")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Activation of mTOR: a culprit of Alzheimer’s disease?
Authors Cai Z, Chen G, He W, Xiao M , Yan LJ, Zhang H, Yan N
Received 12 October 2014
Accepted for publication 11 December 2014
Published 9 April 2015 Volume 2015:11 Pages 1015—1030
DOI https://doi.org/10.2147/NDT.S75717
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Zhiyou Cai,1 Guanghui Chen,1 Wenbo He,1 Ming Xiao,2 Liang-Jun Yan3
1Department of Neurology, Renmin Hospital, Hubei University of Medicine, Shiyan Renmin Hospital, Shiyan, Hubei Province, People’s Republic of China; 2Department of Anatomy, Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China; 3Department of Pharmacology and Neuroscience, University of North Texas Health Science Center, Fort Worth, TX, USA
Abstract: Alzheimer’s disease (AD) is characterized by cognitive impairment in clinical presentation, and by β-amyloid (Aβ) production and the hyper-phosphorylation of tau in basic research. More highlights demonstrate that the activation of the mammalian target of rapamycin (mTOR) enhances Aβ generation and deposition by modulating amyloid precursor protein (APP) metabolism and upregulating β- and γ-secretases. mTOR, an inhibitor of autophagy, decreases Aβ clearance by scissoring autophagy function. mTOR regulates Aβ generation or Aβ clearance by regulating several key signaling pathways, including phosphoinositide 3-kinase (PI3-K)/protein kinase B (Akt), glycogen synthase kinase 3 [GSK-3], AMP-activated protein kinase (AMPK), and insulin/insulin-like growth factor 1 (IGF-1). The activation of mTOR is also a contributor to aberrant hyperphosphorylated tau. Rapamycin, the inhibitor of mTOR, may mitigate cognitive impairment and inhibit the pathologies associated with amyloid plaques and neurofibrillary tangles by promoting autophagy. Furthermore, the upstream and downstream components of mTOR signaling are involved in the pathogenesis and progression of AD. Hence, inhibiting the activation of mTOR may be an important therapeutic target for AD.
Keywords: Alzheimer’s disease, mammalian target of rapamycin, rapamycin, β-amyloid, neurofibrillary tangles, signaling
Introduction
Alzheimer’s disease (AD), characterized by age-related neurodegenerative disorder, makes clear the progression of cognitive impairment in clinical presentation and the two classical hallmarks of β-amyloid (Aβ) accumulation and aberrant hyperphosphorylated tau in pathology.1,2 Procedural age is the greatest risk factor for AD since most patients with AD are more than 65 years old.3,4 Although extensive research in AD has been undertaken over the past few decades, the pathogenesis of AD is still not completely understood. Current therapeutic intervention for AD cannot stop cognitive impairment from progressing in spite of temporarily slowing the worsening of dementia and improving the quality of life. So far, seeking out effective ways to stop the progression of AD and prevent its onset has been a thorny issue.
The mammalian target of rapamycin (mTOR) is a 289-kD serine/threonine multidomain protein with a kinase domain and a FKBP12 binding domain, regulating many physiological processes. mTOR coordinates or interacts with the upstream signal components, including insulin, growth factors, AMPK, PI-3K/Akt, and glycogen synthase kinase 3 (GSK-3).5–8 More and more studies have found the involvement of the mTOR dysregulation in many diseases, such as aging,9,10 tumor and cancer,11,12 diabetes,12 obesity,13 cardiovascular disease,14,15 and neurodegenerative diseases.16,17 Compelling evidence has shown that the activation of mTOR signaling is a contributor to AD progression and intersects with AD pathology and clinical manifestation.18,19 mTOR signaling is closely associated with the presence of two hallmarks of the disease (Aβ plaques and neurofibrillary tangles [NFTs]) and cognitive impairment in clinical presentation, respectively.20–22 Therefore, the development of mTOR inhibitors may also be useful for the prevention and treatment of AD.23
This review focuses on the roles of mTOR in AD cognitive clinical manifestation and the pathology of amyloid plaques and NFTs. It is also discussed that mTOR inhibitor rapamycin delays cognitive impairment and retards the pathology of amyloid plaques and NFTs. The theoretical basis that inhibiting mTOR induces autophagy enhancement and benefits the treatment of AD is provided here. Finally, whether inhibiting mTOR could be a valid therapeutic means to limit AD pathology is reviewed.
Mammalian target of rapamycin
mTOR, also known as the mechanistic target of rapamycin or FK506 binding protein 12-rapamycin associated protein 1 (FRAP1), is a 289-kD serine/threonine protein kinase that is closely related to the regulation of many physiological processes such as maintaining cell growth, proliferation and survival, and regulating protein synthesis and transcription via a wide variety of cellular signals.24,25 mTOR is present in two main types of molecular complex: mTOR complexes 1 and 2 (mTORC1 and mTORC2).26
mTOR signaling has been regulated by its upstream components, including insulin, growth factors (such as IGF-1), LKB1/AMPK, PI3K/Akt, GSK-3β, IKKβ, MAPK, and p53.27–31 Activated mTORC1 also has a number of downstream biological effects including translation of mRNA via the phosphorylation of downstream targets (4E-BP1 and p70S6 Kinase), suppression of autophagy (Atg13, ULK1), ribosome biogenesis, and activation of transcription leading to mitochondrial metabolism or adipogenesis. mTORC2 regulates ion transport and growth via SGK1 phosphorylation and controls cytoskeletal dynamics via activating (PKC) protein kinase C.
Therefore, aberrant mTOR signaling is closely related to many disease states.32,33 mTOR signaling intersects with AD pathology in several respects, suggesting its potential role as a contributor to the neurodegenerative process. Scientific evidence has supported that the activation of mTOR signaling plays a critical role in the pathogenesis of AD while the activation of mTOR signaling contributes to Aβ generation and the formation of NFTs.20
The activation of mTOR: a trigger for Aβ generation and failure of Aβ clearance?
The most striking pathogenic hallmark recognized by the scientific community for AD is the production and deposition of Aβ.34,35 Genetic evidence indicates that the main factor of Aβ generation is based on mutations either in the precursor protein for Aβ (β-amyloid precursor protein [APP]) or in presenilin-1 (PS1) or presenilin-2 (PS2).36,37 It is well accepted that the production of Aβ is the enzymatic processes responsible for the metabolism of APP, sequentially cleaved by two membrane-bound endoproteases: β- and γ-secretase.38,39 β-secretase is considered to be the rate-limiting enzyme in Aβ generation, while γ-secretase, a multisubunit enzyme composed of the proteins APH1, PEN2, nicastrin, and presenilin (PS1 or PS2), is the final endoprotease that generates the peptide Aβ.40,41
Increasing evidence highlights that the activation of mTOR is an enhancer of Aβ generation and deposition (Figure 1).20,42 mTOR, an inhibitor of autophagy, decreases the Aβ clearance of the autophagy/lysosome system which accounts for the clearance of abnormal proteins.20,43 mTOR also modulates the metabolism of APP by regulating β- and γ-secretase.23,44 In addition, mTOR may interact with several key signaling pathways and regulate Aβ generation or Aβ clearance, including PI3-K/Akt,45,46 GSK-3,32 AMPK,47 and insulin/IGF-1.43
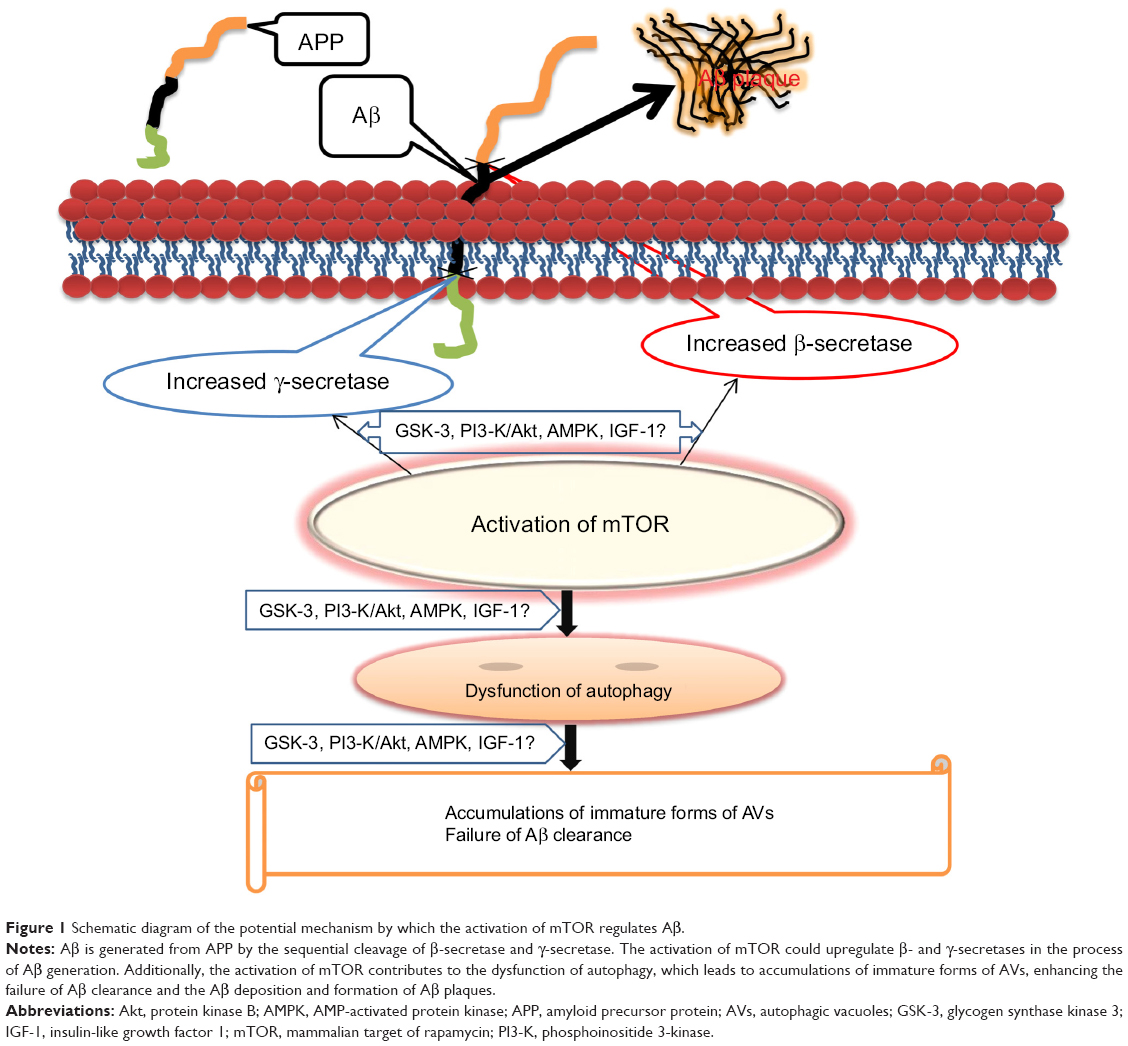 | Figure 1 Schematic diagram of the potential mechanism by which the activation of mTOR regulates Aβ. |
Activation of mTOR induces the failure of Aβ clearance
A variety of research studies have proved that the activation of mTOR leads to the failure of Aβ removal from the brain since the dysfunction of autophagy triggered by mTOR facilitates the process of Aβ generation and weakens its clearance.22,44,48
Autophagy, the molecular machinery for self-digestion, is an essential catabolic process in response to a multitude of physiological and pathological situations.49,50 It is well known that autophagy is an intracellular degradation system that delivers cytoplasmic components to lysosome and degrades cellular components through autophagy/lysosomal pathway to alternatively remove unnecessary cellular constituents.50,51 In the normal physiological function of cell metabolism, autophagy upholds the balance between the synthesis, degradation, and subsequent recycling of cellular products, playing an important role in maintaining cellular homeostasis, cell survival, differentiation, and development.52,53 Although numerous studies have revealed the nature and role of autophagy since the 1960s, many questions about the actual processes and mechanisms still remain. In particular, its role in some diseases still stays in the exploratory or research stage status. Yet it has been well recognized that autophagy plays a crucial role in many pathological processes, such as cancer,54,55 liver and kidney diseases,54,56,57 immune diseases,58,59 pathogen infection,60 aging and neurodegeneration,61–63 including Huntington’s, Alzheimer’s, and Parkinson’s diseases.
An analysis of the molecular mechanisms has shown that the induction of autophagy is a neuroprotective response and that defective autophagy is a favorable factor for neurological damage in most neurological disorders.32,64,65 Autophagy is the main mechanism in neurodegeneration, and autophagy deficiency is a major contributor to abnormal protein aggregation such as the aggregates of Aβ and tau in AD, the aggregates of α-synuclein in Parkinson’s disease, and the Huntingtin aggregates in Huntington’s disease.66,67 Distinctively, inhibition of mTOR activity and induction of autophagy ameliorate the pathogenic aggregates of misfolded proteins and delay the process of neurodegeneration in Huntington’s, Alzheimer’s, Parkinson’s disease, and Lewy body diseases.68,69
Several research findings point to the notion that a chronic deterioration of the autophagy/lysosome pathway is an important factor in the failure of Aβ clearance from the AD brain, while the autophagy/lysosome system determines the outcome of Aβ in the AD brain.23,46,48,70 Additionally, mTOR, inhibitor of autophagy, is closely associated with the levels of Aβ. Accordingly, inhibition of mTOR activity induces autophagy, lessens the aggregates of Aβ, and enhances the process of Aβ clearance.23
Normally, Aβ is most degraded by the autophagy/lysosome pathway that can participate in protein quality control and in the removal of aberrant forms of protein. The immaturity of autophagolysosomes plays a significant role in a massive accumulation of autophagic vacuoles (AVs) that may be sites of Aβ generation.71,72 Failure of Aβ clearance, generating from the accumulation of AVs that were colocalized within Aβ deposits, subsequently leads to the occurrence of Aβ accumulation and the formation of amyloid plaque in AD, which deteriorates from impaired clearance of AVs.72 It is recognized that inducing or inhibiting autophagy by regulating mTOR signaling induces corresponding changes in AVs proliferation and Aβ generation.65,73 In 3xTg-AD mice without the formation of amyloid plaques and tangles, autophagy induction via rapamycin ameliorates cognitive deficits, implying that rapamycin will be beneficial for the early treatment of AD.74 Recent research has demonstrated that chronic rapamycin intervention retards the progression of Alzheimer’s-like deficits and decreases Aβ levels through autophagy enhancement in the human amyloid precursor protein (PDAPP) mouse model, while rapamycin maintains proteostasis by the upregulation of classical chaperones/heat shock proteins (HSPs) in the brains of rapamycin-fed PDAPP mice.70
mTOR modulates APP process
The metabolic process of APP determines the onset of the amyloid pathogenic formation in AD as a result of sequential APP proteolysis.75 APP is a single-chain transmembrane protein and is metabolized by a series of sequential proteases under extensive posttranslational modification such as phosphorylation, glycosylation, and tyrosine sulfation.76,77 The metabolic process of APP occurs mainly by two pathways: 1) nonamyloidogenic process involving sequential cleavage by α-secretase and γ-secretase,78 2) amyloidogenic process sequentially cleaved by β-secretase and γ-secretase.79 γ-secretase is a large multisubunit complex whose components include presenilin that has been identified as a major genetic risk factor for AD.
The involvement of mTOR signaling in the pathogenesis of AD is the regulation of autophagy, while the autophagy/lysosome pathway is an important regulator of the processing of APP.44,80 In vitro study notes that it is a vital Aβ-generating pathway via increasing mature APLP1 (amyloid precursor-like protein 1) APP that is degraded through the autophagy/lysosome pathway.81 Recent findings suggest that inhibition of mTOR signaling alters APP processing by autophagosome accumulation in insulin-resistant conditions.44 Thus, mTOR modulates the APP process via autophagy intermediary. β- and γ-secretases are two essential enzymes for Aβ generation. mTOR may regulate postsecretase APP-CTF catabolism via autophagy/lysosomal proteolysis to influence the Aβ generation.82
mTOR regulates Aβ generation via some signaling pathways
Biomolecular evidence highlights the notion that the importance of both traditional and newly recognized interaction between mTOR and several signaling pathways has regulated Aβ generation to date, such as PI3-K/Akt, GSK-3, insulin/IGF-1, AMPK, and p70S6K.32,83
PI3-K/Akt/mTOR
The PI3-K/Akt/mTOR signaling pathway has a central function in the regulation of crucial metabolism,84,85 cell growth and proliferation,86,87 apoptosis,88 and secretion.89 Protein kinase B (PKB, also known as Akt) acts as a central intersection between phosphoinositide 3-kinase (PI3-K) and mTOR by phosphorylating a variety of substrates.90,91 Based on its crucial role in regulating critical cellular functions, it is highly plausible that the deregulation of PI3-K/Akt/mTOR participates in numerous disease disorders.92–96 Recent research demonstrates that the abnormal PI3-K/Akt/mTOR signaling pathway has been shown to contribute to the occurrence and development of AD.5 Aberrant activation of neuronal PI3-K/Akt/mTOR signaling is an early pathogenesis in the brains of AD individuals and a major candidate for pathophysiological change of Aβ product.5 Insulin and IGF-1 may rescue and normalize aberrant PI3-K/Akt/mTOR signaling against the development of amyloid pathology and cognitive impairment.5,97
GSK-3/mTOR
GSK-3, a multifunctional serine/threonine protein kinase, regulates numerous signaling pathways involved in a series of cellular processes, from glycogen metabolism to cell cycle regulation and proliferation. Excessive GSK-3 activation has recently been identified as an important factor in the onset of numerous diseases, including diabetes,98 bipolar disorder,99,100 cancer,98,101 and neurodegeneration diseases.102,103 The roles of GSK-3 in AD have been supported by the accumulating pathologic evidence, indicated in the generation of multiple pathological lesions,104,105 including Aβ production, the formation of neurofibrillary tangles, and neuron loss.106,107 An increasing amount of literature points out that GSK-3 is involved in the PI3K/Akt/mTOR signaling pathway.108–111 GSK-3/mTOR signaling pathway not only modulates neuronal cell proliferation, migration, and plasticity,32,112,113 but also regulates glucose uptake and glucose transporter.114,115 Both PI3-K/Akt and GSK-3 signaling pathways are important signaling machinery regulating the coming and leaving of Aβ in the pathogenesis of AD.96,116 Recent in vivo and in vitro studies show that an inhibitor of GSK-3, L803-mts, reduces Aβ deposits, delays cognitive impairment, and restores lysosomal acidification and the activity of mTOR, which is an effective target activated by GSK-3 but inhibited by impaired lysosomal acidification,117 implying that inhibition of GSK-3 rehabilitates lysosomal acidification that successively improves Aβ clearance via restoration of aberrant mTOR signaling and activation of autophagy.
AMPK/mTOR
AMP-activated protein kinase (AMPK), a key energy enzyme, regulates cellular metabolism to maintain energy homeostasis in response to a fall in intracellular ATP levels. The structure and function of AMPK has been regulated by ADP levels.118,119 AMPK is activated when cellular ADP levels increase coping with changes in cellular energy status.119 Numerous research results related to AMPK have been implicated in many kinds of pathological processes such as diabetes,120,121 obesity,122,123 cancer,124,125 aging,126 and neurodegenerative diseases.127,128 AMPK and mTOR act as a common regulator of autophagy through direct phosphorylation of Ulk since the Atg1/Ulk complex plays an essential role in the initiation of autophagy.129,130 Specifically, AMPK directly modulates Ulk1 through phosphorylation of Ser317 and Ser777, which results in autophagy enhancement. The activation of mTOR inhibits phosphorylation of Ulk1 Ser757 and cuts off the interaction between Ulk1 and AMPK, weakening autophagy.129 Moreover, molecular biological investigations into the role of AD have shown that both AMPK and mTOR participate in the regulation of the Aβ level.20,47,131 Through autophagy enhancement, the activation of AMPK limits the generation of Aβ.47,132 Conversely, the activation of mTOR is likely to promote Aβ production.20,23 Based on the data that the Aβ level in the AD brain is determined by the overall functional status of autophagy and that AMPK activation inhibits mTOR signaling activity to facilitate autophagy and promotes lysosomal degradation of Aβ,42,43,72 it is mainly through autophagy that the AMPK/mTOR signaling may regulate the Aβ level.
Insulin/IGF-1/mTOR
Insulin/insulin-like growth factor 1 (IGF-1) signaling is an important biochemical pathway that regulates glucose storage and uptake, cell growth and mitosis, protein synthesis, lipid synthesis, and the activity of numerous enzymes.133 Numerous studies have revealed that conduction disturbance of insulin/IGF-1 signaling has an intimate connection with the pathological process of many diseases related to metabolic disorders, such as aging,134–135 cancer,136,137 diabetes,138,139 and neurodegenerative disorders.140,141 Many data have suggested that conduction disturbance of insulin/IGF-1 signaling is an important mechanism of Aβ generation and deposition, including the increased Aβ level induced by hyperglycemia,142–144 the Aβ generation elicited by hyperinsulinemia,145,146 the APP modulation by hyperinsulinemia or hyperglycemia,143,147–149 and the failure of Aβ clearance triggered by the receptor for advanced glycation end products (RAGE) that is thought to be a primary transporter of Aβ across the blood–brain barrier (BBB) into the brain from the systemic circulation.150–153
Insulin/IGF-1 signaling regulates mTOR signaling by controlling the TSC GTPase activator function toward the Rheb GTPase.154 The insulin/IGF (IR, IGF-1R, IRS-1, PTEN, Akt, GSK3α, and GSK3β) to mTOR (TSC2, mTOR, p70S6K, and RPS6) signaling pathway is essential for the growth, proliferation, and survival of cells.155,156 More studies are in favor of the negative intermodulation between mTOR and the insulin/IGF-1 signaling pathway.157,158 It is clarified that the mTOR signaling pathway is activated by inhibiting the activity of insulin/IGF-1 signaling key components, including insulin receptor, insulin receptor substrate 1 (IRS-1), and IGF-1R.156,159–161 Furthermore, mTOR can induce insulin resistance by phosphorylation of IRS-1 on serine307 residues.162 Clearly, the insulin/IGF-1/mTOR signaling pathway is a negative cycle loop that regulates a variety of pathophysiological features.
It is well recognized that insulin and rapamycin (the inhibitor of mTOR) play a neuroprotective role in many neurological disorders,163,164 especially in neurodegeneration including AD.6,165 The striking similarity is the results from the insulin intervention and the inhibition of mTOR by rapamycin that rescue cognitive impairment and retard Aβ pathology in AD animal models.22,23,166,167 Hence, delaying the process of AD via regulation of the insulin/IGF-1/mTOR signaling pathway may be a promising intervention.140,168
Nevertheless, considerable evidence exists to show that insulin can enhance the activation of mTOR via stimulation of 4EBP1 binding to dimeric mTOR complex 1,169 and mediated by the Akt/PKB substrate PRAS40 (proline-rich Akt/PKB substrate 40 kDa).170 From in vivo and in vitro studies, it seems that mTOR activation has a neuroprotective property in the pathogenesis of AD, ameliorating Aβ pathology.117 Thus, the roles of insulin/IGF-1/mTOR signaling in AD remain elusive under these contradictory findings. Maybe, different results will emerge under different species, different diseases, and different experimental conditions.
The activation of mTOR: an enhancer of the hyperphosphorylation of tau?
Tau protein is a highly soluble microtubule-associated protein (MAP) that stimulates tubulin assembly into microtubules and stabilizes microtubules in the brain.171 It is well established that abnormal hyperphosphorylation of tau has been linked to the pathogenesis of AD (major components of paired helical filaments and neurofibrillary tangles), although the mechanism of the hyperphosphorylation is still not fully understood.172,173 The abnormal hyperphosphorylation of tau leads to massive detachment, which consequently weakens the stability of microtubules in nerve cells. This microtubule instability is one of the main causes of the symptoms of AD. Numerous scientific data have proved that abnormal tau hyperphosphorylation plays a central role in the formation of neurofibrillary tangles, which is a leading cause of neuronal death in AD.171,174 Tau phosphorylation is dynamically regulated by tau kinases and tau phosphatases, including GSK-3β,175,176 cyclin-dependent protein kinase 5 (cdk5), cAMP-dependent protein kinase,177,178 stress-activated protein kinases (SAPK1c/JNK1, SAPK2a/p38α, SAPK2b/p38β, SAPK3/p38γ, and SAPK4/p38δ),179,180 and mTOR.164,181
Compelling scientific results support the critical role of mTOR in the tau-related pathological progress, implying that the activity status of mTOR determines the abnormal hyperphosphorylation of tau, the onset of paired helical filaments, and the formation of NFTs.164,181–183 The activation of mTOR signaling promotes tau pathology, while inhibiting mTOR signaling slows down the progress of tau pathology.181
A recent study has shown that mTOR activated in diabetic condition accelerates the extent of tau hyperphosphorylation and promotes the occurrence of AD by impairing insulin signaling.164 Consistent with the discovery of the above, rapamycin (an inhibitor of mTOR, by inhibiting mTOR activity) reduces tau phosphorylation at Ser214 through the regulation of cAMP-dependent kinase, while tau phosphorylation at Ser214 may prepare tau for further phosphorylation by other kinases.184
It has been evidenced that mTOR signaling pathway has a close link to tau phosphorylation and the formation of PHFs and NFTs through autophagy function. Rapamycin-induced autophagy may enhance the clearance of the hyperphosphorylated tau.184 Inhibition of mTOR by rapamycin retards cognitive deficits and reduces the abnormal hyperphosphorylation of tau by autophagy enhancement in AD models.23,183 mTOR, coupled with PI3-K signaling, regulates protein phosphatase 2A and GSK-3-dependent phosphorylation of tau, while GSK-3β, antagonized by protein phosphatase 2A, regulates tau phosphorylation at many sites.111 It may be an effective therapeutic target for AD that mTOR regulates tau phosphorylation by controlling autophagy pathway.
The upstream and downstream components of mTOR signaling in AD
As shown in Table 1, the upstream and downstream components of the mTOR signaling pathway are involved in the pathogenesis and progression of AD.6,32,164 Considerable work has been dedicated to elucidating that mTOR-related signaling components have been identified as potential biomarkers of cognitive impairments in the clinical diagnosis of AD and as a critical target for a therapeutic program in AD.185,186
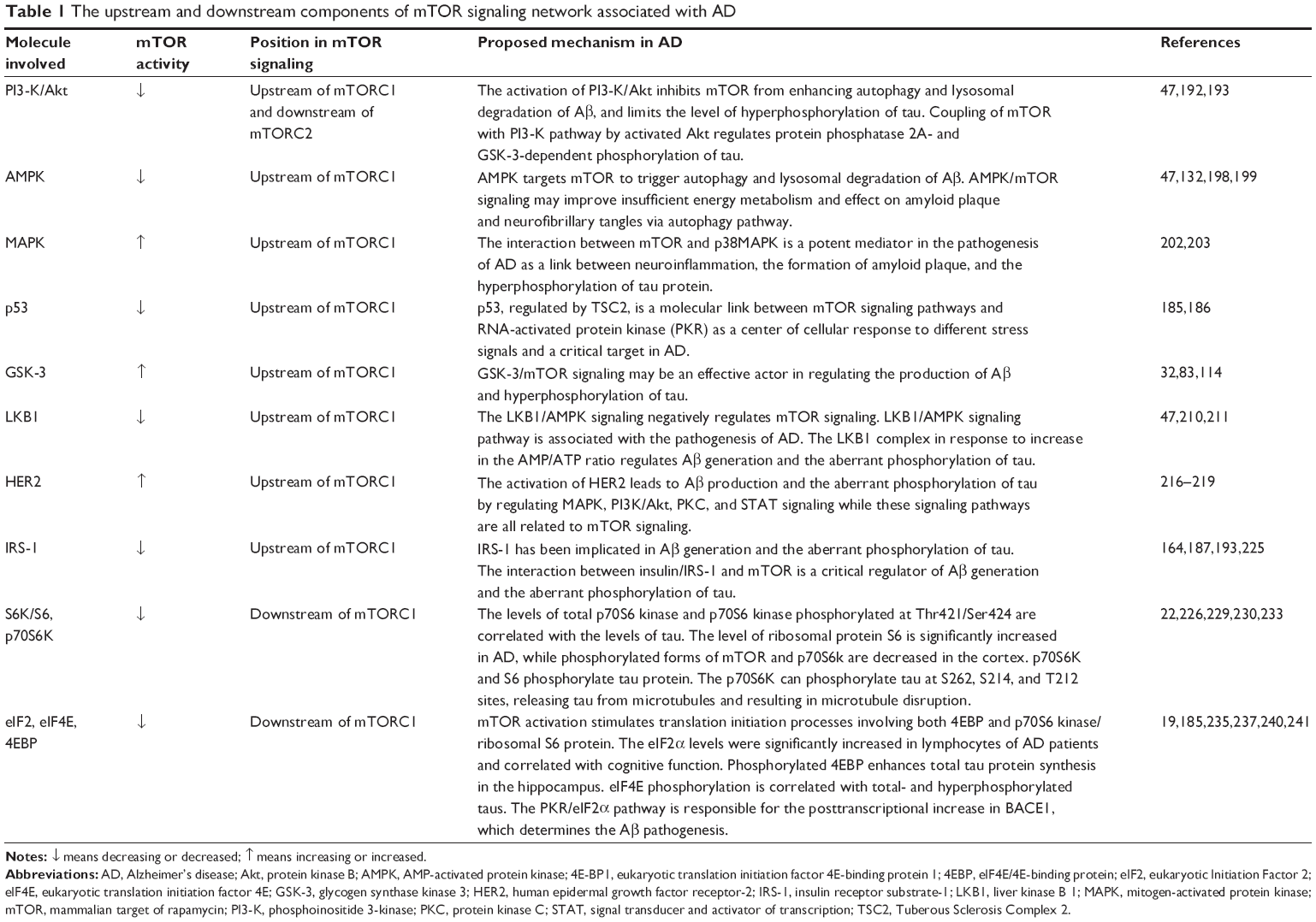 | Table 1 The upstream and downstream components of mTOR signaling network associated with AD |
The upstream components of mTOR signaling associated with AD
mTORC is regulated by numerous signaling components, including PI3K/Akt, AMPK, MAPK, p53, GTPase, LKB1, ERBB2, IRS-1, PTEN, GSK-3, insulin/IGF-1, and AMPK.32,83,187,188 It has been found that several upstream components of mTOR signaling (PI3K/Akt, AMPK, GSK-3, insulin/IGF-1, and AMPK) play a critical role in the regulation of Aβ generation and the aberrant phosphorylation of tau.5,32,47,187 In fact, there are many specific mechanisms that are also well addressed about the status of the mTOR upstream components in AD.
PI3-K/Akt
mTOR is a member of the PIKK (PI3-K-related Kinase) family, present in two distinct multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Activation of the PI3-K/Akt regulates mTORC1 by phosphorylating the tuberous sclerosis complex 2 (TSC2), blocking TSC2 from forming a heterodimer with tuberous sclerosis complex 1 (TSC1), and phosphorylating PRAS40, separating PRAS40 from mTOR. Preclinical research evidence also supports that coupling of mTOR with the PI3-K/Akt pathway regulates protein phosphatase 2A- and GSK-3-dependent phosphorylation of tau.111 The most striking is that the activation of PI3-K/Akt acts on mTOR to enhance autophagy and lysosomal degradation of Aβ, and limits the level of the hyperphosphorylation of tau.47,131,189 According to the previous findings that both PI3-K/Akt and mTOR signaling are effective regulators in the pathogenesis of AD,46,190,191 it is obvious that the PI3-K/Akt/mTOR signaling or the interaction between PI3-K/Akt and mTOR has a critical effect on the development and progression of AD pathology.47,192,193
AMPK
AMP-activated protein kinase (AMPK) maintains cellular energy homeostasis and acts as a metabolic master switch regulating many intracellular systems. Emerging studies have demonstrated that the AMPK signaling is closely associated with the major hallmarks of AD (insufficient energy metabolism, amyloid plaque, and NFTs).194–197 As an activator of autophagy function, AMPK activation retards the progress of AD pathology in respect to mTOR that plays a fundamental role in regulating autophagy state.47,132,198,199 AMPK targets mTOR to trigger autophagy and lysosomal degradation of Aβ.47 It is likely that the AMPK/mTOR signaling may improve insufficient energy metabolism and effect on amyloid plaque and NFTs via the autophagy pathway.
MAPK
Accumulating evaluation of p38MAPK significance involved in the pathophysiology and pathogenesis of AD has evidenced that the MAPK signaling may be a great contributor to the development of AD.200 The activation of p38MAPK brings about mTOR inactivation and leads to the induction of autophagy.201 The activation of mTOR decreases the activity of p38MAPK and reduces autophagy.202 It seems that the interaction between mTOR and p38MAPK is a potent mediator in the pathogenesis of AD as a link between neuroinflammation, the formation of amyloid plaque, and the hyperphosphorylation of tau proteins.203 Accordingly, inhibition of mTOR via MAPK may potentially prevent neurodegeneration from occurring in AD.
p53
p53 (protein 53 or tumor protein 53) is a tumor suppressor protein that plays a major role in preventing tumor development. It responds to a range of potentially oncogenic stresses by activating protective mechanisms, most notably cell cycle arrest and apoptosis. It has been noted that the level of p53 increases in AD and that p53 induces phosphorylation of human 2N4R tau at the tau-1/AT8 epitope in HEK293a cells.204 Thus p53 and mTOR are both associated with the progress of neurodegenerative disorders in AD. It has been evidenced that p53, regulated by TSC2, is a molecular link between the mTOR signaling pathways and RNA-activated protein kinase (PKR) as a center of cellular response to different stress signals and a critical target in AD.185,186
GSK-3
GSK-3 participates in a wide range of signal transduction cascades involving cellular processes, ranging from glycogen metabolism, gene transcription, protein translation to cytoskeletal organization, cell cycle regulation, and proliferation. GSK-3 plays a crucial role in the hyperphosphorylation of tau and neurofibrillary lesions since GSK-3 phosphorylates tau in most serine and threonine residues.111 The production and aggregation of Aβ is promoted by GSK-3 but reduced by pharmacological inhibition.117 It may be an integrating link between amyloid pathology and tauopathies via the activation of GSK-3.205,206 Identified as an upper regulator of mTOR,32,83,114 GSK-3/mTOR signaling may be an effective actor to regulate the production of Aβ and the hyperphosphorylation of tau.
LKB1
LKB1, a primary upstream kinase of AMPK, is a necessary element in cell metabolism that is required for maintaining energy homeostasis. The LKB1/AMPK signaling negatively regulates mTOR signaling.207–209 The LKB1 complex, in response to an increase in the AMP/ATP ratio, regulates Aβ generation and the aberrant phosphorylation of tau.47,210,211 Accordingly, it is possible that the LKB1/AMPK/mTOR signaling acts as a controller of Aβ generation and the aberrant phosphorylation of tau.
HER2
HER2 (Human Epidermal Growth Factor Receptor 2), a member of the epidermal growth factor receptor (EGFR/ErbB) family, has been shown to play an important role in the pathogenesis and progression of AD, and it has evolved to become an important biomarker and target of therapy for AD.212–215 Comprehensive research literature suggests that the activation of HER2 leads to Aβ production and the aberrant phosphorylation of tau through the regulation of MAPK, PI3K/Akt, protein kinase C (PKC), and signal transducer and activator of transcription (STAT) signaling, while these signaling pathways have all related to mTOR signaling.216–219
IRS-1
Insulin receptor substrate 1 (IRS-1) serves an important biological function for both metabolic and mitogenic pathways from the insulin and insulin-like growth factor-1 (IGF-1) receptors to downstream signaling pathways, including PI3K/Akt,220 mTOR, the stress kinase c-jun N-terminal kinase (JNK), and MAPK/ERK.221,222 Both IRS-1 and its downstream signaling pathways have been implicated in the pathogenesis and progression of Aβ generation and the aberrant phosphorylation of tau.187,223,224 Many studies have also suggested that the interaction between insulin/IRS-1 and mTOR is a critical regulator of Aβ generation and the aberrant phosphorylation of tau.164,187,193,225
The downstream components of mTOR signaling associated with AD
The eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) and ribosomal protein S6 kinase 1 (S6K1) are well-known downstream targets of mTORC1 via an interaction between raptor and a TOR signaling (TOS) motif in S6K and 4EBP. Numerous findings have evidenced that downstream mTOR signaling might be causally linked to AD and deregulation of downstream mTOR signaling could be a novel mechanism for AD.226,227
S6/S6K/p70S6K1
The p70 ribosomal S6 kinase (p70S6K) and S6, the most well-known downstream components of mTORC1, can phosphorylate tau protein and regulate Aβ production.227–229 The levels of total p70S6K and p70S6K phosphorylated at Thr421/Ser424 are significantly correlated with the levels of both total tau and PHF-tau rather than at Thr389,230 whereas zinc induces rapamycin-dependent p70S6K phosphorylation at Thr421/Ser424 and Thr389.231 The p70S6K can also phosphorylate tau at S262, S214, and T212 sites, releasing tau from microtubules and resulting in microtubule disruption.226 The level of p-p70S6K was significantly correlated with p-tau at S262, S214, and T212. These suggested that p70S6K is closely related to tau pathology in AD.232 In AD patients, the level of ribosomal protein S6 is significantly increased, and phosphorylated forms of mTOR and p70S6k decreased in the cortex.22 Moreover, the level of phosphorylated p70S6k is significantly decreased in the lymphocytes of Alzheimer’s patients, and correlated with Mini Mental Status Examination (MMSE) scores, while the decline of cognition in AD patients corresponds to the decrease in p70S6k levels.22,233
4EBP1
The activation of mTORC1 leads to the phosphorylation of two main downstream components, 4EBP1 and S6K1. 4EBP1 inhibits the initiation of protein translation by binding and inactivating eIF4E (eukaryotic translation initiation factor 4E). mTORC1 can phosphorylate 4EBP1 at multiple sites to dissociate eIF4E from 4EBP1, inhibiting 4EBP1 on eIF4E-dependent translation initiation. The eukaryotic initiation factor 2alpha (eIF2α) levels are significantly increased in the lymphocytes of AD patients and significantly correlated with cognitive and memory test scores.19 Similarly, increased phosphorylated eIF2α exists in AD patients’ brains and may account for cognitive impairment by decreasing synaptic plasticity.234,235 Phosphorylation of eIF2α is associated with the degeneration of neurons in AD due to the involvement in the autophagy process.236 The mTOR/eIF2α pathway is responsible for a posttranscriptional increase in BACE1, which determines the Aβ pathogenesis.237–239 A dramatic increase in phosphorylated eIF4E has occurred in the late stages of neurofibrillary changes. The level of eIF4E phosphorylation is markedly consistent with total and hyperphosphorylated tau, implying that the increase in eIF4E phosphorylation contributes to the formation of neurofibrillary changes.240 Phosphorylated 4EBP, the substrates of mTOR, enhances tau protein synthesis in the hippocampus. Taken together, levels of mTOR and its downstream targets 4EBP1, eIF2, and eIF4E have a close association with tau pathology.241
Conclusion and perspective
This review summarizes the roles of mTOR in the pathogenesis of AD and the advancements that the upstream and downstream components of mTOR signaling are involved in a wide variety of AD pathogenesis. The activation of mTOR enhances Aβ generation and deposition since the activation inhibits the autophagy/lysosome system, accounting for the Aβ generation and clearance. The activation of mTOR also modulates APP turnover via increasing β- and γ-secretase. mTOR may regulate Aβ generation via interaction with several key signaling pathways such as PI3-K/Akt, GSK-3, AMPK, and insulin/IGF-1. The activation of mTOR promotes the occurrence of tau pathology, whereas the inhibition of mTOR signaling retards the progress of tau pathology. It has been evidenced that the components of mTOR signaling are associated with the pathogenesis and progression of AD. These findings have implicated a profound clinical application for further development of interventions by inhibiting mTOR activation for the treatment and prevention of AD.
mTOR signaling plays a central role in maintaining protein homeostasis, and negatively regulates the autophagy/lysosome system. The activation of mTOR is involved in the pathogenesis of AD, and inhibiting mTOR activity by rapamycin will rescue cognitive impairments and retard the progression of AD pathology. However, several studies point out that the activation of mTOR may benefit the recovery of AD pathology. Inhibiting mTOR activity seems to be a nonneuroprotective property and induces detrimental outcomes. Rapamycin increases Aβ generation by reducing the activity of a disintegrin and metallopeptidase domain-10 (ADAM-10), an important α-secretase candidate that inhibits Aβ generation.242 Herein, the activation of mTOR may undertake a dual function in different situations. In acute stress, the activation of mTOR may be neuroprotective, and in chronic conditions, the activation of mTOR will be harmful. The activation of mTOR will benefit AD by slowing its pathologies, but whether it is an enemy remains uncertain. Thus, basic and clinical research is necessary to further clarify the roles of the mTOR activation in AD pathogenesis.
Acknowledgment
This work was supported by an award from the Hubei Province Health and Family Planning Scientific Research Project (WJ2015MB219) and the Nature Science Foundation of Shiyan Renmin Hospital to Dr Zhiyou Cai.
Disclosure
The authors report no conflicts of interest in this work.
References
Shahpasand K, Uemura I, Saito T, et al. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J Neurosci. 2012; 32(7):2430–2441. | ||
Salkovic-Petrisic M, Osmanovic J, Grünblatt E, Riederer P, Hoyer S. Modeling sporadic Alzheimer’s disease: the insulin resistant brain state generates multiple long-term morphobiological abnormalities including hyperphosphorylated tau protein and amyloid-beta. J Alzheimers Dis. 2009;18(4):729–750. | ||
Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21(2):372–381. | ||
Harman D. Alzheimer’s disease pathogenesis: role of aging. Ann N Y Acad Sci. 2006;1067:454–460. | ||
O’Neill C. PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol. 2013;48(7):647–653. | ||
Gouras GK. mTOR: at the crossroads of aging, chaperones, and Alzheimer’s disease. J Neurochem. 2013;124(6):747–748. | ||
Ferrer I, Barrachina M, Puig B. Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 2002;104(6):583–591. | ||
Kaper F, Dornhoefer N, Giaccia AJ. Mutations in the PI3K/PTEN/TSC2 pathway contribute to mammalian target of rapamycin activity and increased translation under hypoxic conditions. Cancer Res. 2006;66(3):1561–1569. | ||
Gharibi B, Farzadi S, Ghuman M, Hughes FJ. Inhibition of Akt/mTOR attenuates age-related changes in mesenchymal stem cells. Stem Cells. 2014;32(8):2256–2566. | ||
Yang F, Chu X, Yin M, et al. mTOR and autophagy in normal brain aging and caloric restriction ameliorating age-related cognition deficits. Behav Brain Res. 2014;264:82–90. | ||
Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 2014;16(3):378–386. | ||
Habib SL, Liang S. Hyperactivation of Akt/mTOR and deficiency in tuberin increased the oxidative DNA damage in kidney cancer patients with diabetes. Oncotarget. 2014;5(9):2542–2550. | ||
Martínez-Martínez E, Jurado-López R, Valero-Muñoz M, et al. Leptin induces cardiac fibrosis through galectin-3, mTOR and oxidative stress: potential role in obesity. J Hypertens. 2014;32(5):1104–1114; discussion 1114. | ||
Chong ZZ, Shang YC, Maiese K. Cardiovascular disease and mTOR signaling. Trends Cardiovasc Med. 2011;21(5):151–155. | ||
Yang Z, Ming XF. mTOR signalling: the molecular interface connecting metabolic stress, aging and cardiovascular diseases. Obes Rev. 2012;(13 Suppl 2):58–68. | ||
Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans. 2013;41(5):1103–1130. | ||
Jiang TF, Zhang YJ, Zhou HY, et al. Curcumin ameliorates the neurodegenerative pathology in A53T alpha-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J Neuroimmune Pharmacol. 2013;8(1):356–369. | ||
Ma T, Hoeffer CA, Capetillo-Zarate E, et al. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One. 2010;5(9):e12845. | ||
Paccalin M, Pain-Barc S, Pluchon C, et al. Activated mTOR and PKR kinases in lymphocytes correlate with memory and cognitive decline in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;22(4): 320–326. | ||
Cai Z, Zhao B, Li K, et al. Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer’s disease? J Neurosci Res. 2012;90(6):1105–1118. | ||
Pozueta J, Lefort R, Shelanski ML. Synaptic changes in Alzheimer’s disease and its models. Neuroscience. 2013;251:51–65. | ||
Lafay-Chebassier C, Paccalin M, Page G, et al. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J Neurochem. 2005;94(1):215–225. | ||
Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5(4): e9979. | ||
Asnaghi L, Bruno P, Priulla M, Nicolin A. mTOR: a protein kinase switching between life and death. Pharmacol Res. 2004;50(6):545–549. | ||
Balasubramanian S, Johnston RK, Moschella PC, Mani SK, Tuxworth WJ Jr, Kuppuswamy D. mTOR in growth and protection of hypertrophying myocardium. Cardiovasc Hematol Agents Med Chem. 2009;7(1):52–63. | ||
Ikenoue T, Hong S, Inoki K. Monitoring mammalian target of rapamycin (mTOR) activity. Methods Enzymol. 2009;452:165–180. | ||
Cleveland-Donovan K, Maile LA, Tsiaras WG, Tchkonia T, Kirkland JL, Boney CM. IGF-I activation of the AKT pathway is impaired in visceral but not subcutaneous preadipocytes from obese subjects. Endocrinology. 2010;151(8):3752–3763. | ||
Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29(21):6860–6870. | ||
Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7(2):e38. | ||
Woiwode A, Johnson SA, Zhong S, et al. PTEN represses RNA polymerase III-dependent transcription by targeting the TFIIIB complex. Mol Cell Biol. 2008;28(12):4204–4214. | ||
Bertrand FE, Spengemen JD, Shelton JG, McCubrey JA. Inhibition of PI3K, mTOR and MEK signaling pathways promotes rapid apoptosis in B-lineage ALL in the presence of stromal cell support. Leukemia. 2005;19(1):98–102. | ||
Maiese K, Chong ZZ, Shang YC, Wang S. mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med. 2013; 19(1):51–60. | ||
Chong ZZ, Shang YC, Wang S, Maiese K. Shedding new light on neurodegenerative diseases through the mammalian target of rapamycin. Prog Neurobiol. 2012;99(2):128–148. | ||
Mann DM, Iwatsubo T, Cairns NJ, et al. Amyloid beta protein (Abeta) deposition in chromosome 14-linked Alzheimer’s disease: predominance of Abeta42(43). Ann Neurol. 1996;40(2):149–156. | ||
Howlett DR, Bowler K, Soden PE, et al. Abeta deposition and related pathology in an APP x PS1 transgenic mouse model of Alzheimer’s disease. Histol Histopathol. 2008;23(1):67–76. | ||
Gandy S, Zhang YW, Ikin A, et al. Alzheimer’s presenilin 1 modulates sorting of APP and its carboxyl-terminal fragments in cerebral neurons in vivo. J Neurochem. 2007;102(3):619–626. | ||
Reznik-Wolf H, Machado J, Haroutunian V, et al. Somatic mutation analysis of the APP and Presenilin 1 and 2 genes in Alzheimer’s disease brains. J Neurogenet. 1998;12(1):55–65. | ||
Pluta R, Furmaga-Jabłońska W, Maciejewski R, Ułamek-Kozioł M, Jabłoński M. Brain ischemia activates beta- and gamma-secretase cleavage of amyloid precursor protein: significance in sporadic Alzheimer’s disease. Mol Neurobiol. 2013;47(1):425–434. | ||
Kounnas MZ, Danks AM, Cheng S, et al. Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron. 2010;67(5):769–780. | ||
Nadler Y, Alexandrovich A, Grigoriadis N, et al. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia. 2008;56(5):552–567. | ||
Li T, Li YM, Ahn K, Price DL, Sisodia SS, Wong PC. Increased expression of PS1 is sufficient to elevate the level and activity of gamma-secretase in vivo. PLoS One. 2011;6(11):e28179. | ||
Chen TJ, Wang DC, Chen SS. Amyloid-beta interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor-induced Arc expression in rat cortical neurons. J Neurosci Res. 2009;87(10):2297–2307. | ||
Pei JJ, Hugon J. mTOR-dependent signalling in Alzheimer’s disease. J Cell Mol Med. 2008;12(6B):2525–2532. | ||
Son SM, Song H, Byun J, et al. Altered APP processing in insulin-resistant conditions is mediated by autophagosome accumulation via the inhibition of mammalian target of rapamycin pathway. Diabetes. 2012;61(12):3126–3138. | ||
Damjanac M, Rioux Bilan A, Paccalin M, et al. Dissociation of Akt/PKB and ribosomal S6 kinase signaling markers in a transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2008;29(2):354–367. | ||
Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14. | ||
Cai Z, Yan LJ, Li K, Quazi SH, Zhao B. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromolecular Med. 2012;14(1):1–14. | ||
Li L, Zhang S, Zhang X, et al. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer’s disease. Curr Alzheimer Res. 2013;10(4):433–441. | ||
Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118(Pt 1):7–18. | ||
Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;(12 Suppl 2):1542–1552. | ||
Periyasamy-Thandavan S, Jiang M, Schoenlein P, Dong Z. Autophagy: molecular machinery, regulation, and implications for renal pathophysiology. Am J Physiol Renal Physiol. 2009;297(2):F244–F256. | ||
Das GC, Hollinger FB. Molecular pathways for glucose homeostasis, insulin signaling and autophagy in hepatitis C virus induced insulin resistance in a cellular model. Virology. 2012;434(1):5–17. | ||
Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823–830. | ||
Cui J, Gong Z, Shen HM. The role of autophagy in liver cancer: molecular mechanisms and potential therapeutic targets. Biochim Biophys Acta. 2013;1836(1):15–26. | ||
Chang Y, Lin J, Tsung A. Manipulation of autophagy by MIR375 generates antitumor effects in liver cancer. Autophagy. 2012;8(12):1833–1834. | ||
Nakagawa S, et al. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur J Pharmacol. 2012;696(1–3):143–154. | ||
Wang JH, Ahn IS, Fischer TD, et al. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology. 2011;141(6):2188–2199; e6. | ||
Harris J, Hope JC, Lavelle EC. Autophagy and the immune response to TB. Transbound Emerg Dis. 2009;56(6–7):248–254. | ||
Chaturvedi A, Pierce SK. Autophagy in immune cell regulation and dysregulation. Curr Allergy Asthma Rep. 2009;9(5):341–346. | ||
Josefsen L, Droce A, Sondergaard TE, et al. Autophagy provides nutrients for nonassimilating fungal structures and is necessary for plant colonization but not for infection in the necrotrophic plant pathogen Fusarium graminearum. Autophagy. 2012;8(3):326–337. | ||
Yang D, Li L, Liu H, et al. Induction of autophagy and senescence by knockdown of ROC1 E3 ubiquitin ligase to suppress the growth of liver cancer cells. Cell Death Differ. 2013;20(2):235–247. | ||
Ma Q, Qiang J, Gu P, Wang Y, Geng Y, Wang M. Age-related autophagy alterations in the brain of senescence accelerated mouse prone 8 (SAMP8) mice. Exp Gerontol. 2011;46(7):533–541. | ||
Graziotto JJ, Cao K, Collins FS, Krainc D. Rapamycin activates autophagy in Hutchinson-Gilford progeria syndrome: implications for normal aging and age-dependent neurodegenerative disorders. Autophagy. 2012;8(1):147–151. | ||
Zemke D, Azhar S, Majid A. The mTOR pathway as a potential target for the development of therapies against neurological disease. Drug News Perspect. 2007;20(8):495–499. | ||
Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy – a novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171(1):87–98. | ||
Yao H, Zhao D, Khan SH, Yang L. Role of autophagy in prion protein-induced neurodegenerative diseases. Acta Biochim Biophys Sin (Shanghai). 2013;45(6):494–502. | ||
Xue Z, Zhang S, Huang L, He Y, Fang R, Fang Y. Upexpression of Beclin-1-dependent autophagy protects against beta-amyloid-induced cell injury in PC12 cells. J Mol Neurosci. 2013;51(1):180–186. | ||
Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11(9):1107–1117. | ||
Spencer B, Potkar R, Trejo M, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009;29(43):13578–13588. | ||
Pierce A, Podlutskaya N, Halloran JJ, et al. Over-expression of heat shock factor 1 phenocopies the effect of chronic inhibition of TOR by rapamycin and is sufficient to ameliorate Alzheimer’s-like deficits in mice modeling the disease. J Neurochem. 2013;124(6):880–893. | ||
Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120(Pt 23):4081–4091. | ||
Boland B, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28(27):6926–6937. | ||
Mizushima N. A (beta) generation in autophagic vacuoles. J Cell Biol. 2005;171(1):15–17. | ||
Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6(9):e25416. | ||
Bandyopadhyay S, Goldstein LE, Lahiri DK, Rogers JT. Role of the APP non-amyloidogenic signaling pathway and targeting alpha-secretase as an alternative drug target for treatment of Alzheimer’s disease. Curr Med Chem. 2007;14(27):2848–2864. | ||
Cordy JM, Hooper NM, Turner AJ. The involvement of lipid rafts in Alzheimer’s disease. Mol Membr Biol. 2006;23(1):111–122. | ||
O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. | ||
Naslund J, Jensen M, Tjernberg LO, Thyberg J, Terenius L, Nordstedt C. The metabolic pathway generating p3, an A beta-peptide fragment, is probably non-amyloidogenic. Biochem Biophys Res Commun. 1994;204(2):780–787. | ||
Soriano S, Lu DC, Chandra S, Pietrzik CU, Koo EH. The amyloidogenic pathway of amyloid precursor protein (APP) is independent of its cleavage by caspases. J Biol Chem. 2001;276(31):29045–29050. | ||
Funderburk SF, Marcellino BK, Yue Z. Cell “self-eating” (autophagy) mechanism in Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):59–68. | ||
Zhou F, van Laar T, Huang H, Zhang L. APP and APLP1 are degraded through autophagy in response to proteasome inhibition in neuronal cells. Protein Cell. 2011;2(5):377–383. | ||
Boland B, Smith DA, Mooney D, Jung SS, Walsh DM, Platt FM. Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J Biol Chem. 2010;285(48):37415–37426. | ||
d’Abramo C, Ricciarelli R, Pronzato MA, Davies P. Troglitazone, a peroxisome proliferator-activated receptor-gamma agonist, decreases tau phosphorylation in CHOtau4R cells. J Neurochem. 2006;98(4):1068–1077. | ||
Dal Col J, Dolcetti R. GSK-3beta inhibition: at the crossroad between Akt and mTOR constitutive activation to enhance cyclin D1 protein stability in mantle cell lymphoma. Cell Cycle. 2008;7(18):2813–2816. | ||
Prada PO, Hirabara SM, de Souza CT, et al. L-glutamine supplementation induces insulin resistance in adipose tissue and improves insulin signalling in liver and muscle of rats with diet-induced obesity. Diabetologia. 2007;50(9):1949–1959. | ||
Syed DN, Adhami VM, Khan MI, Mukhtar H. Inhibition of Akt/mTOR signaling by the dietary flavonoid fisetin. Anticancer Agents Med Chem. 2013;13(7):995–1001. | ||
Battelli C, Cho DC. mTOR inhibitors in renal cell carcinoma. Therapy. 2011;8(4):359–367. | ||
Vazquez de la Torre A, Junyent F, Folch J, Pelegri C, et al. PI3 k/akt inhibition induces apoptosis through p38 activation in neurons. Pharmacol Res. 2013;70(1):116–125. | ||
Zang L, Xue B, Lu Z, et al. Identification of LRP16 as a negative regulator of insulin action and adipogenesis in 3T3-L1 adipocytes. Horm Metab Res. 2013;45(5):349–358. | ||
Yin G, Li LY, Qu M, Luo HB, Wang JZ, Zhou XW. Upregulation of AKT attenuates amyloid-beta-induced cell apoptosis. J Alzheimers Dis. 2011;25(2):337–345. | ||
Rokutanda S, Fujita T, Kanatani N, et al. Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev Biol. 2009; 328(1):78–93. | ||
Honda K, Sawada H, Kihara T, et al. Phosphatidylinositol 3-kinase mediates neuroprotection by estrogen in cultured cortical neurons. J Neurosci Res. 2000;60(3):321–327. | ||
Que J, Lian Q, El Oakley RM, Lim B, Lim SK. PI3 K/Akt/mTOR-mediated translational control regulates proliferation and differentiation of lineage-restricted RoSH stem cell lines. J Mol Signal. 2007;2:9. | ||
Lloberas N, Cruzado JM, Franquesa M, et al. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol. 2006;17(5):1395–1404. | ||
Haga S, Ozaki M, Inoue H, et al. The survival pathways phosphatidylinositol-3 kinase (PI3-K)/phosphoinositide-dependent protein kinase 1 (PDK1)/Akt modulate liver regeneration through hepatocyte size rather than proliferation. Hepatology. 2009;49(1):204–214. | ||
Kitagishi Y, Kobayashi M, Kikuta K, Matsuda S. Roles of PI3K/AKT/GSK3/mTOR pathway in cell signaling of mental illnesses. Depress Res Treat. 2012;2012:752563. | ||
Solano DC, Sironi M, Bonfini C, Solerte SB, Govoni S, Racchi M. Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 2000;14(7):1015–1022. | ||
Amar S, Belmaker RH, Agam G. The possible involvement of glycogen synthase kinase-3 (GSK-3) in diabetes, cancer and central nervous system diseases. Curr Pharm Des. 2011;17(22):2264–2277. | ||
Valvezan AJ, Klein PS. GSK-3 and Wnt signaling in neurogenesis and bipolar disorder. Front Mol Neurosci. 2012;5:1. | ||
Rowe MK, Wiest C, Chuang DM. GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci Biobehav Rev. 2007;31(6):920–931. | ||
Martinez A, Castro A, Dorronsoro I, Alonso M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med Res Rev. 2002;22(4):373–384. | ||
Giese KP. GSK-3: a key player in neurodegeneration and memory. IUBMB Life. 2009;61(5):516–521. | ||
Cai Z, Zhao Y, Zhao B. Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Curr Alzheimer Res. 2012;9(7):864–879. | ||
Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis. 2006;9(3 Suppl):309–317. | ||
Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B. Divergent roles of GSK3 and CDK5 in APP processing. Biochem Biophys Res Commun. 2003;312(4):922–929. | ||
Hu S, Begum AN, Jones MR, et al. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis. 2009;33(2):193–206. | ||
Huang HC, Klein PS. Multiple roles for glycogen synthase kinase-3 as a drug target in Alzheimer’s disease. Curr Drug Targets. 2006;7(11):1389–1397. | ||
Zhang YC, Cheng T, Yuan WP. PI3K/AKT/mTOR | ||
Jin Y, Sui HJ, Dong Y, et al. Atorvastatin enhances neurite outgrowth in cortical neurons in vitro via up-regulating the Akt/mTOR and Akt/GSK-3beta signaling pathways. Acta Pharmacol Sin. 2012;33(7):861–872. | ||
Vigneron F, Dos Santos P, Lemoine S, et al. GSK-3beta at the crossroads in the signalling of heart preconditioning: implication of mTOR and Wnt pathways. Cardiovasc Res. 2011;90(1):49–56. | ||
Meske V, Albert F, Ohm TG. Coupling of mammalian target of rapamycin with phosphoinositide 3-kinase signaling pathway regulates protein phosphatase 2A- and glycogen synthase kinase-3-dependent phosphorylation of Tau. J Biol Chem. 2008;283(1):100–109. | ||
Lee ES, Lee JO, Lee SK, et al. Caffeic acid phenethyl ester accumulates beta-catenin through GSK-3beta and participates in proliferation through mTOR in C2C12 cells. Life Sci. 2009;84(21–22):755–759. | ||
Sarkar S, Krishna G, Imarisio S, Saiki S, O’Kane CJ, Rubinsztein DC. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum Mol Genet. 2008;17(2):170–178. | ||
Buller CL, Loberg RD, Fan MH, et al. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol. 2008;295(3):C836–C843. | ||
Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18(4):1437–1446. | ||
Ma T, Tzavaras N, Tsokas P, Landau EM, Blitzer RD. Synaptic stimulation of mTOR is mediated by Wnt signaling and regulation of glycogen synthetase kinase-3. J Neurosci. 2011;31(48):17537–17546. | ||
Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288(2):1295–1306. | ||
Xiao B, Sanders MJ, Underwood E, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472(7342): 230–233. | ||
Jin X, Townley R, Shapiro L. Structural insight into AMPK regulation: ADP comes into play. Structure. 2007;15(10):1285–1295. | ||
Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9(5):407–416. | ||
Andreasen AS, Kelly M, Berg RM, Moller K, Pedersen BK. Type 2 diabetes is associated with altered NF-kappaB DNA binding activity, JNK phosphorylation, and AMPK phosphorylation in skeletal muscle after LPS. PLoS One. 2011;6(9):e23999. | ||
O’Neill HM, Holloway GP, Steinberg GR. AMPK regulation of fatty acid metabolism and mitochondrial biogenesis: implications for obesity. Mol Cell Endocrinol. 2013;366(2):135–151. | ||
Shin E, Shin S, Kong H, et al. Dietary aloe reduces adipogenesis via the activation of AMPK and suppresses obesity-related inflammation in obese mice. Immune Netw. 2011;11(2):107–113. | ||
Dupont J, Reverchon M, Cloix L, Froment P, Rame C. Involvement of adipokines, AMPK, PI3K and the PPAR signaling pathways in ovarian follicle development and cancer. Int J Dev Biol. 2012;56(10–12):959–967. | ||
Priebe A, Tan L, Wahl H, et al. Glucose deprivation activates AMPK and induces cell death through modulation of Akt in ovarian cancer cells. Gynecol Oncol. 2011;122(2):389–395. | ||
Niemann B, Pan R, Teschner M, Boening A, Silber RE, Rohrbach S. Age and obesity-associated changes in the expression and activation of components of the AMPK signaling pathway in human right atrial tissue. Exp Gerontol. 2013;48(1):55–63. | ||
Vingtdeux V, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011;121(3):337–349. | ||
Ju TC, Chen HM, Lin JT, et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J Cell Biol. 2011;194(2):209–227. | ||
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. | ||
Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22(2):132–139. | ||
Chong ZZ, Shang YC, Wang S, Maiese K. A critical kinase cascade in neurological disorders: PI 3-K, Akt, and mTOR. Future Neurol. 2012;7(6):733–748. | ||
Vingtdeux V, Chandakkar P, Zhao H, d’Abramo C, Davies P, Marambaud P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J. 2011;25(1):219–231. | ||
Katic M, Kahn CR. The role of insulin and IGF-1 signaling in longevity. Cell Mol Life Sci. 2005;62(3):320–343. | ||
Deelen J, Uh HW, Monajemi R, et al. Gene set analysis of GWAS data for human longevity highlights the relevance of the insulin/IGF-1 signaling and telomere maintenance pathways. Age (Dordr). 2013;35(1):235–249. | ||
Lehrbach NJ, Castro C, Murfitt KJ, Abreu-Goodger C, Griffin JL, Miska EA. Post-developmental microRNA expression is required for normal physiology, and regulates aging in parallel to insulin/IGF-1 signaling in C. elegans. RNA. 2012;18(12):2220–2235. | ||
Anisimov VN, Berstein LM, Popovich IG, et al. Central and peripheral effects of insulin/IGF-1 signaling in aging and cancer: antidiabetic drugs as geroprotectors and anticarcinogens. Ann N Y Acad Sci. 2005;1057:220–234. | ||
Heni M, Hennenlotter J, Scharpf M, et al. Insulin receptor isoforms A and B as well as insulin receptor substrates-1 and -2 are differentially expressed in prostate cancer. PLoS One. 2012;7(12):e50953. | ||
Bender SB, McGraw AP, Jaffe IZ, Sowers JR. Mineralocorticoid receptor-mediated vascular insulin resistance: an early contributor to diabetes-related vascular disease? Diabetes. 2013;2(2):313–319. | ||
Fowlkes JL, Bunn RC, Thrailkill KM. Contributions of the insulin/insulin-like growth factor-1 axis to diabetic osteopathy. J Diabetes Metab. 2011;1(3). | ||
Giuffrida ML, Tomasello F, Caraci F, Chiechio S, Nicoletti F, Copani A. Beta-amyloid monomer and insulin/IGF-1 signaling in Alzheimer’s disease. Mol Neurobiol. 2012;46(3):605–613. | ||
Salminen A, Kaarniranta K. Insulin/IGF-1 paradox of aging: regulation via AKT/IKK/NF-kappaB signaling. Cell Signal. 2010;22(4):573–577. | ||
Kim B, Backus C, Oh S, Feldman EL. Hyperglycemia-induced tau cleavage in vitro and in vivo: a possible link between diabetes and Alzheimer’s disease. J Alzheimers Dis. 2013;34(3):727–739. | ||
Lin B, Ginsberg MD, Busto R. Hyperglycemic but not normoglycemic global ischemia induces marked early intraneuronal expression of beta-amyloid precursor protein. Brain Res. 2001;888(1):107–116. | ||
Liu LP, Yan TH, Jiang LY, et al. Pioglitazone ameliorates memory deficits in streptozotocin-induced diabetic mice by reducing brain beta-amyloid through PPARgamma activation. Acta Pharmacol Sin. 2013;34(4):455–463. | ||
Luchsinger JA. Adiposity, hyperinsulinemia, diabetes and Alzheimer’s disease: an epidemiological perspective. Eur J Pharmacol. 2008;585(1):119–129. | ||
Craft S, Newcomer J, Kanne S, et al. Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol Aging. 1996;17(1):123–130. | ||
Jimenez-Palomares M, Ramos-Rodriguez JJ, Lopez-Acosta JF, et al. Increased Abeta production prompts the onset of glucose intolerance and insulin resistance. Am J Physiol Endocrinol Metab. 2012;302(11):E1373–E1380. | ||
Farris W, Leissring MA, Hemming ML, Chang AY, Selkoe DJ. Alternative splicing of human insulin-degrading enzyme yields a novel isoform with a decreased ability to degrade insulin and amyloid beta-protein. Biochemistry. 2005;44(17):6513–6525. | ||
Hiltunen M, Khandelwal VK, Yaluri N, et al. Contribution of genetic and dietary insulin resistance to Alzheimer phenotype in APP/PS1 transgenic mice. J Cell Mol Med. 2012;16(6):1206–1222. | ||
Yan SD, Chen X, Fu J. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382(6593):685–691. | ||
Chen X, Walker DG, Schmidt AM, Arancio O, Lue LF, Yan SD. RAGE: a potential target for Abeta-mediated cellular perturbation in Alzheimer’s disease. Curr Mol Med. 2007;7(8):735–742. | ||
Yan SS, Chen D, Yan S, Guo L, Du H, Chen JX. RAGE is a key cellular target for Abeta-induced perturbation in Alzheimer’s disease. Front Biosci (Schol Ed). 2012;4:240–250. | ||
Srikanth V, Maczurek A, Phan T, et al. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging. 2011;32(5):763–777. | ||
Avruch J, Hara K, Lin Y, et al. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006;25(48):6361–6372. | ||
Floyd S, Favre C, Lasorsa FM, et al. The insulin-like growth factor-I-mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol Biol Cell. 2007;18(9):3545–3555. | ||
Sharma N, Castorena CM, Cartee GD. Tissue-specific responses of IGF-1/insulin and mTOR signaling in calorie restricted rats. PLoS One. 2012;7(6):e38835. | ||
Zhang C, Wendel AA, Keogh MR, Harris TE, Chen J, Coleman RA. Glycerolipid signals alter mTOR complex 2 (mTORC2) to diminish insulin signaling. Proc Natl Acad Sci U S A. 2012;109(5): 1667–1672. | ||
Hwang SL, Li X, Lee JY, Chang HW. Improved insulin sensitivity by rapamycin is associated with reduction of mTOR and S6K1 activities in L6 myotubes. Biochem Biophys Res Commun. 2012;418(2):402–407. | ||
Bose SK, Shrivastava S, Meyer K, Ray RB, Ray R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J Virol. 2012;86(11):6315–6322. | ||
Ueno M, Carvalheira JB, Tambascia RC, et al. Regulation of insulin signalling by hyperinsulinaemia: role of IRS-1/2 serine phosphorylation and the mTOR/p70 S6K pathway. Diabetologia. 2005;48(3):506–518. | ||
Gual P, Gremeaux T, Gonzalez T, Le Marchand-Brustel Y, Tanti JF. MAP kinases and mTOR mediate insulin-induced phosphorylation of insulin receptor substrate-1 on serine residues 307, 612 and 632. Diabetologia. 2003;46(11):1532–1542. | ||
Xin-Long C, Zhao-Fan X, Dao-Feng B, Wei D. mTOR partly mediates insulin resistance by phosphorylation of insulin receptor substrate-1 on serine(307) residues after burn. Burns. 2011;37(1):86–93. | ||
De Felice FG. Alzheimer’s disease and insulin resistance: translating basic science into clinical applications. J Clin Invest. 2013;123(2):531–539. | ||
Ma YQ, Wu DK, Liu JK. mTOR and tau phosphorylated proteins in the hippocampal tissue of rats with type 2 diabetes and Alzheimer’s disease. Mol Med Rep. 2013;7(2):623–627. | ||
Craft S, Cholerton B, Baker LD. Insulin and Alzheimer’s disease: untangling the web. J Alzheimers Dis. 2013;(33 Suppl 1):S263–S275. | ||
Long-Smith CM, Manning S, McClean PL. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer’s Disease. Neuromolecular Med. 2013;15(1):102–114. | ||
Han X, Ma Y, Liu X, et al. Changes in insulin-signaling transduction pathway underlie learning/memory deficits in an Alzheimer’s disease rat model. J Neural Transm. 2012;119(11):1407–1416. | ||
Morris JK, Burns JM. Insulin: an emerging treatment for Alzheimer’s disease dementia? Curr Neurol Neurosci Rep. 2012;12(5):520–527. | ||
Wang L, Rhodes CJ, Lawrence JC Jr. Activation of mammalian target of rapamycin (mTOR) by insulin is associated with stimulation of 4EBP1 binding to dimeric mTOR complex 1. J Biol Chem. 2006;281(34):24293–24303. | ||
Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316–323. | ||
Zhang Y, Tian Q, Zhang Q, Zhou X, Liu S, Wang JZ. Hyperphosphorylation of microtubule-associated tau protein plays dual role in neurodegeneration and neuroprotection. Pathophysiology. 2009;16(4):311–316. | ||
Delacourte A, Defossez A. Alzheimer’s disease: tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J Neurol Sci. 1986;76(2–3):173–186. | ||
Bancher C, Lassmann H, Budka H, et al. Neurofibrillary tangles in Alzheimer’s disease and progressive supranuclear palsy: antigenic similarities and differences. Microtubule-associated protein tau antigenicity is prominent in all types of tangles. Acta Neuropathol. 1987;74(1):39–46. | ||
Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15(23):2321–2328. | ||
Canudas AM, Gutierrez-Cuesta J, Rodriguez MI. Hyperphosphorylation of microtubule-associated protein tau in senescence-accelerated mouse (SAM). Mech Ageing Dev. 2005;126(12):1300–1304. | ||
Li T, Paudel HK. Glycogen synthase kinase 3beta phosphorylates Alzheimer’s disease-specific Ser396 of microtubule-associated protein tau by a sequential mechanism. Biochemistry. 2006;45(10):3125–3133. | ||
Liu Y, Su Y, Sun S, et al. Human tau may modify glucocorticoids-mediated regulation of cAMP-dependent kinase and phosphorylated cAMP response element binding protein. Neurochem Res. 2012;37(5):935–947. | ||
Jicha GA, Weaver C, Lane E, et al. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J Neurosci. 1999;19(17):7486–7494. | ||
Buee-Scherrer V, Goedert M. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases in intact cells. FEBS Lett. 2002;515(1–3):151–154. | ||
Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, Cohen P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409(1):57–62. | ||
Caccamo A, Magri A, Medina DX, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell. 2013;12(3):370–380. | ||
Taga M, Mouton-Liger F, Paquet C, Hugon J. Modulation of oxidative stress and tau phosphorylation by the mTOR activator phosphatidic acid in SH-SY5Y cells. FEBS Lett. 2011;585(12):1801–1806. | ||
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285(17):13107–13120. | ||
Liu Y, Su Y, Wang J, et al. Rapamycin decreases tau phosphorylation at Ser214 through regulation of cAMP-dependent kinase. Neurochem Int. 2013;62(4):458–467. | ||
Morel M, Couturier J, Lafay-Chebassier C, Paccalin M, Page G. PKR, the double stranded RNA-dependent protein kinase as a critical target in Alzheimer’s disease. J Cell Mol Med. 2009;13(8A):1476–1488. | ||
Damjanac M, Page G, Ragot S, et al. PKR, a cognitive decline biomarker, can regulate translation via two consecutive molecular targets p53 and Redd1 in lymphocytes of AD patients. J Cell Mol Med. 2009;13(8B):1823–1832. | ||
O’Neill C, Kiely AP, Coakley MF, Manning S, Long-Smith CM. Insulin and IGF-1 signalling: longevity, protein homoeostasis and Alzheimer’s disease. Biochem Soc Trans. 2012;40(4):721–727. | ||
Cheng HC, Kim SR, Oo TF, et al. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci. 2011;31(6):2125–2135. | ||
Maiese K, Chong ZZ, Shang YC, Wang S. Targeting disease through novel pathways of apoptosis and autophagy. Expert Opin Ther Targets. 2012;16(12):1203–1214. | ||
Pignataro G, Capone D, Polichetti G, et al. Neuroprotective, immunosuppressant and antineoplastic properties of mTOR inhibitors: current and emerging therapeutic options. Curr Opin Pharmacol. 2011;11(4):378–394. | ||
Wu X, Kihara T, Akaike A, Niidome T, Sugimoto H. PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochem Biophys Res Commun. 2010;393(3):514–518. | ||
Yuan M, Pino E, Wu L, Kacergis M, Soukas AA. Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J Biol Chem. 2012;287(35):29579–29588. | ||
Nemoto T, Yanagita T, Satoh S, et al. Insulin-induced neurite-like process outgrowth: acceleration of tau protein synthesis via a phosphoinositide 3-kinase~mammalian target of rapamycin pathway. Neurochem Int. 2011;59(6):880–888. | ||
Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. 2009;380(1):98–104. | ||
Won JS, Im YB, Kim J, Singh AK, Singh I. Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem Biophys Res Commun. 2010;399(4):487–491. | ||
Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem J. 2011;434(3):503–512. | ||
Yoshida H, Goedert M. Phosphorylation of microtubule-associated protein tau by AMPK-related kinases. J Neurochem. 2012;120(1):165–176. | ||
Salminen A, Kaarniranta K, Haapasalo A, Soininen H, Hiltunen M. AMP-activated protein kinase: a potential player in Alzheimer’s disease. J Neurochem. 2011;118(4):460–474. | ||
Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I. Abeta-induced formation of autophagosomes is mediated by RAGE-CaMKKbeta-AMPK signaling. Neurobiol Aging. 2012;33(5):1006; e11–e23. | ||
Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology. 2010;58(3):561–568. | ||
Tang G, Yue Z, Talloczy Z, Goldman JE. Adaptive autophagy in Alexander disease-affected astrocytes. Autophagy. 2008;4(5): 701–703. | ||
Hulmi JJ, Silvennoinen M, Lehti M, Kivela R, Kainulainen H. Altered REDD1, myostatin, and Akt/mTOR/FoxO/MAPK signaling in streptozotocin-induced diabetic muscle atrophy. Am J Physiol Endocrinol Metab. 2012;302(3):E307–E315. | ||
Li JS, Yao ZX. Modulation of FGF receptor signaling as an intervention and potential therapy for myelin breakdown in Alzheimer’s disease. Med Hypotheses. 2013;80(4):341–344. | ||
Hooper C, Meimaridou E, Tavassoli M, Melino G, Lovestone S, Killick R. p53 is upregulated in Alzheimer’s disease and induces tau phosphorylation in HEK293a cells. Neurosci Lett. 2007;418(1):34–37. | ||
Hernandez F, Gomez de Barreda E, Fuster-Matanzo A, Lucas JJ, Avila J. GSK3: a possible link between beta amyloid peptide and tau protein. Exp Neurol. 2010;223(2):322–325. | ||
Hernandez F, de Barreda EG, Fuster-Matanzo A, Goni-Oliver P, Lucas JJ, Avila J. The role of GSK3 in Alzheimer disease. Brain Res Bull. 2009;80(4–5):248–250. | ||
Dong LX, Sun LL, Zhang X, et al. Negative regulation of mTOR activity by LKB1-AMPK signaling in non-small cell lung cancer cells. Acta Pharmacol Sin. 2013;34(2):314–318. | ||
Nguyen HB, Babcock JT, Wells CD, Quilliam LA. LKB1 tumor suppressor regulates AMP kinase/mTOR-independent cell growth and proliferation via the phosphorylation of Yap. Oncogene. 2012;32(35):4100–4109. | ||
Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6(1): 91–99. | ||
Lee S, Wang JW, Yu W, Lu B. Phospho-dependent ubiquitination and degradation of PAR-1 regulates synaptic morphology and tau-mediated Abeta toxicity in Drosophila. Nat Commun. 2012;3:1312. | ||
Wang JW, Imai Y, Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J Neurosci. 2007;27(3):574–581. | ||
Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Golde TE, Koo EH. Abeta42-lowering nonsteroidal anti-inflammatory drugs preserve intramembrane cleavage of the amyloid precursor protein (APP) and ErbB-4 receptor and signaling through the APP intracellular domain. J Biol Chem. 2003;278(33):30748–30754. | ||
Ryu J, Yu HN, Cho H, et al. Neuregulin-1 exerts protective effects against neurotoxicities induced by C-terminal fragments of APP via ErbB4 receptor. J Pharmacol Sci. 2012;119(1):73–81. | ||
Fleck D, Garratt AN, Haass C, Willem M. BACE1 dependent neuregulin processing: review. Curr Alzheimer Res. 2012;9(2):178–183. | ||
Slack BE, Breu J, Muchnicki L, Wurtman RJ. Rapid stimulation of amyloid precursor protein release by epidermal growth factor: role of protein kinase C. Biochem J. 1997;327(Pt 1):245–249. | ||
Mori N, Kyo S, Nakamura M, et al. Expression of HER-2 affects patient survival and paclitaxel sensitivity in endometrial cancer. Br J Cancer. 2010;103(6):889–898. | ||
Gao M, Patel R, Ahmad I, et al. SPRY2 loss enhances ErbB trafficking and PI3K/AKT signalling to drive human and mouse prostate carcinogenesis. EMBO Mol Med. 2012;4(8):776–790. | ||
Mut M, Lule S, Demir O, Kurnaz IA, Vural I. Both mitogen-activated protein kinase (MAPK)/extracellular-signal-regulated kinases (ERK) 1/2 and phosphatidylinositide-3-OH kinase (PI3K)/Akt pathways regulate activation of E-twenty-six (ETS)-like transcription factor 1 (Elk-1) in U138 glioblastoma cells. Int J Biochem Cell Biol. 2012;44(2):302–310. | ||
Nunes M, Shi C, Greenberger LM. Phosphorylation of extracellular signal-regulated kinase 1 and 2, protein kinase B, and signal transducer and activator of transcription 3 are differently inhibited by an epidermal growth factor receptor inhibitor, EKB-569, in tumor cells and normal human keratinocytes. Mol Cancer Ther. 2004;3(1):21–27. | ||
Bocker D, Verspohl EJ. Role of protein kinase C, PI3-kinase and tyrosine kinase in activation of MAP kinase by glucose and agonists of G-protein coupled receptors in INS-1 cells. Int J Exp Diabetes Res. 2001;2(3):233–244. | ||
Strle K, Broussard SR, McCusker RH, et al. C-jun N-terminal kinase mediates tumor necrosis factor-alpha suppression of differentiation in myoblasts. Endocrinology. 2006;147(9):4363–4373. | ||
Madhala-Levy D, Williams VC, Hughes SM, Reshef R, Halevy O. Cooperation between Shh and IGF-I in promoting myogenic proliferation and differentiation via the MAPK/ERK and PI3K/Akt pathways requires Smo activity. J Cell Physiol. 2012;227(4):1455–1464. | ||
Wang P, Jiang S, Cui Y, et al. The n-terminal 5-MER peptide analogue P165 of amyloid precursor protein exerts protective effects on SH-SY5Y cells and rat hippocampus neuronal synapses. Neuroscience. 2011;173:169–178. | ||
Talbot K, Wang HY, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122(4):1316–1338. | ||
Nistico R, Cavallucci V, Piccinin S, et al. Insulin receptor beta-subunit haploinsufficiency impairs hippocampal late-phase LTP and recognition memory. Neuromolecular Med. 2012;14(4):262–269. | ||
Pei JJ, Bjorkdahl C, Zhang H, Zhou X, Winblad B. p70 S6 kinase and tau in Alzheimer’s disease. J Alzheimers Dis. 2008;14(4):385–392. | ||
Pei JJ, Sjogren M, Winblad B. Neurofibrillary degeneration in Alzheimer’s disease: from molecular mechanisms to identification of drug targets. Curr Opin Psychiatry. 2008;21(6):555–561. | ||
Pei JJ, Gong CX, An WL, et al. Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2, and p70 S6, similar to that in Alzheimer’s disease. Am J Pathol. 2003;163(3):845–858. | ||
Shang YC, Chong ZZ, Wang S, Maiese K. Prevention of beta-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI 3-K/mTOR pathway, Bad, and Bcl-xL. Aging (Albany NY). 2012;4(3):187–201. | ||
An WL, Cowburn RF, Li L, et al. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am J Pathol. 2003;163(2):591–607. | ||
Bjorkdahl C, Sjogren MJ, Winblad B, Pei JJ. Zinc induces neurofilament phosphorylation independent of p70 S6 kinase in N2a cells. Neuroreport. 2005;16(6):591–595. | ||
Pei JJ, An WL, Zhou XW, et al. P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett. 2006;580(1):107–114. | ||
Paccalin M, Al Khidir F, Barc SP, et al. Peripheral p70S6k levels and emotional memory in patients with Alzheimer’s disease. Neurosci Lett. 2006;410(3):162–164. | ||
Choi Y, Kim HS, Shin KY, et al. Minocycline attenuates neuronal cell death and improves cognitive impairment in Alzheimer’s disease models. Neuropsychopharmacology. 2007;32(11):2393–2404. | ||
Kim HS, Choi Y, Shin KY, et al. Swedish amyloid precursor protein mutation increases phosphorylation of eIF2alpha in vitro and in vivo. J Neurosci Res. 2007;85(7):1528–1537. | ||
Chang RC, Wong AK, Ng HK, Hugon J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport. 2002;13(18):2429–2432. | ||
Mouton-Liger F, Paquet C, Dumurgier J, et al. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim Biophys Acta. 2012;1822(6):885–896. | ||
Moreno JA, Radford H, Peretti D, et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature. 2012;485(7399):507–511. | ||
Devi L, Ohno M. Phospho-eIF2alpha level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS One. 2010;5(9):e12974. | ||
Li X, An WL, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport. 2004;15(14):2237–2240. | ||
Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005;272(16):4211–4220. | ||
Zhang S, Salemi J, Hou H, et al. Rapamycin promotes beta-amyloid production via ADAM-10 inhibition. Biochem Biophys Res Commun. 2010;398(3):337–341. |
 [Recent studies on PI3K/AKT/mTOR signaling pathway in hematopoietic stem cells]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2013;21(1):245–249. Chinese.
[Recent studies on PI3K/AKT/mTOR signaling pathway in hematopoietic stem cells]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2013;21(1):245–249. Chinese.
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.