual (1:1) Micelle, or TsAbs Micelle). All values of the area under the receiver operating characteristics curve at 0~72 h (AUC0-72) and from time 0 to infinity (AUC0-inf) for Micelle alone or those micelles armed with BsAbs, Dual BsAbs, or TsAbs were 30~40% lower than that for Docetaxel except that for Anti-FAP Micelle which was nearly equal to that for Docetaxel. A shorter terminal half-life (T1/2) and lower steady state volume of distribution (Vss) were only seen for Dual (1:1) Micelle and TsAbs Micelle, and insignificant differences were detected in the clearance (CL) among all formulations examined. From these results, we concluded that all micelle formulations regardless of whether or not they were modified without or with TsAbs, BsAbs, or Dual BsAbs presented rapid distribution to tissues or organs, including tumors, resulting in less residence times in and fast clearance from the blood circulation.
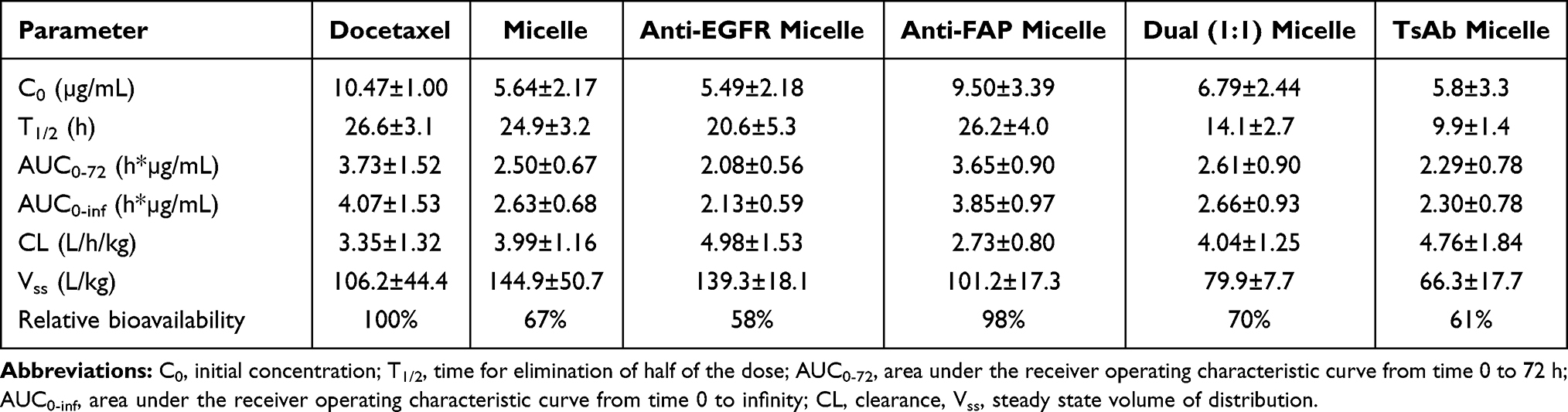 |
Table 1 Pharmacokinetic Parameters of Docetaxel (DTX) After Intravenous (IV Bolus Dose 10 Mg/Kg) to SD Rats (n = 3) of Docetaxel, Micelle, Anti-EGFR Micelle, Anti-FAP Micelle, Dual (1:1), and TsAb Micelle |
In vivo Antitumor Efficacy in Tumor-Bearing Mice
The antitumor effects of Docetaxel, Micelle, Anti-EGFR Micelle, Anti-FAP Micelle, Dual (1:1) Micelle, or TsAbs Micelle were evaluated on a 1:1 co-culture of EGFR-overexpressing MIA PaCa-2 (MIA PaCa-2/EGFR) and FAP-overexpressing WS1 (WS1/FAP) models. Results shown in Figure 7A clearly demonstrate that Anti-EGFR Micelle, Anti-FAP Micelle, Dual (1:1) Micelle, TsAbs Micelle, and Docetaxel all efficaciously inhibited the growth of MIA PaCa-2/EGFR:WS1/FAP 1:1 co-culture tumors after treatment with a regimen of 5 mg/kg for Q3*4. The percent of tumor volumes remaining after treatment with Docetaxel, Micelle, Anti-EGFR Micelle, Anti-FAP Micelle, TsAbs Micelle, and Dual (1:1) Micelle calculated with respect to that for PBS treatment group (at 100%) were 74.7%±103.4%, 39.9%±10.0%, 37.5%±10.3%, 41.1%±14.7%, 39.9%±6.9%, and 39.3%±16.4%, respectively. All of the BsAbs, Dual (1:1), and TsAbs modified DTX-loaded mPEG-lsbPMs formulations showed the greatest antitumor effects which were similar, but were significantly suppressed compared with those of the negative control of PBS (p < 0.05). There was no significant difference among those lsbPMs non-covalently binding with BsAbs or TsAbs indicating that both targeting antibody arms (anti-FAP and anti-EGFR) could similarly promote lsbPMs binding to those tumor cells over-expressing EGFR and those tumor-associated fibroblasts over-expressing FAP. This resulted in a similar efficiency on tumor suppression. Furthermore, the weight change profiles of all treatment groups illustrated in Figure 7B demonstrate that there was greater weight loss in the Docetaxel treatment group than for any of the BsAbs, Dual (1:1), or TsAbs modified DTX-loaded mPEG-lsbPMs formulations, indicating that all of them induced less systemic toxicity than did Docetaxel. Thus, it was concluded that treatment with DTX-loaded mPEG-lsbPMs formulations modified with or without BsAbs, Dual (1:1), and TsAbs for inhibiting tumor growth was enhanced compared with that for Docetaxel but showed fewer signs of adverse effects.
 |
Figure 7 (A) Tumor volume profile of nude mice bearing Mia PaCa-2:WS1/fibronectin activation protein (FAP) (1:1) tumors treated with intravenous administration (5 mg/kg Q3D*4) of docetaxel, micelle, anti-epidermal growth factor receptor (EGFR) micelle, anti-FAP micelle, dual (1:1), and trispecific antibody (TsAb) micelle. (B) Body weight changes in tumor-bearing mice. *p < 0.05 compared with PBS group. |
Biodistribution Assessment in Tumor-Bearing Mice
Biodistribution of DTX in major organs was assessed in MIA PaCa-2/EGFR:WS1/FAP (1:1)-bearing BALB/c nude mice at 2 and 8 h after IV injections of all treatments, and the results are illustrated in Figure 8A and B, respectively, for 2 and 8 h. At 2 h, DTX had obviously accumulated in the heart, spleen, lungs, kidneys, and tumor, but showed a lower amount in the liver. This also indicates that similar accumulations of DTX in the heart, liver, and tumor were shown for all treatments except that for Micelle alone. Furthermore, with the exception of Micelle alone, Anti-FAP Micelle, Anti-EGFR Micelle, Dual (1:1) Micelle, and TsAbs Micelle showed higher biodistribution amounts to the spleen, lungs, and kidneys than those for Docetaxel. At 8 h after the injection, the accumulated amount of DTX in the tumor was further observed to have increased for all treatments, and the biodistribution to the other organs maintained a similar pattern trend and difference as those seen at 2 h. Among them, accumulations of DTX in the tumor after administration of Dual (1:1) Micelle were slightly higher than those for Docetaxel at 2 and 8 h.
 |
Figure 8 Tissue distributions of docetaxel (DTX) after administration of docetaxel and micelle alone, anti-fibronectin activation protein (FAP) micelle, anti-epidermal growth factor receptor (EGFR) micelle, dual (1:1) micelle, trispecific antibody (TsAb) micelle at a dose of 5 mg/kg into Mia PaCa-2:WS1/FAP (1:1) tumor-bearing nu/nu mice (n = 4 or 5) at 2 (A) and 8 h (B). |
Discussion
To enable NCs for dual targeting to reach tumors and the tumor environment, non-covalently bound dual BsAbs (anti-mPEG/antitumor antigen and anti-mPEG/anti-TAF) and a TsAb (anti-mPEG/antitumor/anti-TAF) to mPEGylated NCs of lecithin-stabilized polymeric micelles designated mPEG-lsbPMs were developed, on the basis of DTX-loaded micelles sterically stabilized by PEG-lipid layer. By utilizing the unique property of lecithin-stabilized polymeric micelles (lsbPMs), the supported lipid shell, composed of lecithin and DSPE-PEG5000 at an appropriate ratio, was fused onto the PM core of the lsbPMs by ultrasonication to implement mPEG onto surface of lsbPMs for non-covalent binding of anti-PEG BsAbs and the TsAb. The particle size of mPEG-lsbPMs was around 100 nm; EE and DL were high (>90%, 5%). The outer shell of the mPEG-lsbPMs comprised lecithin and DSPE-PEG5000 in a specific molar ratio of 94:5. mPEG chains provided the large steric hindrance to prevent the opsonization and evade the reticuloendothelial system (RES).23
The formulation with high surface coverage and a high concentration of PEG-lipids (5% molar ratio) formed lateral pressure between the overcrowded PEG, which extended the PEG chains to a semi-linear brush-like structure, while PEG-lipids at a <5% molar ratio in the mPEG- lsbPMs were expected to have a mushroom-like structure.24 Furthermore, while the PEG contents were more than 5%, the PEG chains arranged in the brush conformation, allowing BsAbs or the TsAb to non-covalently bind to the methoxy end of PEG chain via the anti-mPEG Fab fragment of antibodies, which enhanced the targeting ability of mPEG-lsbPMs to the tumor and TAFs via the antitumor scFv fragment and anti-TAF scFv fragment of the BsAbs and TsAb.
The in vitro cellular binding study as shown in Figure 3 demonstrated that unmodified DIO-loaded mPEG-lsbPMs (labeled Micelle) offered the least amount of binding to the co-culture of two cell lines (HT-29 and MIA PaCa-2) with WS1/FAP at three different ratios (1:4, 1:1, and 4:1). It also illustrated that the binding of TsAb Micelle and Dual (1:1) Micelle to the coculture of two cell lines (HT-29 and MIA PaCa-2) with WS1/FAP at three different ratios (1:4, 1:1, and 4:1) was equal or higher than that for those Micelles modified with single individual BsAbs (Anti-EGFR/anti-mPEG and Anti-FAP/anti-mPEG). This indicates that modification of micelles with dual-targeting resulted in better cellular affinity of those NCs leading to subsequent uptake by tumor cells or TAFs to potentially enhance the chemotherapeutic efficacy. Furthermore, the in vitro cell viability study as shown in Figure 4 revealed that DTX-loaded mPEG-lsbPMs modified with TsAb or dual BsAbs resulted in higher cytotoxicity against EGFR-overexpressing MIA PaCa-2 and HT-29 cells co-cultured with FAP-overexpressing TAFs at various ratios. It was confirmed that active targeting to both tumor and TAF-specific antigens was able to increase the affinity of DTX-loaded mPEG-lsbPMs toward tumor cells and TAFs leading to successive uptake by tumor cells or TAFs to enhance the chemotherapeutic efficacy against antigen-positive cancer cells.
The in vivo tumor-inhibition study, as shown by Figure 6, conclusively illustrated that enhanced inhibition of tumor growth with fewer signs of systemic toxicity was observed for those treatments with DTX-loaded mPEG-lsbPMs formulations modified with or without BsAbs, Dual (1:1), and TsAb. The greater efficacy in tumor growth inhibition might be attributed to an increase in the tumor and TAF affinity leading to successive uptake by macropinocytosis and caveolin-mediated endocytosis as preferable pathways as revealed in a previous report.8 Furthermore, the slightly higher tumor accumulation of BsAbs- and TsAbs-DTX-loaded mPEG-lsbPMs might be the result from decrease of DTX being pumped out by P-glycoprotein (P-gp) from tumor cells. Additionally, the lower weight loss observed with BsAbs, Dual (1:1), or TsAb-modified DTX-loaded mPEG-lsbPMs compared with the Docetaxel treatment group might be attributed to greater elimination from the kidneys, as revealed in the PK studies (Figure 6). Furthermore, although there was a greater amount of docetaxel biodistributed to lung, kidney, and spleen than that to tumor as shown by the in vivo biodistribution study (Figure 8), a slight weight loss was observed for administration of all formulations except solvent-based docetaxel. It indicates that toxicity related to higher concentration of docetaxel biodistributed to those vital organs was not an issue in this study. Overall, it could be concluded that the suppression of tumor growth with the treatment of DTX-loaded mPEG-lsbPMs formulations modified with or without BsAbs, Dual (1:1), and TsAbs was not inferior to the treatment of Docetaxel but most importantly showed fewer signs of adverse effects.
Conclusions
In conclusion, non-covalently bound dual BsAbs (anti-tumor antigen/anti-mPEG; anti-FAP/anti-mPEG) and TsAb (anti-tumor antigen/anti-FAP/anti-mPEG) onto mPEGylated lsbPMs might potentially be able to enhance anticancer efficacy with less systemic toxicity. Dual BsAbs/TsAbs-modified lsbPMs were developed and utilized the specific binding affinity of BsAbs/TsAbs to achieve the tumor-selective accumulation, which resulted in higher antitumor efficacy against antigen-positive tumors. These BsAbs/TsAbs targeting delivery systems could be a promising platform, applied to not only cancer therapy but also other chronic diseases.
Institutional Review Board Statement
This animal experiment was approved by the Institutional Animal Care and Use Committee of Taipei Medical University (Approval No.: LAC-2017-0334) in compliance with the Taiwanese Animal Welfare Act.
Acknowledgments
This work was financially supported by the Ministry of Science and Technology, Taiwan (MOST 106-2320-B-038-014 and MOST 107-2314-B-038-035) and Jin-lung-yuan Foundation (2020-2021).
Disclosure
The authors declare no conflict of interest.
References
1. Maruyama K, Yuda T, Okamoto A, Kojima S, Suginaka A, Iwatsuru M. Prolonged circulation time in vivo of large unilamellar liposomes composed of distearoyl phosphatidylcholine and cholesterol containing amphipathic poly(ethylene glycol). Biochim Biophys Acta. 1992;1128(1):44–49. doi:10.1016/0005-2760(92)90255-T
2. Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99:28–51.
3. Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–146. doi:10.1016/j.jconrel.2010.08.027
4. Fukuda A, Tahara K, Hane Y, et al. Comparison of the adverse event profiles of conventional and liposomal formulations of doxorubicin using the FDA adverse event reporting system. PLoS One. 2017;12(9):e0185654. doi:10.1371/journal.pone.0185654
5. Xing M, Yan F, Yu S, Shen P. Efficacy and cardiotoxicity of liposomal doxorubicin-based chemotherapy in advanced breast cancer: a meta-analysis of ten randomized controlled trials. PLoS One. 2015;10:e0133569. doi:10.1371/journal.pone.0133569
6. Kao CH, Wang JY, Chuang KH, et al. One-step mixing with humanized anti-mPEG bispecific antibody enhances tumor accumulation and therapeutic efficacy of mPEGylated nanoparticles. Biomaterials. 2014;35:9930–9940. doi:10.1016/j.biomaterials.2014.08.032
7. Chen I-J, Cheng Y-A, Kai-Wen H, et al. Bispecific antibody (HER2 ×mPEG) enhances anti-cancer effects by precise targeting and accumulation of mPEGylated liposomes. Acta Biomaterialia. 2020;111:386–397. doi:10.1016/j.actbio.2020.04.029
8. Su CY, Chen M, Chen LC, et al. Bispecific antibodies (anti-mPEG/anti-HER2) for active tumor targeting of docetaxel (DTX)-loaded mPEGylated nanocarriers to enhance the chemotherapeutic efficacy of HER2-overexpressing tumors. Drug Deliv. 2018;25:1066–1079. doi:10.1080/10717544.2018.1466936
9. Cheng YA, Chen IJ, Su YC, et al. Enhanced drug internalization and therapeutic efficacy of PEGylated nanoparticles by one-step formulation with anti-mPEG bispecific antibody in intrinsic drug-resistant breast cancer. Biomater Sci. 2019;7:3404–3417. doi:10.1039/C9BM00323A
10. Yu S, Li A, Liu Q, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol. 2017;10(1):78. doi:10.1186/s13045-017-0444-9
11. Chiavenna SM, Jaworski JP, Vendrell A. State of the art in anti-cancer mAbs. J Biomed Sci. 2017;24:15. doi:10.1186/s12929-016-0311-y
12. Kojima Y, Acar A, Eaton EN, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107:20009–20014. doi:10.1073/pnas.1013805107
13. Lijnen P, Petrov V. Transforming growth factor-beta 1-induced collagen production in cultures of cardiac fibroblasts is the result of the appearance of myofibroblasts. Methods Find Exp Clin Pharmacol. 2002;24:333–344. doi:10.1358/mf.2002.24.6.693065
14. Erdogan B, Webb DJ. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans. 2017;45:229–236. doi:10.1042/BST20160387
15. Torosean S, Flynn B, Axelsson J, et al. Nanoparticle uptake in tumors is mediated by the interplay of vascular and collagen density with interstitial pressure. Nanomed. 2013;9:151–158. doi:10.1016/j.nano.2012.07.002
16. Eikenes L, Bruland OS, Brekken C, Davies Cde L. Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res. 2004;64:4768–4773. doi:10.1158/0008-5472.CAN-03-1472
17. Tao L, Huang G, Wang R, et al. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci Rep. 2016;6:38408. doi:10.1038/srep38408
18. Hofheinz RD, Al-Batran SE. Stehle, Stromal antigen targeting by a humanised monoclonal antibody: an early Phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003;26:44–48. doi:10.1159/000069863
19. Scott AM, Wiseman G, Welt S, et al. I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9:1639–1647.
20. Wang LC, Lo A, Scholler J, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–166. doi:10.1158/2326-6066.CIR-13-0027
21. Niedermeyer J, Kriz M, Hilberg F, et al. Targeted disruption of mouse fibroblast activation protein. Mol Cell Biol. 2000;20:1089–1094. doi:10.1128/MCB.20.3.1089-1094.2000
22. Chen LC, Chen YC, Su CY, Hong CS, Ho HO, Sheu MT. Development and characterization of self-assembling lecithin-base d mixed polymeric micelles containing quercetin in cancer treatment and an in vivo pharmacokinetic study. Int J Nanomed. 2016;11:1557–1566.
23. Chen YC, Su CY, Jhan HJ, Ho HO, Sheu MT. Physical characterization and in vivo pharmacokinetic study of self-assembling amphotericin B-loaded lecithin-based mixed polymeric micelles. Int J Nanomedicine. 2015;10:7265–7274. doi:10.2147/IJN.S95194
24. Allen C, Dos Santos N, Gallagher R, et al. Controlling the physical behavior and biological performance of liposome formulations through use of surface grafted poly (ethylene glycol). Bio Sci Rep. 2002;22(2):225–250. doi:10.1023/A:1020186505848
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.