")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
A toxic organic solvent-free technology for the preparation of PEGylated paclitaxel nanosuspension based on human serum albumin for effective cancer therapy
Authors Yin T, Dong L, Cui B, Wang L, Yin L, Zhou J, Huo M
Received 19 July 2015
Accepted for publication 28 October 2015
Published 11 December 2015 Volume 2015:10(1) Pages 7397—7412
DOI https://doi.org/10.2147/IJN.S92697
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Tingjie Yin,* Lihui Dong,* Bei Cui, Lei Wang, Lifang Yin, Jianping Zhou, Meirong Huo
State Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Abstract: Clinically, paclitaxel (PTX) is one of most commonly prescribed therapies against a wide range of solid neoplasms. Despite its success, the clinical applicability of PTX (Taxol®) is severely hampered by systemic toxicities induced by Cremophor EL. While attempts to bypass the need for Cremophor EL have been developed through platforms such as Abraxane™, nab™ relies heavily on the use of organic solvents, namely, chloroform. The toxicity introduced by residual chloroform poses a potential risk to patient health. To mitigate the toxicities of toxic organic solvent-based manufacture methods, we have designed a method for the formulation of PTX nanosuspensions (PTX-PEG [polyethylene glycol]-HSA [human serum albumin]) that eliminates the dependence on toxic organic solvents. Coined the solid-dispersion technology, this technique permits the dispersion of PTX into PEG skeleton without the use of organic solvents or Cremophor EL as a solubilizer. Once the PTX-PEG dispersion is complete, the dispersion can be formulated with HSA into nanosuspensions suitable for intravenous administration. Additionally, the incorporation of PEG permits the prolonged circulation through the steric stabilization effect. Finally, HSA-mediated targeting permits active receptor-mediated endocytosis for enhanced tumor uptake and reduced side effects. By eliminating the need for both Cremophor EL and organic solvents while simultaneously increasing antitumor efficacy, this method provides a superior alternative to currently accepted methods for PTX delivery.
Keywords: human serum albumin, nanosuspension, paclitaxel, polyethylene glycol, solid-dispersion technology
Introduction
Paclitaxel (PTX) is a clinically accepted front-line therapy against a wide range of solid neoplasms since 1990. It works by promoting microtubule polymerization from tubulin heterodimers, but inhibiting microtubule destruction. This leads to the induction of cell apoptosis at the late G2/M phase.1 Despite its wide use, the practical application of PTX is still limited by its poor aqueous solubility and low therapeutic index.2 In its commercial formulations (Taxol®, Bristol-Myers Squibb, New York, NY, USA), PTX is formulated in a 50:50 mixture of Cremophor EL and dehydrated ethanol.3,4 Unfortunately, Cremophor EL has been attributed to significant side effects, such as hypersensitivity, neurotoxicity, nephrotoxicity, and cardiotoxicity when intravenously (IV) administrated at a large dose.5,6
Because of the inherent problems associated with Cremophor EL, a number of alternative formulations including liposomes,7 lipid emulsions,8 polymeric micelles,9 cyclodextrin complexes,10 polymer–PTX conjugates,11 nanosuspensions,12 and nanoparticles (NPs)13 have been investigated to circumvent the need for Cremophor EL. Among these alternatives, nanosuspensions in particular offer unique advantages. Some examples include increased solubility for hydrophobic drugs, greater bioavailability in vivo, improved particle accumulation through the enhanced permeability and retention (EPR) effect, excellent applicability to most hydrophobic drugs, and suitability for industrial scale-up.14 Consequently, nanosuspension has been widely used as an effective drug delivery system for hydrophobic anticancer drugs.
While nanosuspensions offer a solution for the chemical formulation of hydrophobic drugs, targeting remains an issue that still must be addressed. Human serum albumin (HSA)-mediated targeting uses one of the major soluble proteins in the blood circulation, HSA, and has been reported as a promising biocompatible method for drug delivery.15 Biologically, HSA functions to maintain colloidal osmotic pressure in the blood, alongside facilitating the transport of several endogenous and exogenous substances.16,17 However, HSA also naturally possesses specific targeting abilities to inflamed and malignant tissues. Once targeted to the tumor site, HSA is efficiently transported into tumor cells through gp60 (a 60 KDa glycoprotein) or SPARC (one secreted protein, acidic and rich in cysteine) receptor-mediated endocytosis.18,19 As a result, HSA has been exploited in a multitude of drug delivery systems due to its natural tumor targeting ability and absence of toxicity or immunogenicity.19
Using nanosuspensions to mediate the chemical formulation of PTX and HSA for tumor targeting, American Bioscience Inc. (Blauvelt, NY, USA) produced the first biodegradable HSA-based PTX nanosuspension, Abraxane™, by nab™. Abraxane™ is the first US Food and Drug Administration (FDA)-approved chemotherapeutic formulation based on nanotechnology.20 To date, the main preparation methods of albumin-based formations can be concluded into four techniques: emulsification,21 thermal gelation,22 desolvation (coacervation),23 and nab™.24 While the first three conventional techniques involve chemical cross-linking processes or heat denaturation, nab™ oxidizes sulfhydryl residues in albumin through homogenization to form new cross-linking disulfide bonds, without denaturing albumin. nab™ provides a unique window of opportunity for albumin-based formulation, as it circumvents the destruction of albumin’s biological characteristic while not requiring the use of chemical crosslinkers. The lack of crosslinkers ensures the absence of aldehyde byproducts, a prominent defect in the conventional preparation methods.25,26
Albeit promising, the use of chloroform in nab™ manufacturing leads to chronic toxicities. In response to the issue of chronic toxicities, we have developed a method for the successful formulation of PTX nanosuspensions that eliminates the dependence on toxic organic solvents during manufacturing. Furthermore, NPs formed using this method still retain their suitability for IV administration. Polyethylene glycol (PEG) is a highly hydrophilic polymer with superb biocompatibility and biodegradability, which has been widely used as a hydrophilic solubilizing regent and pharmacokinetic tailor.27,28 Therefore, PEG was employed to effectively disperse PTX.29,30 In detail, PTX is first co-melted with PEG to form a solid dispersion, which was then mixed with the HSA solution and passed under a high pressure jet to form the PTX-PEG-HSA nanosuspension with a narrow size distribution. Because of the “steric stabilization” effect of PEG, PTX-PEG-HSA nanosuspensions remain invisible to the reticuloendothelial system (RES). As a result, PTX-PEG-HSA nanosuspensions exhibit significantly prolonged circulation and enhanced tumor accumulation while mitigating collateral damage to noncancerous tissues. To confirm these advantages, the properties of the PTX-PEG-HSA nanosuspension such as drug-loading (DL) capacity, pH, osmotic pressure, compatible stability, drug release, and cell growth inhibition against MDA-MB-231 cells were characterized in vitro. Furthermore, the in vivo antitumor efficacy and systemic toxicity of PTX-PEG-HSA were further evaluated in MDA-MB-231 tumor-bearing nude mice in comparison to PTX-HSA and Taxol®.
Materials and methods
Materials
HSA with a molecular weight (MW) of 65,000 Da was purchased from Yumin Biotech Co., Ltd. (Shanghai, People’s Republic of China). PTX and PEG (MW 6 kDa) were purchased from Sunve Pharmaceutical Co., Ltd. (Shanghai, People’s Republic of China) and Taihua Natural Plant Pharmaceutical Co., Ltd. (Xi’an, Shanxi Province, People’s Republic of China), respectively. Diazepam was obtained from Nanjing Xiandao Chemical Co., Ltd. (Jiangsu, People’s Republic of China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma-Aldrich (St Louis, MO, USA). The Cy5.5 N-hydroxysuccinimide ester (Cy5.5-NHS) and fluorescein isothiocyanate (FITC) were obtained from Beijing Fanbo Science and Technology Co., Ltd. (Beijing, People’s Republic of China). HPLC-grade reagents were used as the mobile phase in HPLC (high-performance liquid chromatography) analysis, and all other reagents were of analytical grade and used without further purification. Distilled and deionized water was used in all experiments.
Cells
MDA-MB-231 cells were provided by Origin Biosciences Inc. (Nanjing, People’s Republic of China). Cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 atmosphere.
Animals
BALB/c nude mice were obtained from Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, People’s Republic of China. All the animals were pathogen-free and allowed to access food and water freely. All animals were treated in accordance with the Guide for Care and Use of Laboratory Animals, approved by China Pharmaceutical University.
Preparation of PTX-PEG-HSA and PTX-HSA nanosuspension
Preparation of PTX-PEG-HSA nanosuspension
First, the solid dispersion technique was used for the preparation of PTX-PEG suspension. PTX (5 mg) was cofused with PEG (10, 15, 20 mg, MW 6,000) in 100 μL of anhydrous ethanol for 1 hour at 60°C under stirring. The anhydrous ethanol was rapidly evaporated and the PTX-PEG dispersion was formed. These cofused samples were named as PEG/PTX (w/w) group at 2/1, 3/1, and 4/1, respectively. As a result, the PTX-PEG solid dispersion was formed with PTX incorporated inside the skeleton of the hydrophilic modifiers.
Subsequently, 45 mg of HSA was dissolved in 10 mL of phosphate-buffered saline (PBS, 0.1 M, pH 7.4). The solution was then mixed with the PTX-PEG solid dispersion under magnetic stirring for 15 minutes. Then, the mixture was premixed for 10 minutes by using a high-speed disperser. Finally, the coarse suspension was homogenized at 20,000 psi for 6 cycles. The resulting solution was filtered through a 0.22 μm pore-sized microfiltration membrane and then lyophilized.
Cy5.5-labeled HSA and FITC-labeled PEG were used to prepare the PTX-HSA(Cy5.5)-PEG(FITC) nanosuspension following the same method described above. To obtain Cy5.5-labeled HSA and FITC-labeled PEG, HSA or PEG was agitated with Cy5.5-NHS or FITC for 8 hours away from light in the corresponding buffer solution.31–33 The mixed solution was then dialyzed and lyophilized.
Confocal laser scanning microscopy (CLSM, LeicaTCS SP5, Leica, Heidelberg, Germany) was used to confirm the successful insertion of PEG into the HSA nanosuspension. The localization of Cy5.5-labeled HSA and FITC-labeled PEG was observed under CLSM excited at 494 and 673 nm, and emitted at 522 and 692 nm for FITC and Cy5.5, respectively. The final images were analyzed using Leica Confocal Software.
Preparation of PTX-HSA nanosuspension
PTX-HSA nanosuspension with a size range of 100–150 nm was prepared as reported.19 Briefly, HSA was dissolved in the water saturated with chloroform. Meanwhile, PTX was dissolved in the consolute solutions of chloroform and ethanol (9:1, v:v). These two solutions were then mixed and homogenized at 20,000 psi for 9 cycles. The resulting colloid was placed under rotary evaporation at 25°C for 15 minutes under reduced pressure and lyophilized.
Characterization of PTX-PEG-HSA nanosuspension
Determination of DL and entrapment efficiency
The DL (wt%) and entrapment efficiency (EE, %) of the PTX-PEG-HSA nanosuspension was calculated by using the following formula:
|
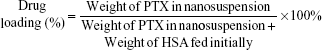
|
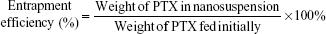
PTX concentrations were measured by HPLC (LC-2010C, Shimadzu Corporation, Kyoto, Japan) equipped with a Lichrospher C18 column (4.6×250 mm, 5 μm). The mobile phase was the mixed solution of methanol and water (75:25, v/v), while the flow rate and detection wavelength were set at 1 mL/min and 227 nm, respectively. The sample-injected volume was 20 μL and the column temperature was maintained at 30°C.
Particle size and zeta potential
The lyophilized PTX-PEG-HSA and PTX-HSA powders were redissolved by 5% (w/v) glucose solution and diluted to 5 mg/mL. The particle size and zeta potential were measured using Malvern Zetasizer Nano-ZS90 (Malvern Instruments, Malvern, UK). All of dynamic light scattering (DLS) measurements were performed at 25°C and at a scattering angle of 90°. The zeta potential values were calculated by the Smoluchowski equation.
pH and osmotic pressure determination
The pH values of PTX-PEG-HSA (dissolved by 5% glucose parenteral solution at 0.5 mg/mL of PTX) and PTX-HSA (dissolved by 5% glucose parenteral solution at 0.5 mg/mL of PTX) were measured using a microelectrode (Radiometer, Copenhagen, Denmark). Determination of osmotic pressure was conducted with an advanced freezing point osmometer obtained from Advanced Instruments Inc (Norwood, MA, USA).
Morphology observation
Transmission electron microscopy (TEM, H-7650, Hitachi, Tokyo, Japan) and atomic force microscopy (AFM, Nano Scope IIIa, Veeco, Santa Barbara, CA, USA) were employed to observe the morphology and size distribution of PTX-PEG-HSA nanosuspension. The samples for TEM imaging were prepared with negative stain. Negative staining of samples was performed as follows: a drop of sample solution was placed onto a copper grid coated with carbon; the sample drop was taped with a filter paper to remove surface water and air-dried for 5 minutes, followed by the application of 0.01% phosphotungstic acid to deposit NPs on the grid. The samples were air-dried before observation. The AFM observation was operated in tapping mode. A drop of properly diluted micelles was placed on the surface of a clean silicon wafer and dried under nitrogen flow at room temperature. The AFM imaging was visualized in contact mode, using high-resonance frequency (F0=129 kHz) pyramidal cantilevers with silicon probes having force constants of 20 Nm. Scan speed was set at 2 Hz. Imaging was processed and the widths of the particles were measured using Nanoscope version 7.3 software.
DSC and WAXD analysis
The dispersion state of PTX in PTX-PEG and PTX-PEG-HSA was evaluated by differential scanning calorimetry (DSC) and wide angle X-ray diffraction (WAXD) analysis. Analysis was conducted using an NETZSCH DSC 204 and an XD-3A powder diffraction meter (Bruker, AXS, Karlsruhe, Germany) with CuKα-radiation, respectively. In this study, the samples including blank HSA, PTX, and the physical mixture of PTX and HSA were used as controls.
Compatible stability of lyophilized PTX-PEG-HSA
To investigate the stability of lyophilized PTX-PEG-HSA upon resuspension, lyophilized powders of the PTX-PEG-HSA nanosuspension were separately resuspended at 0.5 mg/mL in 0.9% sodium chloride parenteral solution and 5% glucose parenteral solution. The solutions were stored at room temperature for 0, 8, 12, 18, or 24 hours, followed by determination of PTX contents as described in the “Determination of DL and entrapment efficiency” section.
In vitro drug release from PTX-PEG-HSA nanosuspension
The release profiles of PTX from the PTX-PEG-HSA nanosuspension and the PTX-HSA nanosuspension were studied using a simple dialysis method. The lyophilized PTX-PEG-HSA or PTX-HSA powders containing 0.5 mg of PTX were dispersed in 1 mL of 5% glucose solution with or without supplement of 10% serum and placed inside a dialysis bag (molecular weight cut off =12,000–14,000 Da). The entire bag was immersed in a beaker containing 150 mL of PBS containing 0.2% Tween 80 and shaken in a 37°C water bath at 100 rpm. At predetermined time intervals, 1 mL of release media was withdrawn and equivalent fresh release medium was added. The amount of PTX released was determined by HPLC analysis as described in the “Determination of DL and entrapment efficiency” section.
In vitro cellular study
Cellular uptake
To evaluate the cellular uptake capacities of the PTX-PEG-HSA nanosuspension, a Coumarin 6 (C6) fluorescence probe was encapsulated into PTX-PEG-HSA and PTX-HSA nanosuspensions accordant to our protocol for the preparation of PTX-loaded nanosuspensions (preparation of PTX-PEG-HSA nanosuspension). The DL of C6 in the both suspensions was approximately 0.1%. Flow cytometry (BD FACSCalibur, Becton Dickinson, San Jose, CA, USA) and CLSM were used to determine the cell uptake capacities of C6-PTX-PEG-HSA and C6-PTX-HSA. Cells were incubated with C6-PTX-PEG-HSA or C6-PTX-HSA (C6 content: 60 μg/mL) for 1 and 4 hours at 37°C. In CLSM analysis, cells were rinsed with PBS three times, followed by the addition of Hoechst 33342 (10 μg/mL) to stain the cell nuclei. The samples were then observed using an Olympus confocal microscope (Olympus FV1000, Olympus Corporation, Tokyo, Japan) at excitation and emission wavelengths of 488 and 530 nm, respectively. In the flow cytometry analysis, all samples were rinsed with PBS three times to remove excess culture media, harvested by trypsinization, and collected in PBS to measure the fluorescence intensity.
Cytotoxicity evaluation
Approximately 100 μL of MDA-MB-231 cells at a concentration of 5×103 cells/well were seeded in each well of a 96-well plate. The plate was incubated at 37°C in a humidified atmosphere with 5% CO2. Subsequently, specific populations of cells were treated with PTX-PEG-HSA, PTX-HSA and Taxol® with increasing doses: 0.001–100 μg/mL for 72 hours. After 72 hours of incubation, the MTT assay was performed using previously reported procedures using a microplate reader (SOFT max® PRO, Molecular Devices Corporation, Sunnyvale, CA, USA).34 The toxicity of samples was expressed as the inhibitory concentration at which 50% of cell growth inhibition was obtained (IC50).
Apoptosis assay
Apoptosis of MDA-MB-231 cells induced by PTX-PEG-HSA nanosuspensions were analyzed by flow cytometry using AnnexinV-FITC/propidium iodide (PI) double staining assay. MDA-MB-231 cells were cultured in 6-well plates at a concentration of 5×104 cells/well and incubated at 37°C in the presence of 5% CO2 for 24 h. Subsequently, the cells were treated with 0.1 μg/mL of PTX-PEG-HSA, PTX-HSA, or Taxol® for 24 hours, followed by three washes with cold PBS. Cells were then harvested, washed, and resuspended in 500 μL of PBS. Afterward, 5 μL of Annexin V-FITC and 5 μL of PI were added and incubated for 15 minutes at room temperature in the dark. Finally, the cell suspension was evaluated by a flow cytometry (BD FACSCalibur, Becton Dickinson).
In vivo antitumor activity
Approximately 1×106 of MDA-MB-231 cells were inoculated subcutaneously in the armpit region of athymic nude mice. Once tumors grew to approximately 50 mm3, mice were randomly divided into four groups and received different injections as follows: 1) saline (the control group, n=5); 2) PTX-PEG-HSA (redissolved in 5% glucose, n=5); 3) PTX-HSA (redissolved in 5% glucose, n=5); 4) Taxol® (n=5). All formulations were administered IV via the tail vein at a PTX dose of 10 mg/kg. Treatments occurred once every 5 days for 15 days. The body weights of all mice were also recorded, and tumor volumes were calculated as (a2×b)/2, where a represents the smallest diameter and b the largest. Tumors were excised from sacrificed mice after 20 days of observation and weighed. The tumor weight inhibition rate (IR) (%) was calculated using the following formula:
|
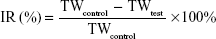
where TWcontrol and TWtest represented the mean tumor weight of the control group and treated groups, respectively. Thereafter, the tumor tissues were fixed in 10% of formalin and embedded in paraffin blocks to conduct hematoxylin and eosin (H&E) staining and terminal deoxynucleotidyl transferase-mediated nicked labeling (TUNEL) assays.35,36 Then the stained tissue sections were observed under a light microscope (Olympus) for histologic examination.
Statistics
Statistical evaluation was performed through a two-tailed Student’s t-test, and one-way analysis of variance. All data are expressed as the mean ± standard deviation (SD), unless otherwise noted. A value of P<0.05 was considered statistically significant.
Results and discussion
Preparation and characterization of PTX-PEG-HSA nanosuspension
Preparation of PTX-PEG-HSA solid suspension
The need to solubilize hydrophobic drugs using organic solvents is an undesirable option from a practical standpoint due to the potential toxicity of the solvents. In this work, we have replaced the need for toxic organic solvents by the solid-dispersion technology in terms of solubilizing PTX. This method was created to further optimize the preparation of PTX-loaded HSA nanosuspensions based on nab™. The PTX powder was co-melted with PEG to form a solid dispersion, which effectively improved the dissolution rate and apparent solubility of PTX. This solid was then mixed with an HSA solution and passed under high pressure, analogous to the operative description used in nab™. Thus, the PTX-PEG-HSA nanosuspension obtained from solid-dispersion technology is free of toxic chloroform residues that limit the clinical application of the original nab™ formulation. In other words, the chronic toxicities of residual chloroform in Abraxane™ can be successfully mitigated using solid-dispersion technology.
We first investigated the optimal weight ratio of PEG to PTX for the preparation of the PTX-PEG solid suspension through co-melting. The solid dispersion technique incorporates PTX inside the hydrophilic skeleton structure of hydrophilic carriers, which allows detection by DSC.37,38 As shown in Figure 1A, all diagrams exhibited the endothermic peak of PEG around 50°C. Furthermore, only the physical mixture of PEG + PTX and the group at 2/1 (PEG/PTX, w/w) obtained the characteristic peaks of PTX in crystalline state around 240°C. The disappearance of characteristic peaks of PTX in groups at 3/1 and 4/1 indicated the successful formation of PTX-PEG solid dispersion with PTX existing in an amorphous state. During the preparation of the solid dispersion, the optimal pharmaceutical parameter was defined as the minimum amount of hydrophilic carriers required for the hydrophobic drugs to be sufficiently dispersed.39 Therefore, the PTX-PEG solid dispersion was first prepared at a weight ratio of 1:3 with respect to PTX:PEG. This mixture was then combined with the HSA solution and homogenized to obtain the PTX-PEG-HSA nanosuspension.
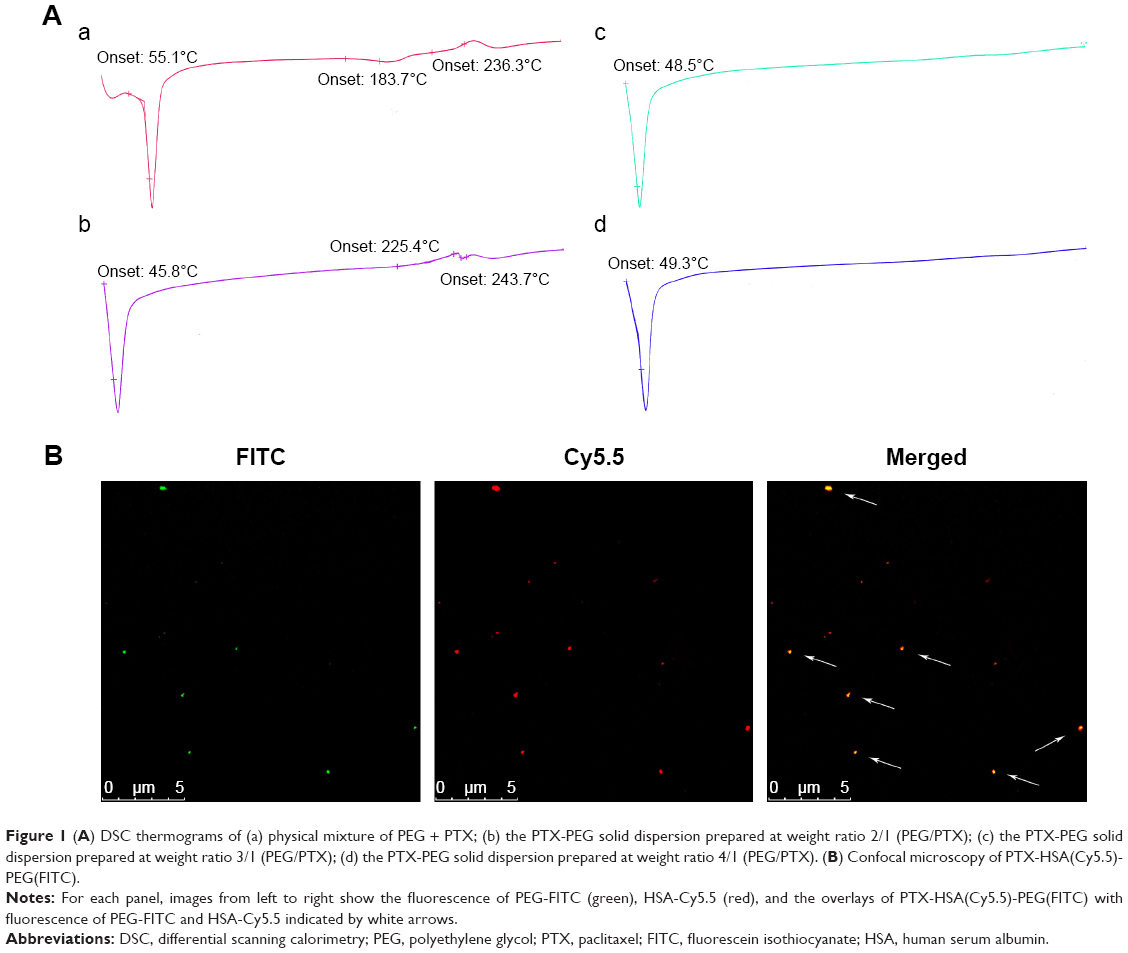 | Figure 1 (A) DSC thermograms of (a) physical mixture of PEG + PTX; (b) the PTX-PEG solid dispersion prepared at weight ratio 2/1 (PEG/PTX); (c) the PTX-PEG solid dispersion prepared at weight ratio 3/1 (PEG/PTX); (d) the PTX-PEG solid dispersion prepared at weight ratio 4/1 (PEG/PTX). (B) Confocal microscopy of PTX-HSA(Cy5.5)-PEG(FITC). |
Furthermore, a double-fluorescent label method was used to confirm the successful attachment of PEG within the HSA-based nanosuspension. In this method, HSA and PEG were first tagged with Cy5.5 and FITC, respectively. Thereafter, the PTX-HSA(Cy5.5)-PEG(FITC) nanosuspension was prepared following the procedures of PTX-PEG-HSA manufacture. The formation of PTX-HSA(Cy5.5)-PEG(FITC) nanosuspension was evaluated visually under a CLSM. As shown in Figure 1B, the merged green/red fluorescence (indicated by white arrows) of PTX-HSA(Cy5.5)-PEG(FITC) nanosuspensions was obtained via confocal microscopy imaging. This result directly demonstrated that PTX-PEG-HSA nanosuspensions could be formed through our toxic organic solvent-free technology (with successful PEG insertion). H-bonding network between PEG and vicinal amino acids of HSA surrounding may play a major role in the PEG/HSA complexation.40
Determination of DL and EE
All of the physicochemical characteristics of PTX-loaded nanosuspensions are listed in Table 1. First, DL capability was determined using HPLC. The DL (wt%) and EE (%) of PTX-PEG-HSA was 9.1±0.2 wt% and 90.5%±0.6%, respectively. PTX-HSA obtained a DL and an EE of 9.2±0.1 wt% and 91.4%±0.5%, respectively. The similar DL and EE observed in these two HSA-based nanosuspensions indicated that the unique drug dispersion by PEG did not affect the specific binding interactions between PTX and HSA.
 | Table 1 Physicochemical characteristics of PTX-PEG-HSA and PTX-HSA nanosuspension |
Particle size and zeta potential evaluation
The particle size and zeta potential of PTX-PEG-HSA and PTX-HSA were measured by DLS. As listed in Table 1, the effective diameters of PTX-PEG-HSA and PTX-HSA were 123.0 and 137.2 nm with polydispersity indices (PDI) of 0.11 and 0.14, respectively, implying a narrow size distribution. In addition, the particle surface of PTX nanosuspensions were negatively charged with zeta potential of approximately −43.6 mV for PTX-PEG-HSA and −39.9 mV for PTX-HSA. The mean particle size distribution usually affects the formulation stability, pharmacokinetics, and in vivo tissue distribution of nanosuspension systems.41 As compared with PTX-HSA, PTX-PEG-HSA nanosuspensions shared a similar zeta potential but had a smaller mean diameter and are less likely to be nonselectively recognized by the RES. This decrease in RES uptake, therefore, allows the particles to achieve long-circulation times in blood by avoiding RES clearance.42 Additionally, the EPR effect was reported to be a significant factor when the size of NPs was approximately 100–200 nm.43 Furthermore, the relatively high zeta potential provides a repelling force between the particles. This electrostatic repulsion acts to the formulation’s advantage to prevent undesired aggregation or precipitation and thus, improves the formulation’s stability.44 Therefore, it is reasonable to conclude that PTX-loaded nanosuspensions with small diameters (123.0 nm) and negatively charged zeta potentials (−43.6 mV) are suitable for targeted tumor therapy through IV injection due to the reasons that have just been listed.
pH and osmotic pressure determination
Both the PTX-PEG-HSA and PTX-HSA nanosuspensions exhibited pH values around 7.4 and osmotic pressures in the range of 300–340 mOsm/kg, which agrees with the optimal measurements for IV injections.
Morphology observation
TEM and AFM were used to visualize the size and morphology of PTX-PEG-HSA particles. As shown in Figure 2, the morphology of both the nanosuspensions was spherical, and the particles were highly dispersed with diameters approximately 100 nm. It was also noted that the particle size observed by TEM and AFM is smaller than that determined by DLS. In the DLS measurement, the hydrated dynamic diameter of NPs was measured, ie, the colloidal hydrate layer is calculated as one part of “diameter”. In the microscopy determination, the colloidal hydrate layer is dried and the observed true diameter decreased. Therefore, it makes sense that the measurements observed with dehydrated samples in TEM and AFM would be smaller than those determined with hydrated samples during DLS.
 | Figure 2 (A) TEM images of free PTX (a), PTX-PEG-HSA (b), and PTX-HSA (c), and (B) the AFM images of PTX-PEG-HSA. |
DSC and WAXD analysis
To investigate the physical state of PTX in the nanosuspensions, DSC and WAXD analysis were conducted for PTX-PEG-HSA with PEG, HSA, PTX, a physical mixture of HSA + PTX, and a physical mixture of HSA + PTX-PEG as controls. DSC diagrams of the six samples are shown in Figure 3A. As illustrated in Figure 3A(a), the endothermic peak at 217.3°C and exothermic peak at 244.5°C were characteristic peaks of PTX in crystalline state, while the exothermic peak at 208.9°C in Figure 3A(b) represented the peak of decomposed HSA. Figure 3A(c) exhibited the endothermic peak of PEG at around 50°C. Additionally, an exothermic peak at 245.8°C was seen for the physical mixture of HSA + PTX (Figure 3A(d)), but was nearly absent for HSA + PTX-PEG (Figure 3A(e)) and PTX-PEG-HSA nanosuspensions (Figure 3A(f)). All results conclude that the PTX were physically encapsulated in these nanosuspensions with reduced crystallinity. The results of WAXD analysis in Figure 3B further confirmed the existing state of PTX loaded in PTX-PEG-HSA nanosuspensions. As shown in Figure 3B(a), PTX had three intense peaks at 5°, 9°, and 12° and small peaks at 15°–30°. The HSA gave a broad and weak peak at 15°–30°. The typical crystal peaks of PTX and HSA were all present in the WAXD diagram of a physical mixture of PTX + HSA. However, the PTX-PEG-HSA nanosuspensions possessed negligible characteristic peaks of PTX and exhibited one obvious peak, similar to that of free HSA, which also demonstrated that the PTX were encapsulated in nanosuspensions with a significantly reduced crystalline state.45
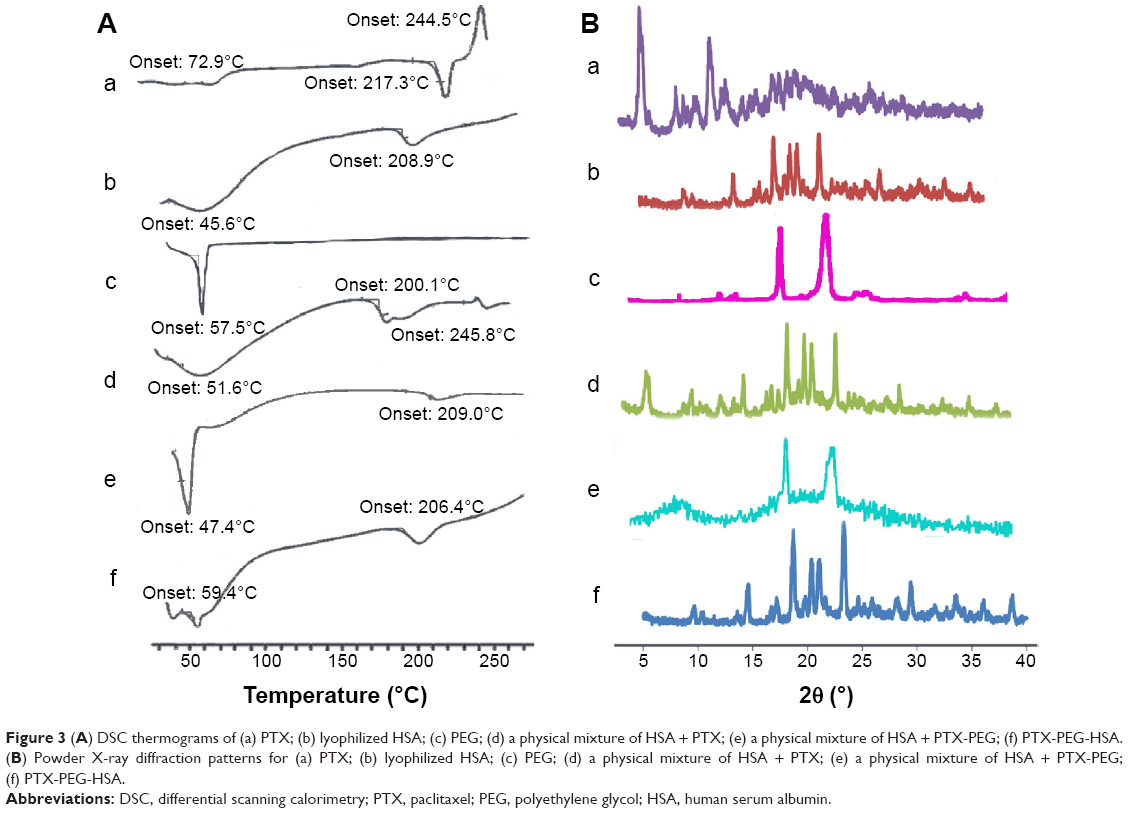 | Figure 3 (A) DSC thermograms of (a) PTX; (b) lyophilized HSA; (c) PEG; (d) a physical mixture of HSA + PTX; (e) a physical mixture of HSA + PTX-PEG; (f) PTX-PEG-HSA. (B) Powder X-ray diffraction patterns for (a) PTX; (b) lyophilized HSA; (c) PEG; (d) a physical mixture of HSA + PTX; (e) a physical mixture of HSA + PTX-PEG; (f) PTX-PEG-HSA. |
Compatible stability for clinical application
The lyophilized powders of this formulation are usually redissolved by 0.9% sodium chloride or 5% glucose injection for clinical application. Therefore, the stability and compatibility of resuspensions are a crucial criterion for formulation design. In this study, PTX-PEG-HSA redissolved in 0.9% sodium chloride or 5% glucose injection was analyzed for the amount of unincorporated PTX and particle size. As listed in Table 2, there was no significant change of the two indexes within 24 hours, suggesting that both the 0.9% sodium chloride and 5% glucose injections can be used for resuspending the PTX-PEG-HSA from lyophilized powders.
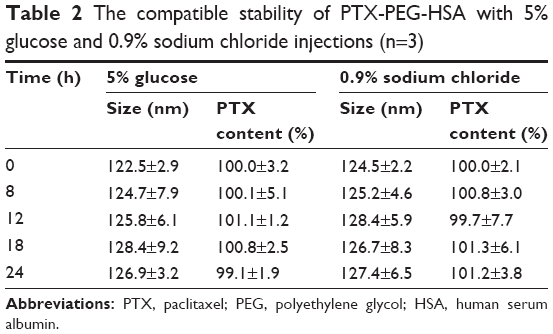 | Table 2 The compatible stability of PTX-PEG-HSA with 5% glucose and 0.9% sodium chloride injections (n=3) |
In vitro drug release
Drug release rate was determined to examine whether a burst release pattern was present, or whether the drug could be slowly dissipated from the polymer matrix for exerting potency. Therefore, PTX-PEG-HSA and PTX-HSA nanosuspensions were incubated in PBS at pH 7.4, and the PTX release profiles were determined. As shown in Figure 4A, both PTX-PEG-HSA and PTX-HSA nanosuspensions exhibited steady and continual release profiles with no initial burst release. The results suggested that PTX molecules were indeed encapsulated into the hydrophobic micropockets of HSA and not lodged on the particle surface. This finding agreed with the results of DSC and WAXD analysis. To simulate the in vivo drug release, the release evaluation of PTX-PEG-HSA and PTX-HSA in 10% serum supplemented medium was also conducted. The results shown in Figure 4B exhibited a similar slow drug release behavior of PTX-PEG-HSA within 24 hours to that obtained in PBS solution and an increased drug release from PTX-HSA as compared with that in PBS solution. These results indicated that PEG in PTX-PEG-HSA could protect NPs from protein interaction and the PTX-PEG-HSA could achieve a less drug preleakage during blood circulation, therefore improving the circulative stability and also mitigating side effects. Another point worth noting is that the amount of PTX released from PTX-HSA was lower than that from PTX-PEG-HSA over the same time intervals in PBS without serum. Within the first 4 days, only approximately 35% of PTX was released from PTX-HSA, while the release amount in PTX-PEG-HSA group reached up to 53%. This result may be attributed to the insertion of PEG within PTX-PEG-HSA nanosuspensions that furthers drug diffusion. Finally, the cumulative PTX release percentage of both preparations reached 80% after 10 days in serum-free media, indicating successful PTX release from designed nanosuspensions.
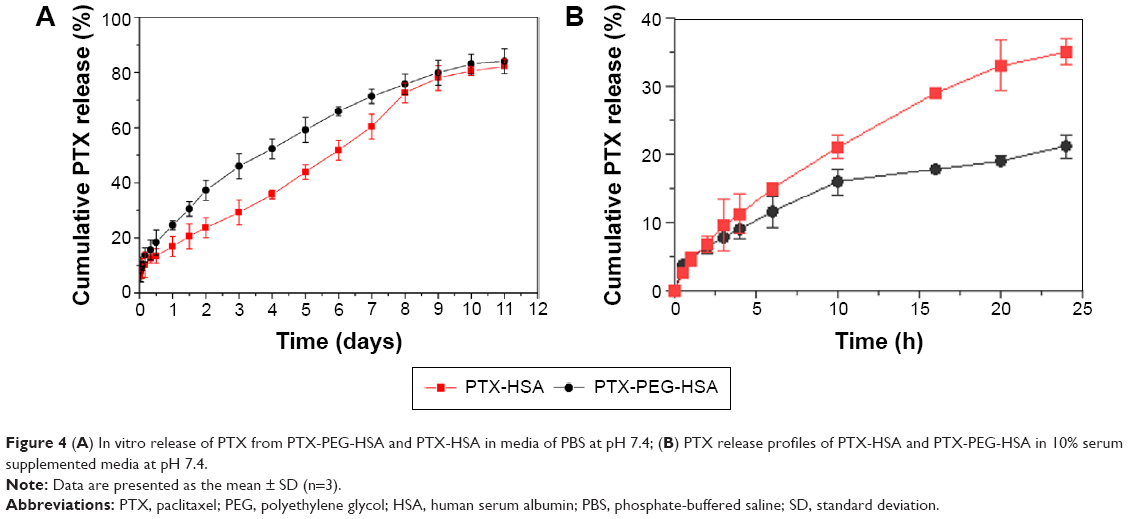 | Figure 4 (A) In vitro release of PTX from PTX-PEG-HSA and PTX-HSA in media of PBS at pH 7.4; (B) PTX release profiles of PTX-HSA and PTX-PEG-HSA in 10% serum supplemented media at pH 7.4. |
In vitro cellular studies
Cellular uptake and intracellular distribution
With confirmed physicochemical properties of PTX-PEG-HSA nanosuspension, investigation into the cellular uptake behavior of the nanosuspension using a C6 as fluorescent marker was conducted. The results from flow cytometry analysis are shown in Figure 5A. MDA-MB-231 cells incubated with the C6-PTX-PEG-HSA and C6-PTX-HSA both exhibited fluorescence of C6 at 1 and 4 hours, indicating a time-dependent cellular internalization mechanism. Meanwhile, the fluorescence intensity in the PTX-PEG-HSA group was approximately equal to that in PTX-HSA group at 1 and 4 h, respectively. This result demonstrates that the PEGylation did not adversely affect the cell uptake of PTX-PEG-HSA, which could be mediated through the 60 KD glycoprotein (gp60) or SPARC receptor on the cell membrane.46 The highly effective cellular internalization of PTX-loaded nanosuspensions was also confirmed via CLSM analysis. The time dependence of endocytosis and strength of intracellular fluorescence is shown in Figure 5B and is in agreement with the results from flow cytometry analysis. In addition, the internalized fluorescence of C6 was distributed throughout the cytoplasm, the optimal location for PTX to induce cytotoxicity. Therefore, all the results here indicated that our designed nanosuspension could be quickly taken into tumor cells and reside in the preferred intracellular location for inducing optimal cytotoxicity.
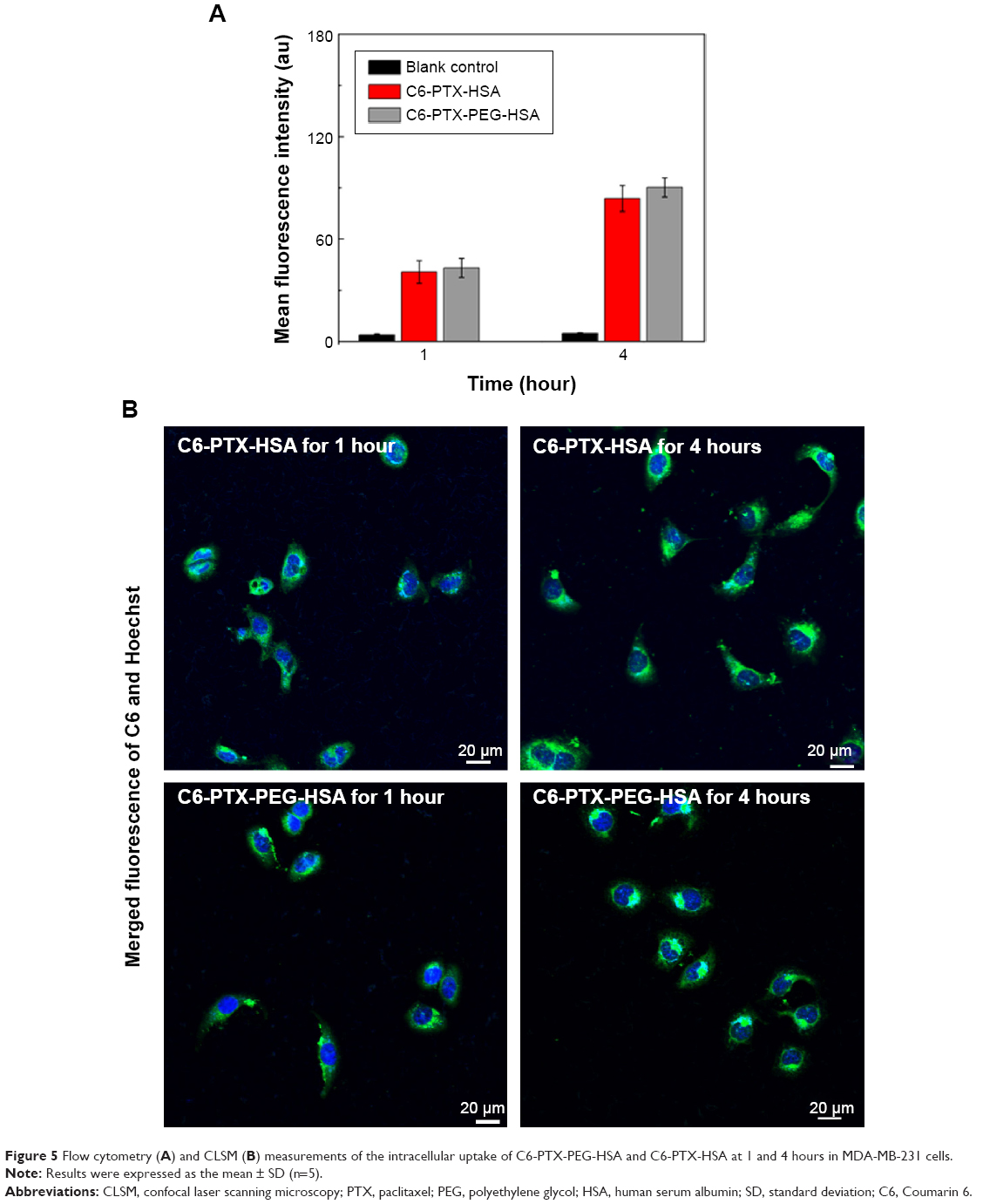 | Figure 5 Flow cytometry (A) and CLSM (B) measurements of the intracellular uptake of C6-PTX-PEG-HSA and C6-PTX-HSA at 1 and 4 hours in MDA-MB-231 cells. |
Cytotoxicity and apoptosis assay
To further investigate whether the highly efficient intracellular PTX delivery platform (PTX-PEG-HSA) could achieve strong cytotoxicity against tumor cells, an MTT assay of PTX-PEG-HSA against MDA-MB-231 cells was performed using PEG-HSA and Taxol® as controls. After 24 hours of incubation, Taxol® had significantly higher IC50 values against MDA-MB-231 cells than PTX-HSA and PTX-PEG-HSA (P<0.001, Figure 6A). This phenomenon could be attributed to a number of reasons. First, Taxol® primarily utilizes passive diffusion to enter the cell, and this is solely dependent on the concentration gradient. On the other hand, the two HSA-based nanosuspensions could be transported into tumor cells through active binding of HSA to the gp60 or SPARC receptor on cell membrane. Furthermore, PTX in the HSA-based nanosuspensions accumulated in the tumor cells, while free PTX was rapidly secreted back outside the cells.47 In addition, the IC50 of the PTX-PEG-HSA group was 1.76-fold lower than that of PTX-HSA group. As stated, the higher cytotoxicity of PTX-PEG-HSA on tumor cells may be attributed to its faster drug release behavior, which has been confirmed by the in vitro drug release experiment.
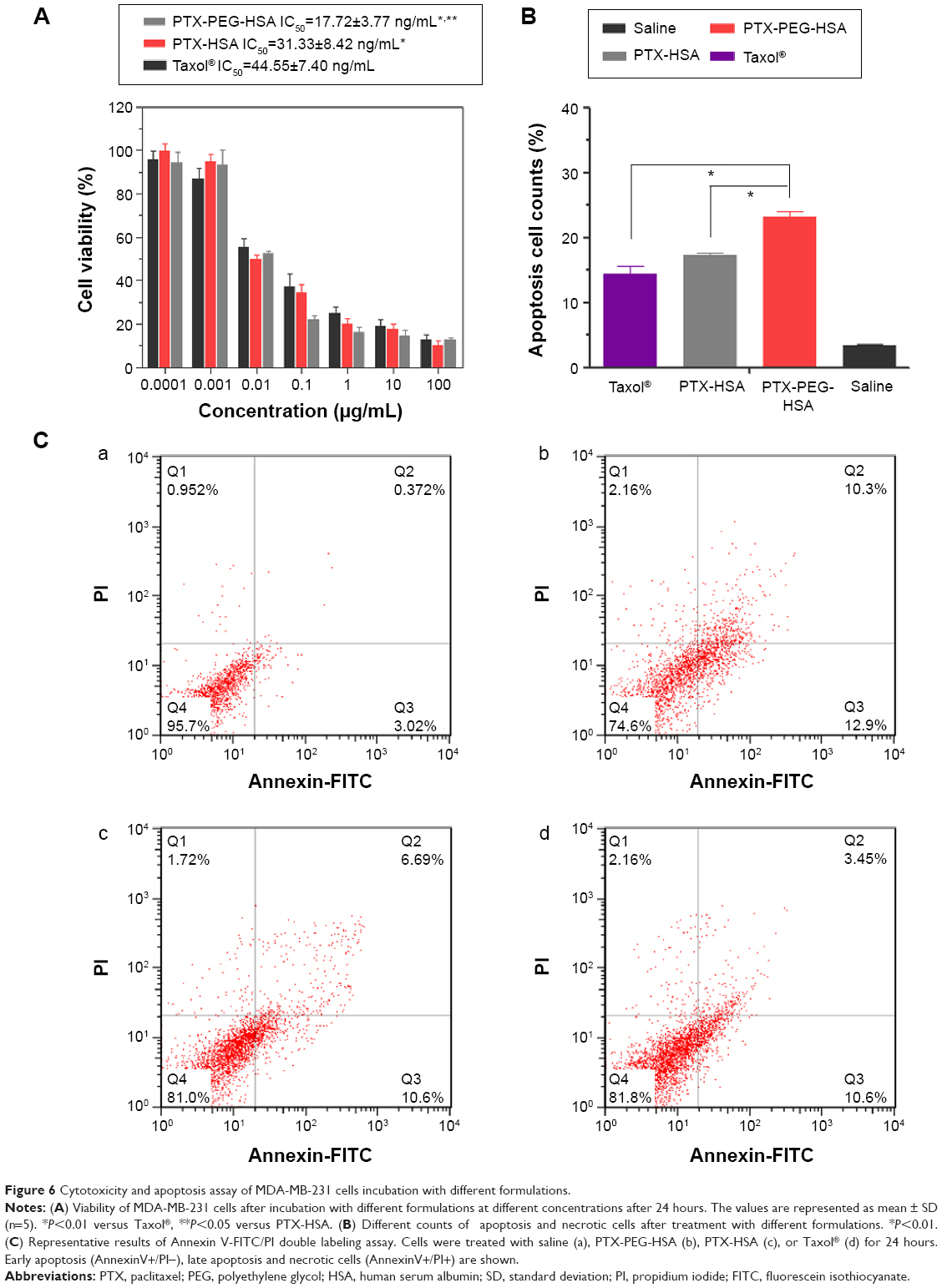 | Figure 6 Cytotoxicity and apoptosis assay of MDA-MB-231 cells incubation with different formulations. |
The significant inhibitory effect of PTX-PEG-HSA on MDA-MB-231 cells was also demonstrated by an AnnexinV-FITC/PI double staining assay, and cell apoptosis was measured via flow cytometry. The representative images obtained from flow cytometry shown in Figure 6C mirrored results from the MTT assay. In detail (Figure 6B), the early apoptosis (Annexin V+/PI−) value of MDA-MB-231 cells treated with PTX-PEG-HSA, PTX-HSA, and Taxol® was 12.9%, 10.6%, and 10.6%, respectively. The percentage of late apoptosis/necrosis cells (AnnexinV+/PI+) was 10.3%, 6.69%, and 3.45%, respectively. These results indicated that PTX-PEG-HSA could induce elevated apoptosis over PTX-HSA and Taxol®.
In vivo antitumor activity
To investigate the in vivo antitumor potential of PTX-PEG-HSA nanosuspensions, the antitumor efficacy was further evaluated on BALB/c nude mice bearing MDA-MB-231 xenografts with PTX-HSA and Taxol® as controls. The changes of tumor volume over 20 days of treatment and the IR (%) values of these PTX formulations calculated on the basis of tumor weight are depicted in Figure 7A and B, respectively. As indicated, all PTX formulations exhibited effective inhibition on tumor growth when compared with saline group. However, the PTX-PEG-HSA nanosuspension achieved the strongest tumor inhibition of all four groups, with an IR value of 89.24%, followed by Taxol® (79.96%) and PTX-HSA (75.08%).
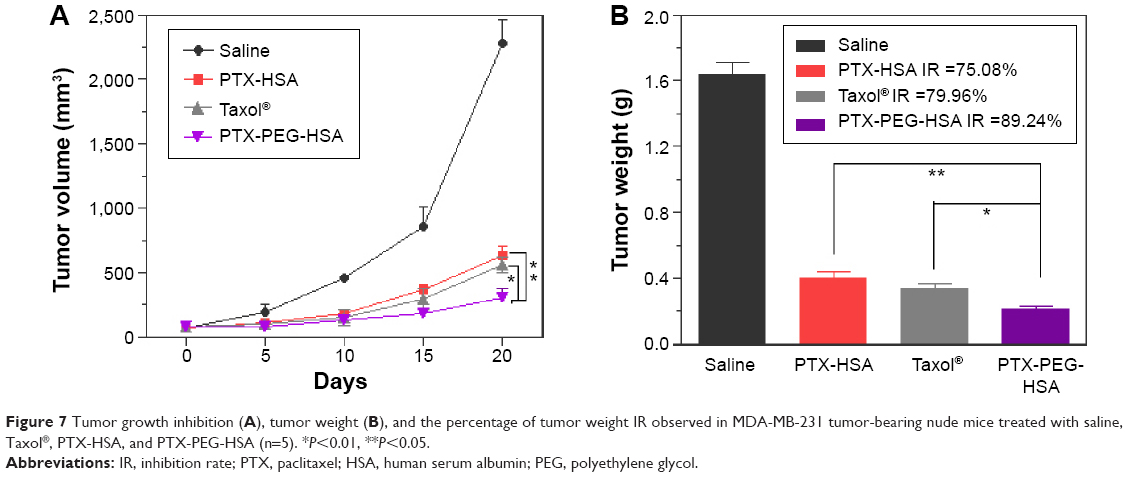 | Figure 7 Tumor growth inhibition (A), tumor weight (B), and the percentage of tumor weight IR observed in MDA-MB-231 tumor-bearing nude mice treated with saline, Taxol®, PTX-HSA, and PTX-PEG-HSA (n=5). *P<0.01, **P<0.05. |
Furthermore, the remarkable antitumor efficacy of the PTX-PEG-HSA nanosuspensions is also well supported by H&E histological evaluation and TUNEL assay. Unlike the tumors in saline control group with histologic characteristics including hyper chromatic nuclei, scant cytoplasm, and closely arranged tumor cells, the tumors of mice treated with PTX-PEG-HSA, PTX-HSA or Taxol® all exhibited cell shrinkage and intercellular blank (Figure 8). Yet, tumors treated with PTX-PEG-HSA exhibited the greatest degree of apoptosis observed through TUNEL assay and the lowest tumor cellularity. The enhanced efficacy of these two HSA-based nanosuspensions over Taxol® might be explained by the combination of EPR effect and HSA binding receptor-mediated cellular internalization, which significantly improved PTX accumulation in tumors.48,49 Moreover, the additional PEGylation of PTX-PEG-HSA prolonged the in vivo half-life of particles in circulation through its unique “stealth function” to prevent RES recognition, thereby leading to a further increased target distribution of PTX into tumors. Finally, the accelerated drug release behavior of PTX-PEG-HSA over PTX-HSA further enhanced the induction of apoptosis. Consequently, a more effective anticancer activity was achieved by PTX-PEG-HSA compared with PTX-HSA.
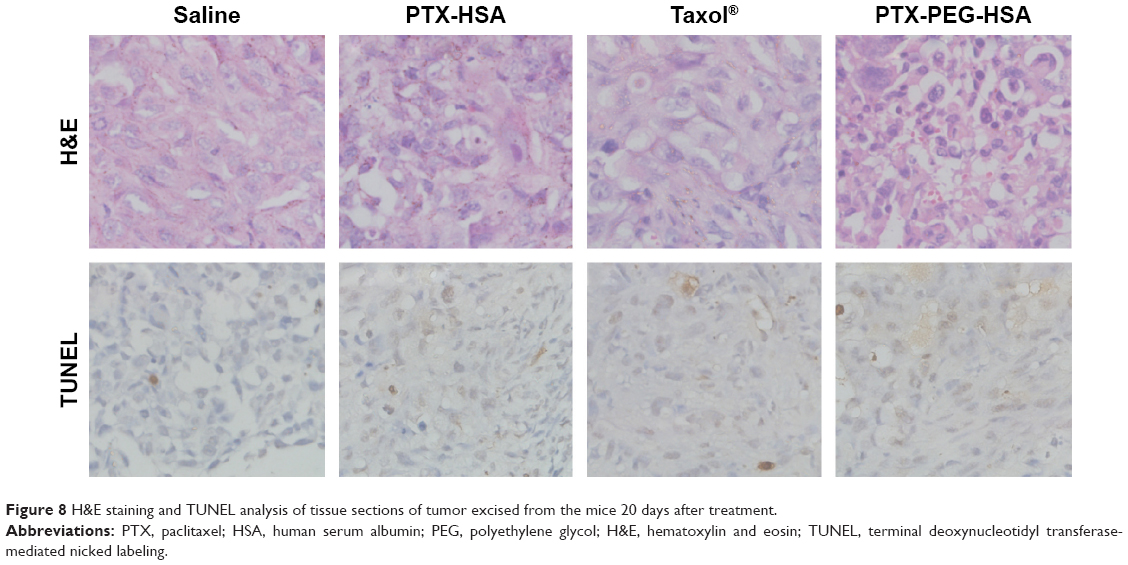 | Figure 8 H&E staining and TUNEL analysis of tissue sections of tumor excised from the mice 20 days after treatment. |
It is known that in vivo toxicity evaluation is essential for systemic drug delivery. Therefore, we investigated animal behavior and body weight during formulation treatments. First, all mice in PTX-PEG-HSA and PTX-HSA groups showed normal behavior during the entire treatment, while the mice treated with Taxol® frequently demonstrated decreased activity and appetite. Additionally, mice in the Taxol® group exhibited significant weight loss (Figure 9). In contrast, mice receiving HSA formulation-based treatments revealed negligible changes in body weight. The obvious side effects of Taxol® treatment have already been well explained by the dose-limiting toxicity of the nonionic surfactant Cremophor® EL alongside the nonspecific distribution of PTX.50 The marked reduction of systemic toxicity achieved by HSA nanosuspensions was mainly due to the effective encapsulation of PTX into the HSA-based particles, which avoided Cremophor EL-related side effects. Furthermore, the well-defined selective accumulation of HSA nanosuspensions in tumor sites through both the EPR and protein-receptor-mediated endocytosis enhanced the tumor specificity and reduced systemic cytotoxicity.51 Additionally, the HSA-based particles demonstrated decreased leakage of PTX into the bloodstream during circulation as compared with Taxol®, which further mitigated side effects.52
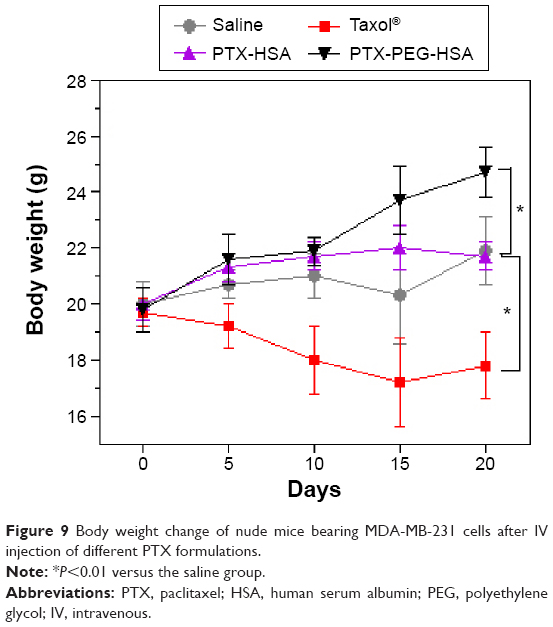 | Figure 9 Body weight change of nude mice bearing MDA-MB-231 cells after IV injection of different PTX formulations. |
Conclusion
The solid-dispersion technology has allowed the formulation of PTX-PEG-HSA, a powerful nanosuspension-based delivery system for PTX that circumvents the need for organic solvents and Cremophor EL. PTX molecules were successfully dispersed by PEG through co-melting, followed by combination, and homogenized with HSA solution. Particles were confirmed to be of a small size (123.0 nm) with smooth spherical morphology. Furthermore, the PTX molecules in PTX-PEG-HSA were dispersed in a molecular or amorphous state. The stability of this formulation was evident through the steady, continued drug release behavior without an initial burst release effect, which suggested the promising potential of PTX-PEG-HSA as a safe systemic PTX delivery platform. The high cellular internalization of PTX-PEG-HSA as demonstrated by flow cytometry and CLSM analysis shows that stable NPs could still be internalized into the cell. In addition, the strong cytotoxicity of PTX-PEG-HSA against MDA-MB-231 cells indicates that encapsulated PTX can be released in the intracellular environment for significant antitumor efficacy. More importantly, in vivo antitumor investigation on nude mice bearing MDA-MB-231 cancer xenografts further confirmed that PTX-PEG-HSA achieved improved anticancer activity and reduced systemic toxicity over PTX-HSA and Taxol®. The use of PEG and HSA receptor-mediated targeting has allowed for prolonged blood circulation and significant increases in tumor uptake, all of which have translated to enhanced antitumor efficacy with decreased collateral damage to healthy tissues. Thus, PTX-PEG-HSA without dependence on toxic organic solvents proves a greater alternative to commonly accepted PTX delivery systems. Future work in this direction should focus on in vivo pharmacokinetics and biodistribution of PTX-PEG-HSA on tumor-bearing mice, as well as evaluation of the safety and activity of the delivery system in a more clinically accurate model.
Acknowledgments
This work was supported by the project of the National Natural Science Foundation of China (number 81102397), the Natural Science Foundation of Jiangsu Province (number BK2012761), Qing Lan Project of Jiangsu Province (number 02432009), Innovative Project for Graduate Cultivation of Jiangsu Province (number CXZZ11-0807), the Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (numbers JKGQ201107, JKPZ2013004), Key New Drug Innovation Project from the Ministry of Science and Technology of China (number 2009ZX09310004), Fostering Plan of University Scientific and Technological Innovation Team of Jiangsu Qing Lan Project (2014), and National Basic Research Program of China (973 Program, number 2009CB903300).
Disclosure
The authors report no conflicts of interest in this work.
References
Jose Merlin JP, Venkadesh B, Hussain R, et al. Paclitaxel loaded poly-d, l-lactide-co-glycolide nanoparticles: enhanced anticancer effect in non-small cell lung carcinoma cell line. Biomed Prevent Nutr. 2013;3(1):1–9. | ||
Tao Y, Han J, Wang X, Dou H. Nano-formulation of paclitaxel by vitamin E succinate functionalized pluronic micelles for enhanced encapsulation, stability and cytotoxicity. Colloid Surf B Biointerfaces. 2013;102:604–610. | ||
Xin H, Chen L, Gu J, et al. Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(ε-caprolactone) nanoparticles: in vitro and in vivo evaluation. Int J Pharm. 2010;402(1–2):238–247. | ||
Sun Y, Yu B, Wang G, et al. Enhanced antitumor efficacy of vitamin E TPGS-emulsified PLGA nanoparticles for delivery of paclitaxel. Colloid Surf B Biointerfaces. 2014;123:716–723. | ||
Sharma S, Verma A, Teja BV, Shukla P, Mishra PR. Development of stabilized Paclitaxel nanocrystals: In-vitro and in-vivo efficacy studies. Eur J Pharm Sci. 2015;69:51–60. | ||
Pawar AP, Vinugala D, Bothiraja C. Nanocochleates derived from nanoliposomes for paclitaxel oral use: preparation, characterization, in vitro anticancer testing, bioavailability and biodistribution study in rats. Biomed Pharmacother. 2014;3502:1–9. | ||
Yan F, Li L, Deng Z, et al. Paclitaxel-liposome-microbubble complexes as ultrasound-triggered therapeutic drug delivery carriers. J Control Release. 2013;166(3):246–255. | ||
Lundberg BB, Risovic V, Ramaswamy M, Wasan KM. A lipophilic paclitaxel derivative incorporated in a lipid emulsion for parenteral administration. J Controll Release. 2003;86(1):93–100. | ||
Rapoport N, Gupta R, Kim YS, O’Neill BE. Polymeric micelles and nanoemulsions as tumor-targeted drug carriers: insight through intravital imaging. J Control Release. 2015;206:153–160. | ||
Bouquet W, Ceelen W, Fritzinger B, et al. Paclitaxel/beta-cyclodextrin complexes for hyperthermic peritoneal perfusion – formulation and stability. Eur J Pharm Biopharm. 2007;66(3):391–397. | ||
Chun C, Lee SM, Kim SY, Yang HK, Song SC. Thermosensitive poly(organophosphazene)-paclitaxel conjugate gels for antitumor applications. Biomaterials. 2009;30(12):2349–2360. | ||
Gao L, Liu G, Kang J, et al. Paclitaxel nanosuspensions coated with P-gp inhibitory surfactants: I. Acute toxicity and pharmacokinetics studies. Colloids Surf B Biointerfaces. 2013;111:277–281. | ||
Schleich N, Sibret P, Danhier P, et al. Dual anticancer drug/superparamagnetic iron oxide-loaded PLGA-based nanoparticles for cancer therapy and magnetic resonance imaging. Int J Pharm. 2013;447(1–2):94–101. | ||
Chin WW, Parmentier J, Widzinski M, Tan EH, Gokhale R. A brief literature and patent review of nanosuspensions to a final drug product. J Pharm Sci. 2014;103(10):2980–2999. | ||
Langer K, Anhorn MG, Steinhauser I, et al. Human serum albumin (HSA) nanoparticles: reproducibility of preparation process and kinetics of enzymatic degradation. Int J Pharm. 2008;347(1–2):109–117. | ||
Ranjbar S, Shokoohinia Y, Ghobadi S, et al. Studies of the interaction between isoimperatorin and human serum albumin by multispectroscopic method: identification of possible binding site of the compound using esterase activity of the protein. Scientific World J. 2013;2013:305081. | ||
Chatpun S, Nacharaju P, Cabrales P. Improving cardiac function with new-generation plasma volume expanders. Am J Emerg Med. 2013;31(1):54–63. | ||
Son S, Song S, Lee SJ, et al. Self-crosslinked human serum albumin nanocarriers for systemic delivery of polymerized siRNA to tumors. Biomaterials. 2013;34(37):9475–9485. | ||
Li C, Li Y, Gao Y, et al. Direct comparison of two albumin-based paclitaxel-loaded nanoparticle formulations: is the crosslinked version more advantageous? Int J Pharm. 2014;468(1–2):15–25. | ||
Ibrahim N, Ibrahim H, Dormoi J, et al. Albumin-bound nanoparticles of practically water-insoluble antimalarial lead greatly enhance its efficacy. Int J Pharm. 2014;464(1–2):214–224. | ||
Crisante F, Francolini I, Bellusci M, Martinelli A, D’Ilario L, Piozzi A. Antibiotic delivery polyurethanes containing albumin and polyallylamine nanoparticles. Eur J Pharm Sci. 2009;36(4–5):555–564. | ||
Yu S, Yao P, Jiang M, Zhang G. Nanogels prepared by self-assembly of oppositely charged globular proteins. Biopolymers. 2006;83(2):148–158. | ||
Langer K, Balthasar S, Vogel V, Dinauer N, von Briesen H, Schubert D. Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int J Pharm. 2003;257(1–2):169–180. | ||
Elzoghby AO, Samy WM, Elgindy NA. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157(2):168–182. | ||
Lluch A, Alvarez I, Munoz M, Segui MA, Tusquets I, Garcia-Estevez L. Treatment innovations for metastatic breast cancer: nanoparticle albumin-bound (NAB) technology targeted to tumors. Crit Rev Oncol Hematol. 2014;89(1):62–72. | ||
Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7(8):1041–1053. | ||
Catauro M, Bollino F, Papale F, Gallicchio M, Pacifico S. Influence of the polymer amount on bioactivity and biocompatibility of SiO2/PEG hybrid materials synthesized by sol-gel technique. Mat Sci Eng C Mater Biol Appl. 2015;48:548–555. | ||
Qiu L, Zheng C, Jin Y, Zhu K. Polymeric micelles as nanocarriers for drug delivery. Expert Opin Ther Patents. 2007;17(7):819–830. | ||
Danhier F, Lecouturier N, Vroman B, et al. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133(1):11–17. | ||
Cui F, Li Y, Zhou S, et al. A comparative in vitro evaluation of self-assembled PTX-PLA and PTX-MPEG-PLA nanoparticles. Nanoscale Res Lett. 2013;8(1):301–308. | ||
Hahnenkamp A, Alsibai W, Bremer C, Holtke C. Optimizing the bioavailability of small molecular optical imaging probes by conjugation to an albumin affinity tag. J Control Release. 2014;186:32–40. | ||
Zhong Y, Yang W, Sun H, et al. Ligand-directed reduction-sensitive shell-sheddable biodegradable micelles actively deliver doxorubicin into the nuclei of target cancer cells. Biomacromolecules. 2013;14(10):3723–3730. | ||
Xie J, Chen K, Huang J, et al. PET/NIRF/MRI triple functional iron oxide nanoparticles. Biomaterials. 2010;31(11):3016–3022. | ||
Wang H, Sun X, Gao F, Zhong B, Zhang YH, Sun Z. Effect of ulinastatin on growth inhibition, apoptosis of breast carcinoma cells is related to a decrease in signal conduction of JNk-2 and NF-kappaB. J Exp Clin Cancer Res. 2012;31:1–13. | ||
Pelzer ES, Harris JE, Allan JA, et al. TUNEL analysis of DNA fragmentation in mouse unfertilized oocytes: the effect of microorganisms within human follicular fluid collected during IVF cycles. J Reprod Immunol. 2013;99(1–2):69–79. | ||
Wu D, Zheng Y, Hu X, Fan Z, Jing X. Anti-tumor activity of folate targeted biodegradable polymer-paclitaxel conjugate micelles on EMT-6 breast cancer model. Mater Sci Eng C Mater Biol Appl. 2015;53:68–75. | ||
Dang Y, Zhu C. Genomic study of the absorption mechanism of cantharidin and its solid dispersion. Colloid Surf A Physicochem Eng Asp. 2012;415:295–301. | ||
Liu X, Lu M, Guo Z, Huang L, Feng X, Wu C. Improving the chemical stability of amorphous solid dispersion with cocrystal technique by hot melt extrusion. Pharm Res. 2012;29(3):806–817. | ||
Lima AA, Soares-Sobrinho JL, Silva JL, et al. The use of solid dispersion systems in hydrophilic carriers to increase benzonidazole solubility. J Pharm Sci. 2011;100(6):2443–2451. | ||
Bekale L, Agudelo D, Tajmir-Riahi HA. The role of polymer size and hydrophobic end-group in PEG-protein interaction. Colloid Surf B Biointerfaces. 2015;130:141–148. | ||
Yadav SK, Mishra S, Mishra B. Eudragit-based nanosuspension of poorly water-soluble drug: formulation and in vitro-in vivo evaluation. AAPS Pharm Sci Tech. 2012;13(4):1031–1044. | ||
Guo S, Huang L. Nanoparticles escaping RES and endosome: challenges for siRNA delivery for cancer therapy. J Nanomater. 2011;2011:11. | ||
Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. | ||
Wu L, Zhang J, Watanabe W. Physical and chemical stability of drug nanoparticles. Adv Drug Deliv Rev. 2011;63(6):456–469. | ||
Huo M, Zhang Y, Zhou J, et al. Synthesis and characterization of low-toxic amphiphilic chitosan derivatives and their application as micelle carrier for antitumor drug. Int J Pharm. 2010;394(1–2):162–173. | ||
Frei E. Albumin binding ligands and albumin conjugate uptake by cancer cells. Diabetol Metab Syndr. 2011;3(1):11. | ||
Gao L, Liu G, Ma J, et al. Paclitaxel nanosuspension coated with P-gp inhibitory surfactants: II. Ability to reverse the drug-resistance of H460 human lung cancer cells. Colloid Surf B Biointerfaces. 2014;117:122–127. | ||
Fang J, Liao L, Yin H, Nakamura H, Shin T, Maeda H. Enhanced bacterial tumor delivery by modulating the EPR effect and therapeutic potential of Lactobacillus casei. J Pharm Sci. 2014;103(10):3235–3243. | ||
Lu J, Huang Y, Zhao W, et al. Design and characterization of PEG-derivatized vitamin E as a nanomicellar formulation for delivery of paclitaxel. Mol Pharm. 2013;10(8):2880–2890. | ||
Gligorov J, Lotz JP. Preclinical pharmacology of the taxanes: implications of the differences. Oncologist. 2004;9(Suppl 2):3–8. | ||
Gong J, Huo M, Zhou J, et al. Synthesis, characterization, drug-loading capacity and safety of novel octyl modified serum albumin micelles. Int J Pharm. 2009;376(1–2):161–168. | ||
ten Tije AJ, Verweij J, Loos WJ. Sparreboom A. Pharmacological effects of formulation vehicles: implications for cancer chemotherapy. Clin Pharmacokinet. 2003;42(7):665–685. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.