Back to Journals » Cancer Management and Research » Volume 12
Abnormal MGMT Promoter Methylation in Gastrointestinal Stromal Tumors: Genetic Susceptibility and Association with Clinical Outcome
Authors Lou L, Zhang W, Li J ![]() , Wang Y
, Wang Y ![]()
Received 25 June 2020
Accepted for publication 25 September 2020
Published 12 October 2020 Volume 2020:12 Pages 9941—9952
DOI https://doi.org/10.2147/CMAR.S269388
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Sanjeev K. Srivastava
Liping Lou,1,* Wendi Zhang,2,* Jun Li,1 Yu Wang1
1Institute of Pathology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Department of Pediatrics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yu Wang
Institute of Pathology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, No. 1095 Jiefang Avenue, Wuhan 430030, Hubei Province, People’s Republic of China
Email [email protected]
Purpose: KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GISTs) represent a heterogeneous subgroup of GISTs that lack KIT or PDGFRA mutations and possess distinct genetic alterations and primary resistance to imatinib. Succinate dehydrogenase (SDH)-deficient GISTs comprise the largest subpopulation of WT GISTs that are characterized by loss-of-function of SDH. O6-methylguanine-DNA methyltransferase (MGMT) is a specific DNA repair enzyme that has been identified as a predictor of positive treatment response to alkylating agents in a variety of cancers. The aim of this study was to evaluate the expression of MGMT and the prevalence of MGMT promoter methylation in GISTs and to determine the association between MGMT promoter methylation and clinicopathological characteristics and clinical outcomes.
Patients and Methods: A heterogeneous cohort of 137 primary GISTs that confirmed by immunohistochemistry and KIT/PDGFRA mutation analysis were retrospectively selected and analyzed for MGMT expression and MGMT promoter methylation using immunohistochemical staining and methylation-specific PCR (MSP). A concordance analysis between MGMT promoter methylation and clinicopathological characteristics and prognosis was also performed.
Results: A total of 44.5% (65/137) of GIST patients displayed loss of MGMT protein expression, and 10.9% (15/137) of these patients exhibited MGMT promoter methylation. However, no significant correlation was observed between the loss of MGMT protein expression and MGMT promoter methylation. WT GISTs possessing an epithelioid or mixed phenotype, particularly those that were SDH-deficient, displayed a markedly higher prevalence of MGMT promoter methylation compared to that in KIT/PDGFRA mutated GISTs. Moreover, MGMT promoter methylation was identified as a potential independent prognostic factor for OS and DFS in patients with GIST.
Conclusion: MGMT promoter methylation is particularly frequent in SDH-deficient GISTs and in WT GISTs possessing an epithelioid/mixed phenotype, and knowledge of this methylation status may offer a novel potential therapeutic option for WT GISTs.
Keywords: gastrointestinal stromal tumor, O6-methylguanine-DNA methyltransferase, wild-type, succinate dehydrogenase deficiency
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the digestive tract, and they harbor mutually exclusive mutations in c-kit receptor tyrosine kinase (KIT) or platelet-derived growth factor receptor alpha (PDGFRA) in approximately 85% of cases.1,2 However, the remaining 10–15% of GISTs are categorized as KIT/PDGFRA wild-type (WT), as they do not harbor any detectable mutations in the KIT and PDGFRA genes.3,4 Recently, several studies have suggested that WT GISTs comprise different subtypes with distinct genetic alterations and can be subdivided into succinate dehydrogenase (SDH)-deficient and non-SDH-deficient groups.5 Among these, SDH-deficient GISTs represent the largest subpopulation of WT GISTs that are characterized by a loss of SDH function,6 while non-SDH-deficient GISTs are comprised of neurofibromatosis type 1 (NF1) -associated, serine/threonine protein kinase B-RAF (BRAF) mutation, RAS gene mutation, and quadruple wild-type GISTs.7–9 Therefore, WT GISTs are currently regarded as a heterogeneous subgroup of tumors that are accompanied by different clinicopathological, genetic, and biological characteristics.10,11 Moreover, WT GISTs often display a primary resistance to targeted tyrosine kinase inhibitor (TKI) treatment using imatinib.12,13 Therefore, further identification of the genetic alterations associated with the pathogenesis of WT GISTs may contribute to the development of new therapeutic strategies and may aid in ameliorating the clinical management of GISTs.
O6-methylguanine-DNA methyltransferase (MGMT) is a specific DNA repair enzyme that has been demonstrated to repair DNA alkylation damage by removing the methyl group from the O6-guanine residues and then transferring them to its cysteine residues.14,15 MGMT promoter methylation-related gene silencing and loss of function have been detected in various cancers, including gliomas,16 lung cancer,17 breast cancer,18 colorectal cancer,19 and gastric cancer.20 Moreover, aberrant MGMT promoter methylation was identified as a predictor of positive treatment response to alkylating agents.21–23 Recently, MGMT promoter methylation has been reported in a few GISTs.24–27 However, the preponderance of MGMT promoter methylation in GISTs possessing different genetic subtypes remains unclear. Therefore, we conducted immunohistochemical staining and methylation-specific PCR (MSP) analyses to identify the expression of MGMT and the prevalence of MGMT promoter methylation in a heterogeneous cohort of 137 GISTs. Additionally, the distinct MGMT promoter methylation status and its association with clinicopathological characteristics and prognosis of patients with GIST were evaluated.
Materials and Methods
Patients and Samples
A total of 137 formalin-fixed paraffin-embedded samples of primary GIST with available clinical and pathological characteristics were retrospectively selected from more than 1000 patients who underwent surgery at Tongji Hospital between January 2013 and July 2019. None of the patients received imatinib treatment prior to surgery. Supportive immunohistochemical and mutational analyses were performed for each case. This study was approved by the Ethics Committee of Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, P.R. China. Written informed consent was obtained from the participants themselves or from their parents. All experiments were conducted in accordance with the World Medical Association Declaration of Helsinki.
Histological and Immunohistochemical Analyses
Histological diagnosis was established by the Department of Pathology at Tongji Hospital according to the current World Health Organization (WHO) classification of tumors. Hematoxylin and eosin-stained (H&E) slides were reviewed independently by 2 experienced gastrointestinal pathologists for the presence of histomorphological features. The tumor risk grade was assessed based on the modified National Institute of Health (NIH) stratification criteria. Immunohistochemistry was performed on 4 μm paraffin sections using an UltraVision Quanto Detection System HRP DAB (Thermo Scientific, CA, USA) for the detection of CD117 (rabbit polyclonal, 1:400 dilution, DAKO, Glostrup, Denmark), DOG-1 (rabbit polyclonal, ready to use, Zhongshan Golden Bridge Biotechnology Co., Ltd., ZGBBT, Beijing), CD34 (rabbit polyclonal, 1:400 dilution, DAKO, Glostrup, Denmark), SMA (mouse polyclonal, 1:400 dilution, DAKO, Glostrup, Denmark), Caldesmon (mouse polyclonal, ZGBBT, Beijing), S-100 (rabbit polyclonal, ready to use, ZGBBT, Beijing), Desmin (mouse polyclonal, ready to use, ZGBBT, Beijing), and Ki67 (rabbit polyclonal, 1:400 dilution, ZGBBT, Beijing) according to the manufacturer’s protocols. Immunostaining for SDHB (succinate dehydrogenase complex subunit B) and MGMT were performed on all samples using the mouse monoclonal antibody anti-SDHB antibody (1:32 dilution, ZGBBT, Beijing) and the mouse monoclonal antibody anti-MGMT antibody (ready to use, ZGBBT, Beijing), regardless of the anatomic site and morphologic subtype.
The expression of the MGMT protein was evaluated in tumor cells using a semi-quantitative scoring method that was based on the percentage of positive cells and the staining intensity. The percentage of positive tumor cells was scored as follows: 0 (<10% nuclear staining), 1 (10–25% nuclear staining), 2 (26–50% nuclear staining), and 3 (>50% nuclear staining). The staining intensity of tumor cells was ranked as follows: 0 (no staining), 1 (weak staining), 2 (moderate staining), and 3 (strong staining). An immunoreactive score (IRS) was generated by multiplying the percentage of positive cells by the staining intensity. Finally, samples with IRS >2 were classified as positive, while samples with IRS ≤2 were classified as negative.
DNA Extraction
For paraffin-embedded specimens, the MagCore Genomic DNA Tissue Kit C401 (RBC Bioscience Corp, Xiamen, China) was used to isolate genomic DNA from 8–10 sections of 10 μm thickness according to the recommendations of the manufacturer. DNA concentrations were quantified using a SMA4000 spectrophotometer (Merinton Instrument, Inc., Beijing, China). Subsequently, 1000 ng of genomic DNA was modified by exposure to a sodium bisulfite solution using the EpiTect Bisulfite kit (Qiagen, Hilden, Germany).
KIT and PDGFRA Gene Mutation Analysis
Mutation analysis of KIT exons 9, 11, 12, 13, 14, and 17 and PDGFRA exons 12,14, and 18 were performed using a commercial kit based on fluorescence PCR and capillary electrophoresis sequencing according to the manufacturer’s protocols (SinoMDgene Technology Co., Ltd., Beijing, China). The amplification was carried out on an ABI 7500 Real-Time PCR System (Applied Biosystems, Foster city, CA, USA) with an initial denaturation at 95°C for 3 min, then 40 cycles of 94°C for 15 s and 60°C for 45 s. The PCR amplification products were identified by direct sequencing using a commercially available kit (SinoMDgene Technology Co., Ltd., Beijing, China) on an ABI 3500DX Genetic Analyzer (Applied Biosystems, USA). Gene sequencing results were compared to the National Center for Biotechnology Information (NCBI) database sequences for KIT (NM_001093772.1) and PDGFRA (NM_006206.4).
MGMT Methylation Assessment by Methylation-Specific PCR (MSP)
After sodium bisulfite conversion, genomic DNA was amplified for the MGMT promoter using a commercially available kit obtained from SinoMDgene Technology Co., Ltd., Beijing, China, according to the manufacturer’s instruments. Each reaction mixture contained 2 μL of bisulfite-treated DNA, 12.5 μL of SYBR Green PCR Master Mix, and 5.5 μL of forward and reverse primers to provide a final volume of 20 μL. The amplification was performed on a Bio-Rad CFX96 Real-Time PCR Detection System instrument (Bio-Rad Laboratories) with an initial denaturation at 95°C for 3 min, then 45 cycles of 95°C for 15 s, 60°C for 45 s, and 72°C for 15 s, and a final extension for 10 min at 72°C. Analysis was performed using the Bio-Rad CFX Manager 3.1 software (Bio-Rad Laboratories).
Statistical Analysis
Contingency tables and associations were estimated using the chi-square (χ2) test and Fisher’s exact test. The Kaplan-Meier method was applied to evaluate the overall survival (OS) and disease-free survival (DFS), and the comparisons of survival curves between groups were assessed using the Log rank test. The prognostic value of MGMT promoter methylation in GIST patients was determined by multivariate analyses using the Cox proportional hazard model. A two-tailed P value < 0.05 was considered as statistically significant.
Results
Clinicopathological Characteristics
A total of 137 patients with GIST were recruited for this study, and these patients included 70 men (51.1%) and 67 women (48.9%) (Supplementary Table 1). The ages of the patients ranged from 5 to 81 years with a mean age of 54.6 years. Tumor size ranged from 4 to 350 mm with a mean size of 66 mm. Metastasis was identified in 6 patients, and recurrence was identified in 9 patients. The primary tumors predominantly occurred in the stomach (50.4%) and small intestine (32.8%), and the liver was the most frequent site of metastases. The most common pathomorphological appearance was spindle cell type (73.0%), and this was followed by epithelioid (20.4%) and mixed (6.6%) cell types (Table 1). A high mitotic rate (>5/50 HPF) was observed in 24 cases (17.5%), and 26 cases (19.0%) exhibited necrosis (Table 1). Based on the NIH risk categories, tumors were categorized as very low-risk (25/137, 18.2%), low-risk (45/137, 32.8%), intermediate risk (18/137, 13.1%), and high risk (49/137, 35.9%). The mean follow-up time was 29.1 months (range, 9–75 months).
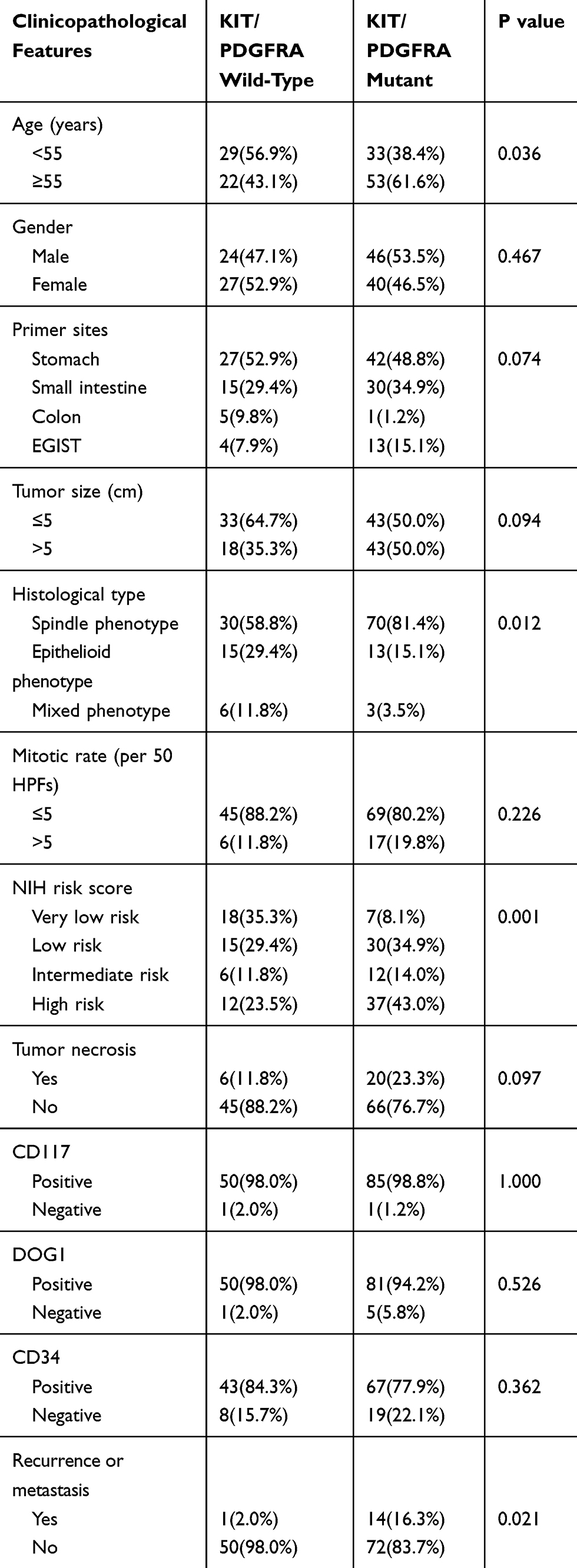 |
Table 1 Clinicopathological Features of 137 Patients with GIST in This Study |
Based on the supporting immunohistochemical analyses, CD117, DOG1, and CD34 antibodies exhibited diffuse positive expression in 135 (98.5%), 131 (95.6%), and 110 (80.3%) cases of GIST (Table 1, Figure 1). Focal positive expressions of SMA, Caldesmon, and desmin were observed in 12.4%, 27.7%, and 2.9% of the tumors. None of the cases were positive for S-100. The Ki-67 index ranged from 1% to 70%, with a mean of 2.2%, 2.9%, 4.9%, and 14.8% in GISTs with very low, low, intermediate, and high risk, respectively. The absence of SDHB expression was noted in 7/139 (5.1%) cases.
 |
Figure 1 H&E staining and immunohistochemistry analysis of GIST specimens. (A) KIT-mutant GISTs showing spindle cell morphology. KIT mutated GISTs with spindle cell morphology showing diffuse positive expression of CD117 (C), DOG1 (E) and SDHB (G) by Immunohistochemistry. (B) SDH-deficient GISTs showing epithelioid morphology. Immunohistochemical staining showing diffuse positive expression of CD117 (D) and DOG1 (F) in SDH-deficient GISTs. (H) SDH-deficient GISTs showing loss expression of SDHB by Immunohistochemistry. Scale bar 100 μm. |
Mutations in KIT and PDGFRA Genes
Among the 137 cases of GISTs included in this study, 75 (54.7%) harbored KIT gene mutations in exons 11 (n=41), 9 (n=12), 13 (n=6), and 17 (n=6), respectively. Of the remaining 62 patients, 11 (8.0%) cases possessed PDGFRA gene mutations, while the remaining 51 (37.3%) cases showed no detectable mutations and were regarded as KIT/PDGFRA WT GISTs. KIT exon 11 mutations appeared as deletions, insertions, point mutations, and complex mutations were detected in 41 (29.9%) tumors. The 12 cases with KIT exon 9 mutations all displayed a 6 bp insertion that resulted in duplication of codons A502 and T503. More than 90% of PDGFRA mutations affecting exons 12, 14, and 18 were associated with epithelioid morphology. Univariate analyses revealed that KIT/PDGFRA WT GISTs exhibited a younger age, a tendency towards epithelioid or mixed morphology, and a lower recurrence or metastasis rate compared to that of KIT/PDGFRA mutated GISTs (Table 1). Furthermore, seven of the WT GISTs were found to possess negative staining for SDHB according to IHC (Figure 1H). Each of these seven cases exhibited positive staining for KIT and DOG1 and possessed typical morphological characteristics that have been recognized as indicative of SDH-deficient GISTs (Figure 1B, D and F). All seven patients with SDH-deficient GIST exhibited tumors that occurred in the stomach, and these patients possessed a mean age of 21 years (range 10–36 years). Their tumors demonstrated a predominantly multinodular growth pattern (5/7) and possessed an epithelioid (5/7) or mixed (2/7) histologic subtype (Table 2). Secondary mutations in KIT that predominantly presented in exons 13, 14, and 17 were observed in 10 out of 15 patients with recurrence or metastasis. All of the mutations are presented in Supplementary Table 1.
 |
Table 2 Clinicopathological Features of 7 Patients with SDH-Deficient GIST |
MGMT Promoter Methylation Analysis
According to MSP, methylation of the MGMT promoter was detected in 10.9% (n=15) of tumors. The mean age of GIST patients with MGMT promoter methylation was 44.1±15.8 (95% CI 35.4–52.9) years, and this was markedly younger than that of patients without MGMT promoter methylation (55.8±12.1 years, 95% CI 53.6–57.9; p=0.001). Further subgroup analysis indicated that the MGMT promoter was methylated in only 3 out of 86 (3.5%) KIT/PDGFRA mutated GISTs and in 12 of 51 (23.5%) WT GISTs. Among the GISTs lacking KIT/PDGFRA mutations, eight out of 44 (18.2%) and four out of 7 (57.1%) cases demonstrated MGMT promoter methylation in the non-SDHB-deficient and SDHB-proficient subgroups. Comparative analyses revealed that WT GISTs, particularly those with SDH-deficiency, demonstrated a markedly higher prevalence of MGMT promoter methylation compared to that of KIT- or PDGFRA-mutant GISTs (P<0.001, Table 3). Moreover, GISTs possessing epithelioid or mixed morphology displayed a higher frequency of MGMT promoter methylation than spindle cell GISTs (P=0.004, Table 3). These findings indicated that MGMT promoter methylation may exhibit a trend toward a higher frequency in epithelioid/mixed-type WT GISTs and in SDH-deficient GISTs. However, no statistically significant relationships were observed between MGMT promoter methylation and patient sex, primary tumor localization, primary tumor size, mitotic rate, risk grade, recurrence, or metastasis in this study.
 |
Table 3 Correlation of MGMT Promoter Methylation with Clinicopathological Parameters |
MGMT Immunohistochemical Expression and Its Correlation with MGMT Promoter Methylation
The expression of the MGMT protein was identified in 15 cases with MGMT promoter methylation and in 122 cases without MGMT promoter methylation according to IHC staining. Positive staining for the MGMT protein was localized primarily within the nucleus of tumor cells (Figure 2E–H). The loss of MGMT protein expression was detected in 61 (44.5%) cases of 137 GISTs (Figure 2A–D, Table 3). Among the 15 cases with methylated MGMT promoter, 5 (33.3%) cases exhibited a loss of MGMT protein expression, and 10 (66.7%) cases demonstrated moderate to high expression of MGMT protein. Meanwhile, 51 (41.8%) cases displayed a loss of MGMT protein expression, and 71 (58.2%) cases exhibited moderate to high expression of MGMT protein among the 122 cases with non-methylated MGMT promoters. However, no significant correlation was observed between the loss of MGMT protein expression and MGMT promoter methylation status (P=0.067, Table 3).
 |
Figure 2 Immunohistochemistry staining of MGMT in GIST specimens. Negative nuclear staining of MGMT in KIT mutated GISTs with spindle cell morphology (A-B, case 57) and SDH-deficient GISTs with epithelioid morphology (C, D, case 87). (E, F) Moderate nuclear staining of MGMT in GISTs (case 7). (G, H) Strong nuclei staining of MGMT in GISTs (case 75). Scale bar (A), (C), (E) and (G) 100 μm; (B), (D), (F) and (H) 50 μm. |
Association Between MGMT Promoter Methylation and Prognosis of Patients with GIST
The Kaplan–Meier method with Log rank test indicated that GIST patients carrying MGMT promoter methylation exhibited longer overall survival (OS) and disease-free survival (DFS) compared to that in patients without MGMT promoter methylation (Figure 3). Moreover, multivariate Cox proportional hazards regression analysis demonstrated that MGMT promoter methylation could be considered as a potential independent prognostic factor for OS (HR=3.711; 95% CI 1.645–8.372; P=0.002) and DFS (HR=3.287; 95% CI 1.169–9.245; P=0.024) in GIST patients. These results suggest that MGMT promoter methylation may predict improved clinical outcomes in patients with GISTs.
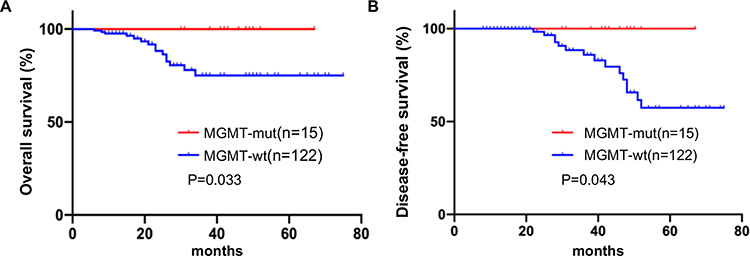 |
Figure 3 Association between MGMT promoter methylation and prognosis of patients with GIST. Kaplan-Meier analysis of OS (A) and DFS (B) in GIST patients according to the MGMT promoter methylation status. |
Discussion
KIT/PDGFRA WT GISTs represent a genetically heterogeneous subgroup of GISTs that lack KIT or PDGFRA mutations.3,10 These WT GISTs exhibit identical clinicopathological, genetic, and biological characteristics that are indistinguishable from those of KIT/PDGFRA mutated GISTs.28 SDH-deficient GISTs are the largest subpopulation of WT GISTs that are characterized by loss-of-function mutations in any of the SDH-subunits A, B, C, or.6,11 Although most WT GISTs exhibit relatively benign clinical behaviors, there are still a few WT GISTs that exhibit malignant potential.28,29 In the present study, we selected a cohort of KIT/PDGFRA-mutated GISTs and WT GISTs, and we found that WT GISTs displayed a tendency to exhibit epithelioid or mixed morphology, and a lower recurrence or metastasis rate compared to these values in KIT/PDGFRA mutated GISTs. Meanwhile, of the WT GISTs examined, 7/51 (13.7%) cases were observed to be negative for SDHB and were identified as SDH-deficient GISTs in our study. Based on the present findings, SDH-deficient GISTs typically share unique clinicopathological features that include gastric primary tumor localization, multinodular growth patterns, and an epithelioid or mixed cell morphological phenotype.30 Lymph node metastasis and local recurrence are also present more frequently in SDH-deficient GISTs.30 However, even with local recurrence or lymph node metastasis, the majority of SDH-deficient GISTs still tend to follow a relatively indolent clinical course in contrast to other GISTs.11,30 Similarly, in this study, we observed that all seven cases occurred exclusively in the stomach and exhibited a predominantly multinodular growth pattern with epithelioid or mixed histologic subtype and a male/female ratio of 1:2.5 with a mean age of 21 years. Additionally, none of the patients developed local recurrence or distant metastasis after surgery, and no patients died of this disease at the longest follow-up of 67 months.
MGMT is a specific DNA repair enzyme that protects against mutagenic and alkylating agent-induced carcinogenesis.31 Aberrant MGMT promoter methylation has been identified as an inheritable epigenetic alteration that occurs in a broad spectrum of human cancers.16–20 To date, MGMT promoter methylation has been reported in only a few GISTs. In two previous studies that were conducted in 2003 and 2008, MGMT promoter methylation was observed to exhibit a comparatively high frequency rate in the study cohort of gastric GISTs, and this contradicts the notion that MGMT methylation is a relatively rare event in GISTs.26,27 Recently, MGMT methylation was detected for the first time in nine SDH-deficient GISTs when these tumors were compared to other pathogenetic GIST subsets, including 15 WT GISTs and 24 KIT/PDGFRA mutated GISTs. These findings indicated that MGMT methylation was clearly enriched in SDH-deficient GISTs and in WT GISTs in comparison to levels in KIT/PDGFRA mutated GISTs.24 However, these results warrant further validation studies using larger GIST series with distinct genetic status and in different populations. Thus, we detected the expression of MGMT and MGMT promoter methylation status in a heterogeneous cohort of GISTs that included 86 KIT/PDGFRA mutant GISTs and 51 WT GISTs through the use of IHC staining and MSP. Our results indicated that 44.5% (65/137) of the GISTs displayed a loss of MGMT protein expression and that 10.9% (15/137) of the GISTs exhibited MGMT promoter methylation. However, no significant correlation was observed between the loss of MGMT protein expression and MGMT promoter methylation status. Interestingly, we found that WT GISTs possessing an epithelioid or mixed phenotype, particularly SDH-deficient ones, exhibited a markedly higher prevalence of MGMT promoter methylation in comparison to that in KIT/PDGFRA mutated GISTs. These findings indicated that MGMT promoter methylation is a less frequent epigenetic alteration in GISTs and that MGMT promoter methylation tends to be more frequent in SDH-deficient GISTs and in WT GISTs with an epithelioid/mixed phenotype. Therefore, the detection of MGMT promoter methylation would only be recommended for WT GISTs. Furthermore, multivariate Cox regression analysis indicated that MGMT promoter methylation could be considered as a potential independent prognostic factor for OS and DFS in GIST patients, suggesting that MGMT promoter methylation may predict better clinical outcomes in patients with GISTs.
Multiple studies have suggested that WT GISTs, including SDH-deficient ones, often exhibit primary resistance to imatinib and lack efficacious therapy options.12,13 This indicates that other molecular genetic alterations may also be involved in the tumorigenesis of this distinct subgroup of GISTs. Recently, methylation of the MGMT promoter has emerged as a predictive factor of positive therapeutic response to alkylating agents.21–23 Based on these premises, it is reasonable to hypothesize that WT GISTs and SDH-deficient GISTs harboring MGMT promoter methylation may benefit from treatment with alkylating agents. Although the current trials studying the effects of the alkylating agents temozolomide and carmustine on GISTs were published with negative results, we still cannot completely deny the possible efficacy of these drugs in the treatment of a subset of WT GISTs, particularly on SDH-deficient GISTs.32–34 Additionally, these negative results are based on studies limited to only a few GIST patients who enrolled in past trials examining sarcomas without selecting the genotype and detecting the SDH status of the tumors. Subsequently, MGMT promoter methylation status was not detected in all cases of these GISTs, with the exception of two cases in which MGMT protein expression was evaluated by IHC.34 Additionally, none of these trials applied Choi criteria to evaluate the GIST response to drug treatment.35 Therefore, further clinical studies are required to validate this hypothesis. Currently, a Phase II trial of temozolomide in metastatic SDH-deficient GIST is ongoing (ClinicalTrials.gov/NCT03556384). In the event of acquiring positive results, this clinical trial may offer a novel potential therapeutic option for WT GISTs.
Conclusion
Our study provides additional evidence that MGMT promoter methylation is particularly frequent in SDH-deficient GISTs and in KIT/PDGFRA WT GISTs with an epithelioid/mixed phenotype. Moreover, GIST patients harboring MGMT promoter methylation exhibited improved clinical outcomes with longer OS and DFS. These findings support the use of MGMT promoter methylation as a prognostic predictor in GISTs, and our findings offer a novel potential therapeutic option for WT GISTs. However, the predictive value of MGMT promoter methylation for the application of alkylating agents as a new treatment strategy in KIT/PDGFRA WT GISTs required further exploration in future clinical studies.
Abbreviations
GISTs, gastrointestinal stromal tumors; WT, wild-type; SDH, succinate dehydrogenase; MGMT, O6-methylguanine-DNA methyltransferase; MSP, methylation-specific PCR; KIT, C-kit receptor tyrosine kinase; PDGFRA, platelet-derived growth factor receptor alpha; NF1, neurofibromatosis type 1; BRAF, serine/threonine protein kinase B-RAF; TKI, tyrosine kinase inhibitor; DFS, disease-free survival; H&E, hematoxylin and eosin-stained; NCBI, National Center for Biotechnology Information; NIH, National Institute of Health; OS, overall survival; WHO, World Health Organization; IRS, immunoreactive score; IHC, immunohistochemistry; HPF, high-power field.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed on the journal to which the article will be submitted; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from National Natural Science Foundation of China (No. 81302114).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Joensuu H, Hohenberger P, Corless CL. Gastrointestinal stromal tumour. Lancet. 2013;382(9896):973–983. doi:10.1016/S0140-6736(13)60106-3
2. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865–878. doi:10.1038/nrc3143
3. Boikos SA, Pappo AS, Killian JK, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors: a report from the national institutes of health gastrointestinal stromal tumor clinic. JAMA Oncol. 2016;2(7):922–928. doi:10.1001/jamaoncol.2016.0256
4. Weldon CB, Madenci AL, Boikos SA, et al. Surgical management of wild-type gastrointestinal stromal tumors: a report from the national institutes of health pediatric and wildtype GIST clinic. J Clin Oncol. 2017;35(5):523–528. doi:10.1200/JCO.2016.68.6733
5. Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108(1):314–318. doi:10.1073/pnas.1009199108
6. Killian JK, Kim SY, Miettinen M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3(6):648–657. doi:10.1158/2159-8290.CD-13-0092
7. Yantiss RK, Rosenberg AE, Sarran L, et al. Multiple gastrointestinal stromal tumors in type I neurofibromatosis: a pathologic and molecular study. Mod Pathol. 2005;18(4):475–484. doi:10.1038/modpathol.3800334
8. Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(6):1769–1776. doi:10.1158/1078-0432.CCR-11-2230
9. Pantaleo MA, Nannini M, Corless CL, et al. Quadruple wild-type (WT) GIST: defining the subset of GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling pathways. Cancer Med. 2015;4(1):101–103. doi:10.1002/cam4.325
10. Nannini M, Biasco G, Astolfi A, et al. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST). J Med Genet. 2013;50(10):653–661. doi:10.1136/jmedgenet-2013-101695
11. Boikos SA, Stratakis CA. The genetic landscape of gastrointestinal stromal tumor lacking KIT and PDGFRA mutations. Endocrine. 2014;47(2):401–408. doi:10.1007/s12020-014-0346-3
12. Lai EC, Lau SH, Lau WY. Current management of gastrointestinal stromal tumors–a comprehensive review. Int J Surg. 2012;10(7):334–340. doi:10.1016/j.ijsu.2012.05.007
13. Marrari A, Wagner AJ, Hornick JL. Predictors of response to targeted therapies for gastrointestinal stromal tumors. Arch Pathol Lab Med. 2012;136(5):483–489. doi:10.5858/arpa.2011-0082-RA
14. Hazra TK, Roy R, Biswas T, et al. Specific recognition of O6-methylguanine in DNA by active site mutants of human O6-methylguanine-DNA methyltransferase. Biochemistry. 1997;36(19):5769–5776. doi:10.1021/bi963085i
15. Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004;4(4):296–307. doi:10.1038/nrc1319
16. Wick W, Weller M, van den Bent M, et al. MGMT testing–the challenges for biomarker-based glioma treatment. Nat Rev Neurol. 2014;10(7):372–385. doi:10.1038/nrneurol.2014.100
17. Zhang Y, Wang R, Song H, et al. Methylation of multiple genes as a candidate biomarker in non-small cell lung cancer. Cancer Lett. 2011;303(1):21–28. doi:10.1016/j.canlet.2010.12.011
18. Asiaf A, Ahmad ST, Malik AA, et al. Protein expression and methylation of MGMT, a DNA repair gene and their correlation with clinicopathological parameters in invasive ductal carcinoma of the breast. Tumour Biol. 2015;36(8):6485–6496. doi:10.1007/s13277-015-3339-9
19. Kimura N, Nagasaka T, Murakami J, et al. Methylation profiles of genes utilizing newly developed CpG island methylation microarray on colorectal cancer patients. Nucleic Acids Res. 2005;33(5):e46. doi:10.1093/nar/gni046
20. Cao Y, Liu H, Li H, et al. Association of O6-methylguanine-DNA methyltransferase. Protein expression with postoperative prognosis and adjuvant chemotherapeutic benefits among patients with stage II or III gastric cancer. JAMA Surg. 2017;152(11):e173120. doi:10.1001/jamasurg.2017.3120
21. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi:10.1056/NEJMoa043331
22. Perry JR, Laperriere N, O’Callaghan CJ, et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl J Med. 2017;376(11):1027–1037. doi:10.1056/NEJMoa1611977
23. Pietrantonio F, Randon G, Romagnoli D, et al. Biomarker-guided implementation of the old drug temozolomide as a novel treatment option for patients with metastatic colorectal cancer. Cancer Treat Rev. 2020;82:101935. doi:10.1016/j.ctrv.2019.101935
24. Ricci R, Martini M, Ravegnini G, et al. Preferential MGMT methylation could predispose a subset of KIT/PDGFRA-WT GISTs, including SDH-deficient ones, to respond to alkylating agents. Clin Epigenetics. 2019;11(1):2. doi:10.1186/s13148-018-0594-9
25. Okamoto Y, Sawaki A, Ito S, et al. Aberrant DNA methylation associated with aggressiveness of gastrointestinal stromal tumour. Gut. 2012;61(3):392–401. doi:10.1136/gut.2011.241034
26. House MG, Guo M, Efron DT, et al. Tumor suppressor gene hypermethylation as a predictor of gastric stromal tumor behavior. J Gastrointest Surg. 2003;7(8):1004–1014. doi:10.1016/j.gassur.2003.08.002
27. Saito K, Sakurai S, Sano T, et al. Aberrant methylation status of known methylation-sensitive CpG islands in gastrointestinal stromal tumors without any correlation to the state of c-kit and PDGFRA gene mutations and their malignancy. Cancer Sci. 2008;99(2):253–259. doi:10.1111/j.1349-7006.2007.00682.x
28. Ding H, Yu X, Yu Y, et al. Clinical significance of the molecular heterogeneity of gastrointestinal stromal tumors and related research: a systematic review. Oncol Rep. 2020;43(3):751–764.
29. Falkenhorst J, Hamacher R, Bauer S. New therapeutic agents in gastrointestinal stromal tumours. Curr Opin Oncol. 2019;31(4):322–328. doi:10.1097/CCO.0000000000000549
30. Nannini M, Urbini M, Astolfi A, et al. The progressive fragmentation of the KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GIST). J Transl Med. 2017;15(1):113. doi:10.1186/s12967-017-1212-x
31. Sharma S, Salehi F, Scheithauer BW, et al. Role of MGMT in tumor development, progression, diagnosis, treatment and prognosis. Anticancer Res. 2009;29(10):3759–3768.
32. Trent JC, Beach J, Burgess MA, et al. A two-arm phase II study of temozolomide in patients with advanced gastrointestinal stromal tumors and other soft tissue sarcomas. Cancer. 2003;98(12):2693–2699. doi:10.1002/cncr.11875
33. Garcia Del Muro X, Lopez-Pousa A, Martin J, et al. A phase II trial of temozolomide as a 6-week, continuous oral schedule in patients with advanced soft tissue sarcoma: a study by the Spanish group for research on sarcomas. Cancer. 2005;104(8):1706–1712. doi:10.1002/cncr.21384
34. Ryan CW, Dolan ME, Brockstein BB, et al. A phase II trial of O6-benzylguanine and carmustine in patients with advanced soft tissue sarcoma. Cancer Chemother Pharmacol. 2006;58(5):634–639. doi:10.1007/s00280-006-0210-0
35. Benjamin RS, Choi H, Macapinlac HA, et al. We should desist using RECIST, at least in GIST. J Clin Oncol. 2007;25(13):1760–1764. doi:10.1200/JCO.2006.07.3411
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.