Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
A Single Cell Atlas of the COPD Lung Identifies Inflammatory Reprogramming in Fibroblasts
Authors Dong L, Chen H, Li F ![]() , Wang H, Zhou H, Cao L, Ye Y, Sun Y
, Wang H, Zhou H, Cao L, Ye Y, Sun Y
Received 17 April 2026
Accepted for publication 26 May 2026
Published 3 June 2026 Volume 2026:21 617662
DOI https://doi.org/10.2147/COPD.S617662
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Zijing Zhou
Lingling Dong,1 Haipin Chen,2 Fei Li,3 Haitao Wang,1 Hongbin Zhou,1 Liming Cao,1 Yao Ye,1 Yilan Sun1
1Geriatric Medicine Center, Department of Pulmonary and Critical Care Medicine, Zhejiang Provincial People’s Hospital (Affiliated People’s Hospital), Hangzhou Medical College, Hangzhou, Zhejiang, People’s Republic of China; 2Department of Hematology-Oncology, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Children and Adolescents’ Health and Diseases, Hangzhou, Zhejiang, People’s Republic of China; 3Department of Medicine, The Warren Alpert Medical School of Brown University, Providence, RI, USA
Correspondence: Yilan Sun, Geriatric Medicine Center, Department of Pulmonary and Critical Care Medicine, Zhejiang Provincial People’s Hospital (Affiliated People’s Hospital), Hangzhou Medical College, No. 158 Shangtang Road, Hangzhou, Zhejiang, 310014, People’s Republic of China, Tel +86-571-87666666, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is characterized by persistent inflammation, structural remodeling, and irreversible airflow limitation, but the cellular mechanisms that sustain chronic inflammatory remodeling remain poorly understood.
Methods: We integrated newly generated single cell transcriptomic data with publicly available datasets, comprising 6 healthy controls and 10 patients with COPD. Gene expression programs, intercellular communication, pseudotime trajectories, and transcription factor regulatory networks were assessed to define fibroblast associated changes in COPD lung tissue.
Results: Fibroblasts exhibited the strongest outgoing signaling activity among lung parenchymal cells and showed the greatest increase in outgoing signaling in COPD. Across multiple fibroblast subpopulations, fibroblasts from COPD lungs acquired a shared proinflammatory and immunoregulatory state associated with inflammatory activation, tissue injury, and fibrotic remodeling. This state was characterized by increased expression of inflammatory mediators and enhanced potential to promote immune cell recruitment and activation. Regulatory analyses further suggested that this inflammatory program was accompanied by extensive remodeling of transcription factor networks in fibroblasts.
Conclusion: These findings identify fibroblasts as key immunoregulatory cells in COPD lung tissue and suggest that fibroblast associated inflammatory programs may contribute to the maintenance of chronic inflammatory remodeling. Fibroblast centered inflammatory pathways may represent potential targets for future mechanistic and translational studies. The diagram illustrates inflammatory reprogramming of fibroblasts in COPD. It is divided into four main processes: 1) Fibrotic remodeling involves TGFβ leading to fibroblast activation and extracellular matrix deposition. 2) Pro-inflammatory and tissue-injurious process involves IL-1β, IL-6, IL33 causing damage to epithelial and endothelial cells. 3) Immune cell recruitment involves CCL, CXCL acting as chemoattractants for macrophages and dendritic cells. 4) Immune cell activation involves CSF1, TNFSF13B activating mast cells, B cells and T cells. The diagram contrasts healthy lungs with COPD-affected lungs, highlighting the impact of these processes.COPD fibroblast reprogramming: fibrosis, immune recruitment, activation.
Keywords: chronic obstructive pulmonary disease, fibroblasts, inflammatory state, immune response, fibrotic remodeling
Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous chronic respiratory disorder characterized primarily by persistent and largely irreversible airflow limitation, with marked variability in symptoms, pathological features, inflammatory responses, and clinical progression among patients. Clinically, COPD commonly manifests as persistent cough, sputum production, and progressive dyspnea.1 It has become the third leading cause of death worldwide, placing a substantial burden on society and the economy.2 The World Health Organization (WHO) predicts that COPD related mortality will continue to rise. However, current clinical strategies for COPD remain limited: diagnosis largely relies on pulmonary function testing, and available pharmacologic therapies can alleviate symptoms but cannot halt disease progression. Early-stage COPD is also difficult to detect, meaning that many patients are not definitively diagnosed until the middle or late stages, when therapeutic outcomes are often suboptimal. A key reason for this is that the core pathogenic mechanisms underlying COPD are still not fully understood. Therefore, elucidating the critical mechanisms driving COPD is of major significance for improving clinical diagnosis and management, with the potential to enhance patient outcomes while substantially reducing the societal and economic burden in the future.
COPD is a chronic inflammatory airway disorder, pathologically characterized predominantly by small airway remodeling, chronic bronchitis, and/or emphysema. Its severity is commonly classified using the GOLD grading system based on post bronchodilator forced expiratory volume in 1 second (FEV1), with airflow limitation categorized from mild to very severe. Both innate and adaptive immune responses contribute to disease initiation and progression.3 In recent years, mechanistic studies of COPD have achieved several major advances, offering renewed hope for the development of disease modifying—and potentially even disease reversing—therapies. In 2020, Conlon et al reported that targeting the lymphotoxin β receptor (LTβR)–WNT/β-catenin signaling axis in lung tissue suppresses noncanonical nuclear factor kappa B (NF-κB) activation in parenchymal cells, disrupts inducible bronchus associated lymphoid tissue(iBALT) formation, and attenuates airway TGF-β signaling, while concurrently limiting epithelial cell death and promoting lung regeneration.4 Another important study by Zhang et al profiled lung tissue from a total of 141 participants with COPD and healthy controls.5 Their analyses revealed that the inflammatory landscape of COPD constitutes a spatially organized, multicellular network in which structural cells acquire pro-inflammatory transcriptional programs and engage in reciprocal signaling with macrophages and other immune cells. Ultimately, the acquisition of “inflamed” states by parenchymal cells, together with subsequent maladaptive remodeling, may drive COPD toward an irreversible trajectory of progressive disease. These studies suggest that COPD progression may be at least partially reversible, and that its key driver may be the establishment and persistence of a chronic inflammatory state rather than inflammation per se, implicating an underrecognized contribution of parenchymal cells, beyond immune cells, to sustaining this state. This may also partly explain why strategies focused primarily on immune cells have not yielded major advances in COPD treatment, whereas directing greater attention toward parenchymal cells may offer new opportunities for therapeutic breakthroughs. However, the precise role of lung parenchymal cells in sustaining this state and the underlying mechanisms remain unclear.
We sought to investigate the role of lung parenchymal cells in coordinating and organizing the local inflammatory microenvironment in COPD lung tissue, as well as the potential mechanisms underlying this process. To this end, we performed single cell transcriptomic profiling on fresh lung tissue samples from 3 healthy controls and 7 patients with COPD, and integrated these data with a previously published single cell dataset from Immunity (2023) comprising 3 healthy controls and 3 patients with COPD. By analyzing the integrated single cell data, we found that fibroblasts exhibited the greatest inflammatory regulatory potential in COPD among lung parenchymal cells. Previous studies have suggested that alterations in fibroblast functional states in COPD are primarily associated with fibrotic processes and aberrant tissue repair, contributing to small airway remodeling, pulmonary vascular remodeling, and alveolar structural destruction, and thereby leading to airflow limitation, pulmonary hypertension, and emphysema in patients with COPD.6,7 Although increasing attention has recently been directed toward the potential immunoregulatory roles of fibroblasts,8 it remains unclear whether this immunoregulatory function plays a central role in the development and persistence of inflammation in COPD and what mechanisms underlie this process.
In this study, analysis of integrated single cell transcriptomic data revealed that fibroblasts were not only the strongest outgoing signaling cells in lung tissue, but also the cell population showing the most pronounced increase in outgoing signaling activity in COPD. Across multiple subpopulations, COPD fibroblasts adopted a shared inflammatory state associated with inflammatory activation, tissue injury, and fibrotic remodeling. This proinflammatory role was reflected not only by increased secretion of inflammatory mediators, but also by enhanced promotion of immune cell chemotaxis and activation. Mechanistically, acquisition of this inflammatory state was further linked to extensive remodeling of transcriptional regulatory networks in fibroblasts. Collectively, these findings identify fibroblasts may as key immunoregulatory cells in COPD, in which extensive transcriptional regulatory remodeling drives a shared inflammatory state that promotes immune activation, tissue injury, and fibrotic remodeling.
Methods
Human Tissues
Human lung tissue samples were primarily collected from Zhejiang Provincial People’s Hospital, and informed consent was obtained from all donors. Based on preoperative pulmonary function test results, healthy controls were defined as patients with normal lung function, whereas the COPD group comprised patients with pulmonary function findings consistent with mild to moderate COPD according to the GOLD classification. Available demographic and clinical characteristics of the included subjects are summarized in Supplementary Table 1. This study was approved by the Institutional Review Board of Zhejiang Provincial People’s Hospital (Approval No. 2025(291)) and was conducted in accordance with all applicable ethical guidelines and regulations governing research involving human participants.
Sample Processing
Immediately after surgical resection, lung tissues were immersed in tissue preservation solution and transported on ice. Under sterile conditions, freshly collected tissues were washed twice with pre chilled RPMI 1640 supplemented with 0.04% BSA. The tissues were then finely minced with surgical scissors into ~0.5mm3 pieces and digested in freshly prepared enzyme solution at 37°C for 30–60min, with gentle inversion every 5–10min. The digestion solution consisted of RPMI 1640 (Corning, Cat#10-040-CVR), 0.04% BSA (MACS, Cat#1000076), and 0.2% Collagenase II (Gibco, Cat#17101015). Following digestion, the cell suspension was filtered through a 40μm cell strainer (BD Falcon, Cat#352340) once or twice and centrifuged at 300×g for 5min at 4°C. The pellet was resuspended in an appropriate volume of medium and mixed with an equal volume of red blood cell lysis buffer (Miltenyi, Cat#130-094-183). After incubating at 4°C for 10min, cells were centrifuged at 300×g for 5min and the supernatant was discarded. The pellet was washed once with medium, centrifuged again at 300×g for 5min, and finally resuspended in 100μL RPMI 1640 containing 0.04%BSA (Corning, Cat#10-040-CVR). Cell concentration and viability were determined by using a Luna-FL automated cell counter (Logos Biosystems, Korea) or by trypan blue exclusion.
Single Cell RNA Library Preparation and Sequencing
Freshly prepared single cell suspensions were adjusted to 700–1200cells/μL. Single cell capture and 3′ transcriptome library preparation were carried out on the MobiNova-100 microfluidic platform following the manufacturer’s protocol for the MobiCube High-Throughput Single-Cell 3′ Transcriptome Kit V2.1 (Cat#PN-S050200301). The resulting libraries were then sequenced on a DNBSEQ-T7 platform using high-throughput sequencing.
Single-Cell RNA-Seq Data Preprocessing
scRNA-seq was performed by Hangzhou Astrocyte Technology Co, Ltd. Raw sequencing data were aligned to the GRCh38 reference genome using the MobiNova official quality-control software MobiVision (version 3.2) with default parameters, followed by cell filtering and gene quantification. The filtered feature–barcode matrix was used for downstream analyses.
Data Acquisition
To minimize potential platform- and technology-related biases in single-cell analyses, we screened datasets in the NCBI Gene Expression Omnibus (GEO) and selected GSE196638 based on the criterion that each sample contained at least 5000 cells. The dataset had been preprocessed by the original authors and was provided as feature–barcode matrices that could be used directly for downstream analyses.
Gene Quantification, Quality Control (QC), and Harmonization
We performed quality control using the Seurat package (version 5.4.0) to remove low-quality cells. Cells were retained if they met the following criteria: 300–6000 detected genes, 1000–60,000 UMIs, and <15% mitochondrial UMI content. Doublets were further identified and removed using DoubletFinder (version 2.0.6). DoubletFinder was applied separately to each sample using PCs 1 to 20, pN=0.25, pK=0.09, and an expected doublet rate of 4.6%. The expected number of doublets was calculated as round (0.0460 × number of cells) for each sample. Cells classified as doublets were removed before downstream integration and analysis. After QC, downstream processing was continued in Seurat with the RNA assay set as default. Gene expression values were log-normalized, and the top 2000 highly variable genes were identified using the variance-stabilizing transformation method. The data were then scaled and subjected to principal component analysis (PCA) using the first 50 principal components. To mitigate batch effects across samples, we applied Harmony (version 1.2.4) to the PCA embeddings, using sample identity as the covariate for integration. The integration performance was visually assessed using UMAP plots before and after Harmony correction, which showed improved mixing of cells from different samples after correction while preserving the major biological structure of the dataset (Figure S1).
Dimensionality Reduction and Clustering Analysis
Dimensionality reduction and clustering analysis were performed using the Seurat R package. The first 15 Harmony-corrected dimensions were selected for downstream analysis. Cell-cell nearest-neighbor graphs were constructed with the FindNeighbors function based on the Harmony reduction. Cell clusters were then identified using the FindClusters function at a resolution of 0.8. The resolution parameter was chosen based on the clustree (version 0.5.1) assessment of cluster stability across multiple resolutions, together with reference to commonly used settings in published single cell studies. Two dimensional visualization was generated with the RunUMAP function using the same Harmony-corrected dimensions. Cluster distributions were displayed with the DimPlot function, with cells colored according to the clustering results.
Marker Gene Identification and Cell Type Annotation
Cluster marker genes were identified using the FindAllMarkers function in the Seurat R package. Only positive markers enriched in each cluster were retained. Genes were required to be detected in at least 25% of cells within a cluster, with a minimum log fold-change threshold of 0.25. Differential expression testing was performed using the Wilcoxon rank-sum test. Cell-type identification was performed primarily by manual annotation, with automated annotation serving as a complementary approach. Automated annotation was conducted using CellTypist, while the final and more accurate cell-type assignments were determined manually based on the expression patterns of canonical marker genes.
Differential Gene Expression (DEG) Analysis and Functional Enrichment
Differentially expressed genes were identified using the FindMarkers function in the Seurat R package with the Wilcoxon rank-sum test. Genes detected in at least 10% of cells and meeting a log fold-change threshold of 0.25 were retained for downstream analysis. Gene Ontology (GO) enrichment analysis was performed using the enrichGO function in the clusterProfiler R package (Version 4.18.4), with annotation from the org.Hs.egdb database. Gene symbols were converted to Entrez IDs using the bitr function, and duplicate mappings were removed by retaining distinct gene symbols. GO enrichment analysis was then conducted for Biological Process terms using the mapped Entrez IDs, with all detected genes used as the background gene set. Multiple testing correction was performed using the Benjamini–Hochberg method, and terms with both adjusted P values and q values below 0.05 were considered significantly enriched. Gene set enrichment analysis (GSEA) was performed for each cell type separately using the fgsea R package (Version 1.36.0). Only cell types containing cells from both comparison groups were included in the analysis. Pre-ranked enrichment analysis was carried out using the fgsea function based on the selected gene set collection, with a minimum gene set size of 10, a maximum gene set size of 500, and 20,000 permutations. Multiple testing correction was performed using the Benjamini–Hochberg method, and adjusted P values were used for downstream interpretation of significantly enriched pathways.
Cell and Cell Interactions
Cell and cell communication analysis was performed using the CellChat R package (Version 2.2.0) to characterize intercellular interaction patterns among annotated cell types. To reduce computational burden, stratified downsampling was applied before analysis, with up to 5000 cells retained for each cell type within each group. For each group, a CellChat object was created using the normalized expression matrix from the RNA assay, with cell identities defined by the annotated cell types. The human ligand–receptor interaction database was used. Signaling genes and ligand–receptor interactions were preprocessed using the subsetData, identifyOverExpressedGenes, and identifyOverExpressedInteractions functions. Communication probabilities were inferred using the computeCommunProb function with the truncated mean method, and interactions involving cell groups with fewer than 10 cells were filtered using the filterCommunication function. Pathway level communication probabilities were calculated using computeCommunProbPathway, and communication networks were summarized using aggregateNet. Network centrality analysis was performed using the netAnalysis_computeCentrality function. For group comparison, CellChat objects were merged using the mergeCellChat function. The overall number and strength of intercellular interactions were compared using the compareInteractions function based on the count and weight measures, respectively. Differential interaction networks were visualized using the netVisual_diffInteraction function with weight scaling enabled. Differences in pathway level information flow were assessed using the rankNet function in comparison mode with stacked visualization and statistical testing enabled. Signaling roles of different cell types were analyzed using the netAnalysis_signalingRole_scatter function. For selected pathways, pathway specific communication networks and ligand–receptor contributions were examined using the netVisual_aggregate and netAnalysis_contribution functions.
Ligand–target regulatory analysis was performed using the nichenetr R package (Version 2.2.1.1) to infer the downstream transcriptional effects of intercellular communication. Differentially expressed genes in receiver cells were identified using the FindMarkers function in the Seurat R package with the Wilcoxon rank-sum test. Genes were filtered using a minimum log fold-change threshold of 0.10, a minimum expression percentage of 10%, and an adjusted P value below 0.05, and the top 300 upregulated genes were selected as the target gene set of interest. Prior knowledge models, including the ligand–target matrix, ligand–receptor network, and weighted signaling networks, were obtained from the nichenetr package. Expressed genes in sender and receiver populations were identified using the get_expressed_genes function with an expression threshold of 10%. Potential ligands were defined based on ligands expressed in sender cells and cognate receptors expressed in receiver cells according to the curated ligand–receptor network. Ligand activity was predicted using the predict_ligand_activities function, and ligands were ranked by Pearson correlation with the receiver gene set. The top 10 ligands were selected for downstream analysis. Ligand–receptor links were extracted from the weighted ligand–receptor network, and the top two receptors for each ligand were retained according to interaction weight. Ligand–target links were inferred using the get_weighted_ligand_target_links function, with up to 250 target genes evaluated for each ligand, and the top two target genes per ligand were retained according to regulatory weight.
Pseudotime Trajectory Analysis
Pseudotime trajectory analysis was performed using the Monocle R package (Version 2.38.0), together with Seurat, Matrix, and dplyr. Fibroblast subcluster cells were extracted from the Seurat object, and, when available, only cells annotated as singlets and non-contaminating cells were retained. A Monocle2 CellDataSet object was constructed from the raw count matrix using the newCellDataSet function with the negative binomial model (expressionFamily = negbinomial.size()), followed by normalization with estimateSizeFactors and dispersion estimation with estimateDispersions. Ordering genes were defined using fibroblast subcluster marker genes identified by the FindAllMarkers function in the Seurat R package, retaining only positive markers with a minimum expression percentage of 25% and a log fold-change threshold of 0.25. These genes were supplied to Monocle2 using the setOrderingFilter function. Dimensionality reduction was then performed using the reduceDimension function with the DDRTree method, with max_components = 2, norm_method = “log”, and num_dim = 30. Cells were ordered along pseudotime using the orderCells function. To define the trajectory direction, the root state was assigned according to the state most enriched for the Adventitial fibroblast subtype.
Transcription Factor Regulatory Network Analysis
Transcription factor regulatory network analysis was performed using the SCENIC workflow in R (Version 1.3.1), primarily based on the SCENIC, RcisTarget, AUCell, Seurat, and Matrix packages. Raw count matrices were extracted from the RNA assay of the Seurat object using the GetAssayData function with the counts layer, and converted to sparse matrices. Genes and cells with zero total counts were removed before downstream analysis. SCENIC analysis was initialized using the initializeScenic function with org = “hgnc” and two human cisTarget ranking databases based on the hg38 genome build and RefSeq r80 gene annotation. The exact databases used were hg38__refseq-r80__10kb_up_and_down_tss.mc9nr.genes_vs_motifs.rankings.feather and hg38__refseq-r80__500bp_up_ and _100bp_down_tss.mc9nr.genes_vs_motifs.rankings.feather, representing motif ranking within ±10kb around the transcription start site and within 500bp upstream to 100bp downstream of the transcription start site, respectively. Gene filtering was performed prior to network inference. The minimum number of cells in which a gene had to be detected was set to the larger of 20 cells or 1% of the total number of cells, and the minimum total count threshold per gene was defined as three times the minimum cell number. Genes passing both thresholds were retained for downstream analysis. The filtered expression matrix was then converted to a dense matrix for SCENIC analysis. Co-expression analysis was performed using the runCorrelation function, followed by regulatory network inference using the runGenie3 function. Co-expression modules were identified using runSCENIC_1_coexNetwork2modules, and regulons were constructed using runSCENIC_2_createRegulons. Regulon activity in individual cells was scored using runSCENIC_3_scoreCells, and regulon activity was binarized using runSCENIC_4_aucell_binarize. Regulon activity scores were extracted using the loadInt function with the “aucell_regulonAUC” slot, and the AUC matrix was obtained using the getAUC function from the AUCell package. The resulting regulon activity matrix was then added back to the Seurat object as a new assay using the CreateAssayObject function, and the SCENIC assay was set as the default assay for downstream analysis.
Results
A Single Cell Atlas of the COPD Lung Reveals Chronic Inflammatory Remodeling
We performed single cell transcriptomic profiling of collected lung tissue samples and integrated the resulting data with the COPD single cell dataset GSE196638 from the GEO database (Figure 1A). After dimensionality reduction and clustering of the integrated single cell transcriptomic data, we annotated cell identities based on canonical marker gene expression and identified 18 major cell populations, including epithelial, endothelial, stromal, and immune cell subsets (Figure 1B). Specifically, these populations included alveolar type I epithelial cells (AT1), alveolar type II epithelial cells (AT2), ciliated and secretory airway epithelial cells (AEC), vascular and lymphatic endothelial cells (EC), fibroblasts, smooth muscle cells (SMC) and pericytes, monocytes (Mono), macrophages (Mac), dendritic cells (DC), mast cells, B cells, plasma cells, non-cytotoxic T cells, cytotoxic T cells, NK cells, and cycling cells, and their annotation was supported by the expected expression of canonical markers (Figure 1C). GO enrichment analysis further corroborated the annotation results by showing functional enrichment patterns consistent with the known biological roles of each cell population (Figure 1D).
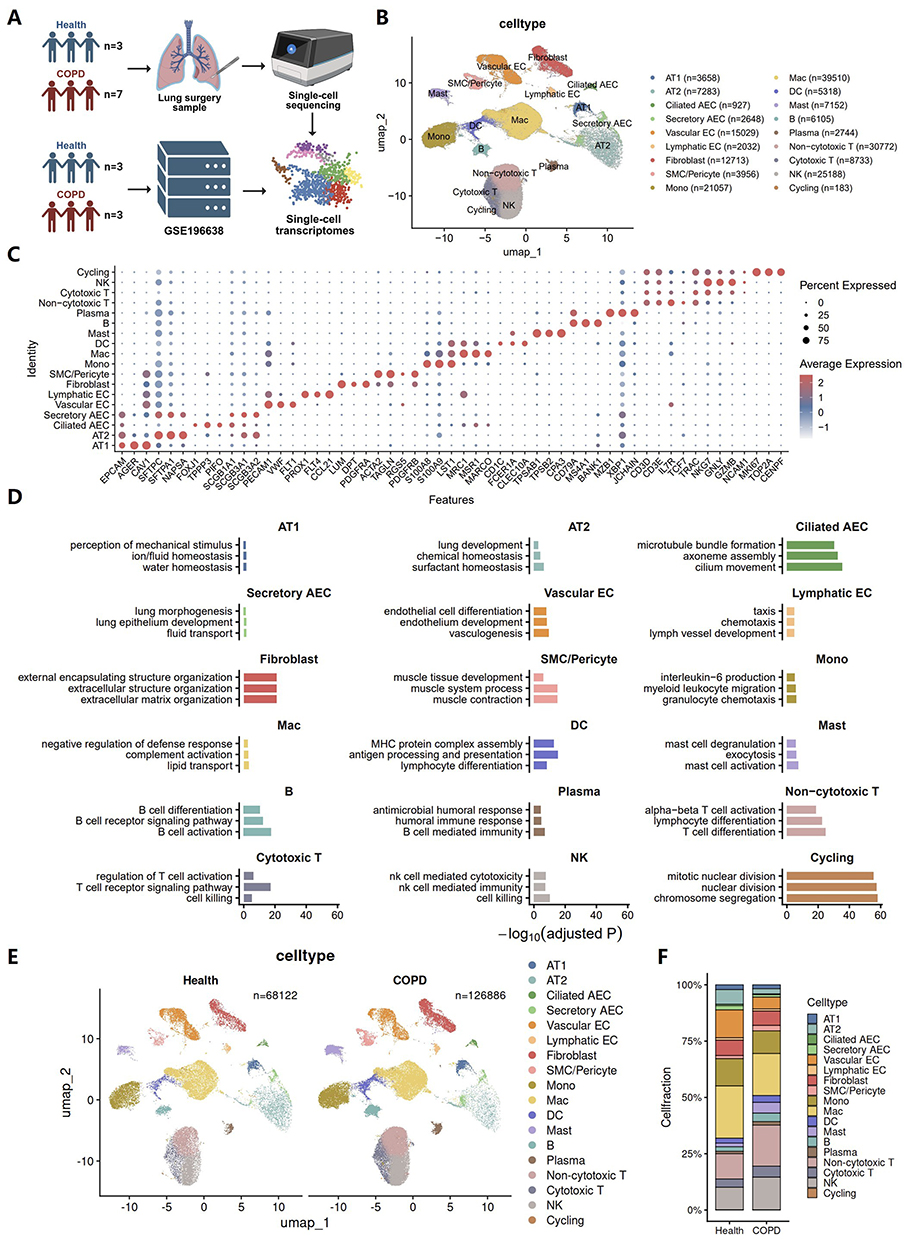 |
Figure 1 A single cell atlas of the COPD lung reveals chronic inflammatory remodeling. (A) Study design for single-cell transcriptomic profiling of fresh lung tissues from healthy controls and patients with COPD, integrated with a published dataset (GSE196638). (B) UMAP embedding of all cells in the integrated dataset, showing the major annotated lung cell populations, with cell numbers indicated for each cell type. (C) Dot plot of representative marker genes used for major cell-type annotation. Dot size indicates the fraction of expressing cells, and color indicates scaled average expression. (D) Gene Ontology enrichment analysis of cell type-specific marker genes for major cell populations. (E) UMAP embeddings split by condition (Health and COPD), showing the distribution of major cell populations in each group. (F) Stacked bar plot showing the relative proportions of major cell populations in healthy and COPD lungs. |
To assess group specific cellular alterations, we visualized the integrated dataset by disease status. UMAP plots showed that both Health and COPD samples contained the same major cell populations with broadly comparable spatial distributions (Figure 1E). Consistent with established findings, the relative abundance of AT2 cells was markedly reduced in the COPD group despite the higher overall cell number, suggesting impaired alveolar epithelial repair and regenerative capacity. Meanwhile, immune cell populations were globally expanded in COPD compared with Health, indicative of a heightened chronic inflammatory milieu (Figures 1F and S2). Together, the single cell atlas and cell proportion changes suggest a heightened inflammatory milieu and tissue injury in COPD lungs.
Inflammatory Reprogramming of Fibroblasts Is Associated with Immune Activation, Tissue Injury, and Fibrotic Remodeling in COPD
To identify upstream regulatory mechanisms within the inflammatory network in COPD, we first performed cell communication analysis on the integrated single cell dataset. The analysis showed that intercellular communication activity was globally increased in COPD lungs (Figure 2A and B). Across both groups, fibroblasts showed the strongest outgoing signaling strength, which was further increased in COPD (Figure 2A). These findings suggest that fibroblasts play an important role in coordinating intercellular activity in the lung and may mediate the development and maintenance of chronic inflammation in COPD. Further quantification of intercellular communication changes in COPD confirmed a marked increase in fibroblast derived signaling toward other cell populations (Figure 2C and D). In addition, the significant enhancement of fibroblast self-communication suggested a potential shift in fibroblast functional state (Figure 2C).
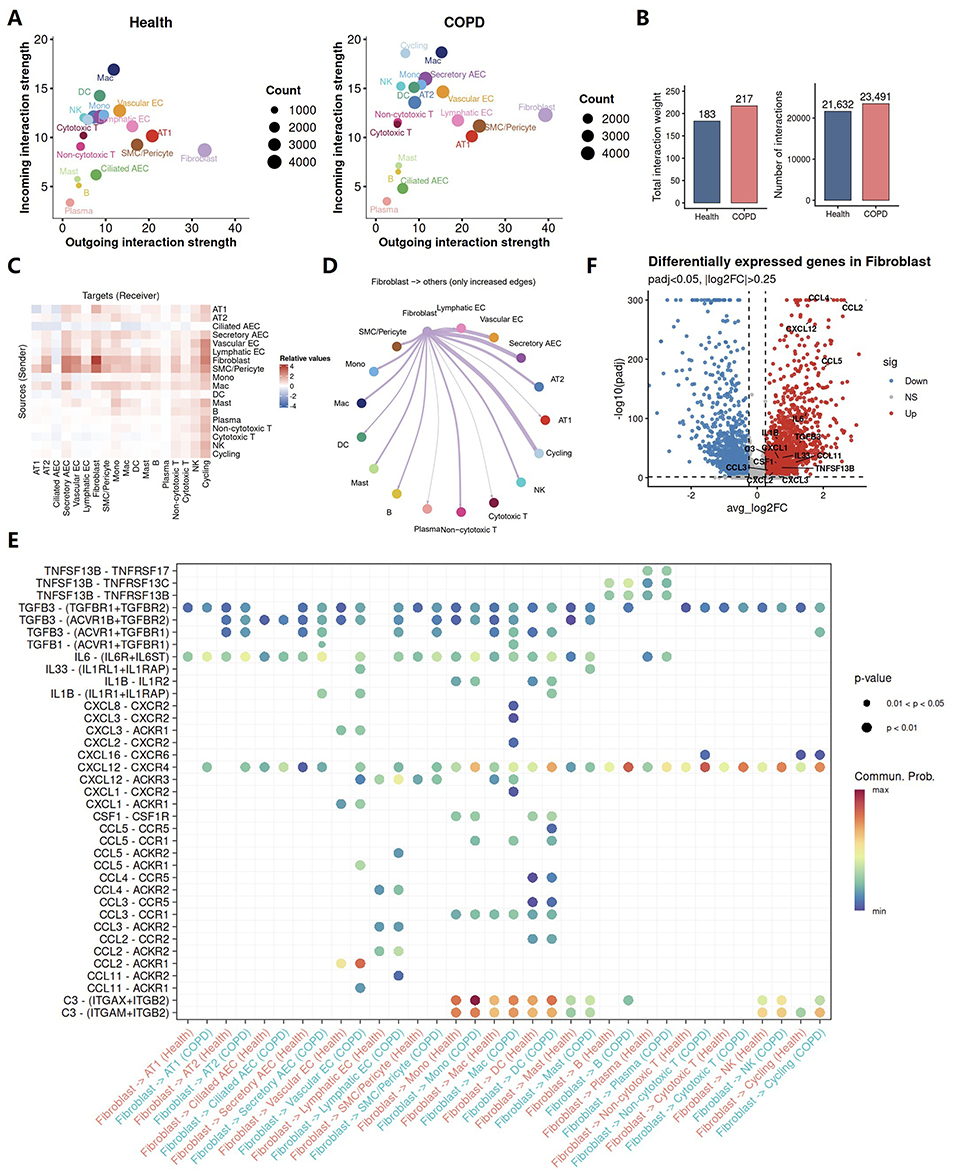 |
Figure 2 Inflammatory reprogramming of fibroblasts is associated with immune activation, tissue injury, and fibrotic remodeling in COPD. (A–E) CellChat analysis of intercellular communication in healthy and COPD lungs. (A) Comparison of outgoing and incoming interaction strength for each major cell population. Dot size indicates cell number. (B) Bar plots showing the total interaction weight and the number of inferred intercellular interactions. (C) Heatmap showing the relative changes in intercellular communication strength among major cell populations in COPD compared with healthy lungs. Red indicates increased communication strength and blue indicates decreased communication strength; deeper color intensity represents a greater magnitude of change. (D) Circle plot showing increased outgoing interactions from fibroblasts to other cell populations in COPD. (E) Bubble plot showing selected ligand-receptor pairs with enhanced signaling between fibroblasts and other lung cell populations in COPD compared with healthy lungs. Dot color indicates communication probability, and dot size indicates statistical significance. (F) Volcano plot showing differentially expressed genes in fibroblasts, with labeled genes indicating fibroblast derived molecules involved in enhanced intercellular communication. |
COPD is characterized by chronic airway inflammation, airway remodeling, and emphysema.9 Fibroblasts have traditionally been regarded as structural effector cells that contribute to airway remodeling through TGFβ signaling and extracellular matrix production,10 rather than as active drivers of disease progression. In our study, fibroblasts in COPD received stronger signals from other lung cell populations, consistent with the conventional view that they respond to the diseased microenvironment and contribute to the fibrotic process. However, an even more prominent change was the marked increase in the signals they sent to other cell populations. These findings suggest that fibroblasts may actively influence intercellular interactions in the lung, rather than merely serving as passive responders, and may therefore contribute to COPD pathogenesis.
However, how fibroblasts in COPD influence the functional states of other cell populations remains unclear. Functional enrichment analysis revealed widespread activation of inflammatory signaling pathways and suppression of metabolism related pathways in COPD fibroblasts (Figure S3), suggesting that fibroblasts may adopt an inflammatory state that is potentially associated with metabolic reprogramming. To further investigate the molecular mechanisms underlying interactions between fibroblasts and other cell populations, we performed ligand and receptor pairing analysis and found that fibroblasts in COPD may communicate extensively with surrounding lung cells through the secretion of a diverse set of molecules, including inflammatory mediators, chemokines, and profibrotic factors (Figure 2E). Consistent with previous studies, TGFβ signaling was markedly enhanced in COPD fibroblasts, further supporting its established role in promoting fibrotic processes and airway remodeling. The inflammatory mediators identified included IL6, IL33, IL1B, TNFSF13B, and C3. Among them, IL6, IL1B, and C3 are key components of the chronic inflammatory microenvironment in COPD and have been linked to inflammatory amplification, immune cell activation, and airway epithelial injury.11–15 IL33 showed markedly enhanced signaling toward endothelial cells and mast cells in our data. IL33 is generally regarded as a cytokine associated with type 2 inflammation, and IL33 targeted therapy has shown potential clinical benefit in a subset of patients with COPD.16,17 In addition, COPD fibroblasts also exhibited enhanced chemotactic effects on multiple immune cell populations through the secretion of chemokines such as CCL and CXCL family members, and may further activate recruited local immune cells, for example through TNFSF13B signaling. TNFSF13B, also known as B cell activating factor (BAFF), is a member of the TNF ligand superfamily and a key regulator of B cell survival, maturation, differentiation, and activation. B cells, by contributing to the formation of iBALT, have been shown to be closely associated with COPD progression and disease severity.4,7,18
To further validate the reliability of the intercellular interaction results described above, we performed differential expression gene analysis of fibroblasts between the disease and control groups. Consistent with the interaction analysis, the inflammatory mediators, chemokines, immune activating factors, and profibrotic molecules described above were all markedly upregulated in COPD fibroblasts (Figure 2F). Together, these findings suggest that fibroblasts, in addition to their structural role, may actively shape the immune microenvironment through their own inflammatory remodeling, thereby sustaining and amplifying tissue inflammation and contributing to the development and progression of irreversible chronic inflammation in COPD.
Fibroblasts Promote Pulmonary Immune Cell Recruitment and Functional Regulation in COPD
As shown above, fibroblasts in COPD may promote the recruitment and activation of inflammatory cells within the lung through the secretion of chemokines and inflammatory mediators. Nevertheless, their precise contribution to inflammatory regulation in COPD and their influence on immune cell function remain incompletely understood.
Based on the results of the intercellular communication analysis described above, CXCL12-CXCR4 signaling was more prominently enhanced between fibroblasts and immune cells in COPD than other chemotactic pathways, with particularly prominent effects on B cells (Figure 2E). As a classical G protein-coupled receptor (GPCR) mediated chemokine signaling axis, CXCL12 signaling has been implicated in pulmonary eosinophilic and neutrophilic inflammation and may contribute to the progression of diseases such as asthma, acute lung injury, and COVID-19.19 Previous studies have suggested that activation of the CXCL12-CXCR4 axis in COPD is mainly linked to fibrotic remodeling.20 A clinical study targeting CXCR4 has suggested that inhibition of this pathway may reduce fibrocyte recruitment, thereby alleviating acute exacerbations of COPD, decreasing mortality, and slowing the decline in lung function (ClinicalTrials NCT01196832).21 However, its contribution to the development of the chronic inflammatory state in COPD is still not fully understood. Among chemokine receptors expressed on B cells in COPD, CXCR4 was the most markedly upregulated (Figure 3A). Moreover, B cell chemotaxis toward lung tissue in COPD appeared to be primarily driven by CXCL12 secreted by fibroblasts and smooth muscle/pericyte populations (Figure 3B). Both differential expression analysis and expression level analysis confirmed the upregulation of CXCL12 in COPD fibroblasts (Figures 2F and 3C). Thus, fibroblasts are involved not only in the chemotactic recruitment of inflammatory cells into the lungs in COPD, but, more importantly, may play a central role in orchestrating this process.
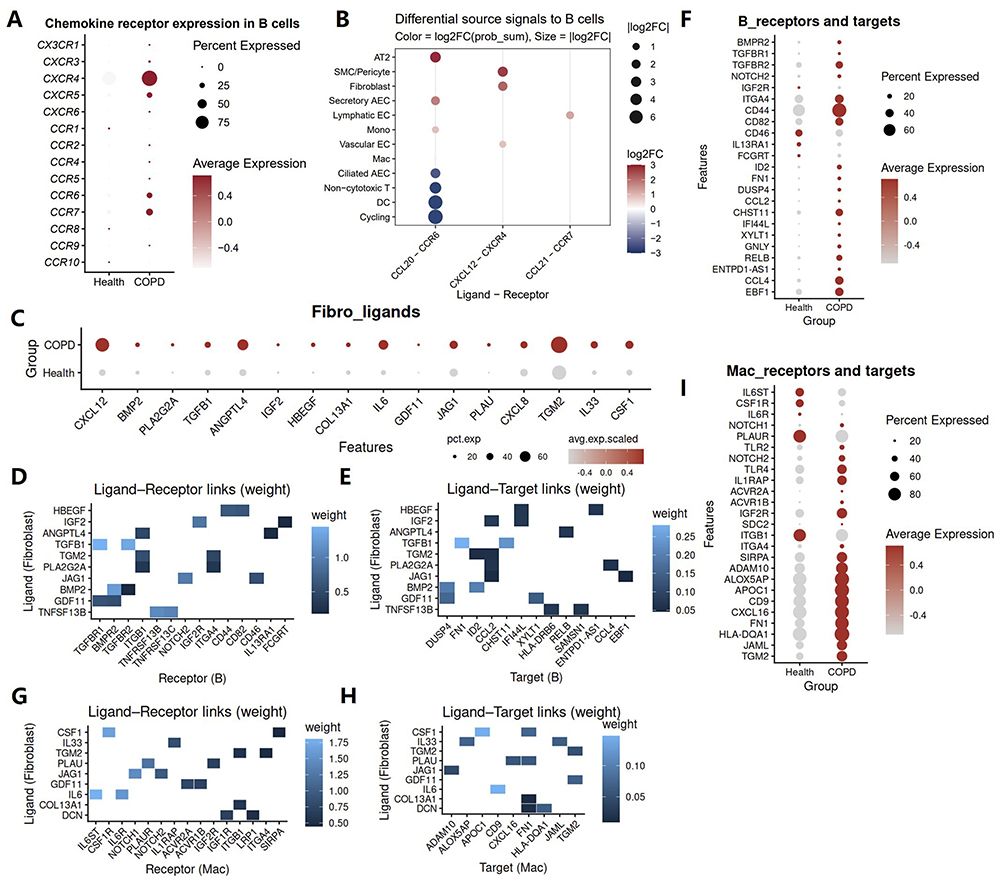 |
Figure 3 Fibroblasts promote pulmonary immune cell recruitment and functional regulation in COPD. (A) Dot plot showing chemokine receptor expression in B cells from healthy and COPD lungs. Dot size indicates the fraction of expressing cells, and color indicates average expression. (B) CellChat analysis showing differential incoming signals to B cells in COPD compared with healthy lungs. Dot color indicates log2 fold change of communication probability, and dot size indicates the absolute log2 fold change. (C) Dot plot showing the differential expression of fibroblast-derived ligand genes between healthy and COPD lungs. (D and E) NicheNet analysis of communication from fibroblasts to B cells. (D) Heatmap showing prioritized ligand–receptor interactions between ligands derived from fibroblasts and receptors expressed on B cells. (E) Heatmap showing prioritized ligand–target links between ligands derived from fibroblasts and predicted downstream target genes in B cells. Color intensity indicates interaction weight. (F) Dot plot showing the differential expression of B cell receptors and target genes associated with signaling from fibroblasts to B cells in COPD compared with healthy lungs. (G and H) NicheNet analysis of communication from fibroblasts to macrophages. (G) Heatmap showing prioritized ligand–receptor interactions between fibroblast derived ligands and receptors expressed on macrophages. (H) Heatmap showing prioritized ligand–target links between fibroblast derived ligands and predicted downstream target genes in macrophages. Color intensity indicates interaction weight. (I) Dot plot showing the differential expression of macrophage receptors and target genes associated with signaling from fibroblasts to macrophages in COPD compared with healthy lungs. |
In addition to their chemotactic effects on inflammatory cells, we further explored whether fibroblasts also contribute to the functional regulation of these cells in COPD. NicheNet analysis revealed that fibroblasts may regulate gene expression in immune target cells through secreted ligands. More specifically, multiple fibroblast derived ligands were predicted to engage receptors on B cells and to influence downstream gene expression programs (Figure 3D and E). Fibroblast regulated B cell target genes were mainly associated with cell differentiation and lineage development (EBF1, ID2), immune regulation (CCL2, ENTPD1-AS1), inflammatory signaling activation (DUSP4, IFI44L, RELB) and extracellular matrix interactions (CHST1, XYLT1) (Figure 3E). Expression level analysis further confirmed the upregulation of the corresponding target genes in COPD B cells (Figure 3F). Beyond lymphocytes, fibroblasts also appeared to regulate innate immune cell function in COPD. Using the same analytical framework, ligand and receptor analysis identified extensive interactions between fibroblast derived ligands and macrophage receptors (Figure 3G). Ligand and target analysis further suggested that these fibroblast derived signals were associated with macrophage gene programs involved in antigen presentation (HLA-DQA1), inflammatory mediator production and immune activation (ALOX5AP, CXCL16), extracellular matrix remodeling and adhesive interactions (FN1, TGM2, JAML, ADAM10, CD9), and lipid metabolic reprogramming (APOC1) (Figure 3H). Consistent with these predictions, macrophages in COPD exhibited increased expression of multiple corresponding receptors and target genes (Figure 3I). These data support the role for fibroblasts in promoting a pro-inflammatory, tissue remodeling macrophage phenotype in COPD.
Overall, these results suggest that fibroblasts may function as immunoregulatory cells that contribute to local immune dysregulation and chronic inflammation in COPD by coordinating both immune cell recruitment and their subsequent functional activation.
Fibroblast Subpopulations in COPD Uniformly Exhibit an Inflammatory State
To further characterize the inflammatory features of fibroblasts in COPD, we performed subtype specific analyses of fibroblast populations. UMAP clustering identified eight fibroblast subpopulations, including inflammatory fibroblasts (Inflammatory), pro-extracellular matrix remodeling/angiogenic fibroblasts (Pro_ECM/Angiogenic), perivascular fibroblasts (Perivascular), stressed fibroblasts (Stressed), adventitial fibroblasts (Adventitial), peribronchial fibroblasts (Peribronchial), myofibroblasts (Myofibro), and high-metabolic fibroblasts (High_Metabolic) (Figure 4A). Marker heatmap analysis confirmed the distinct transcriptional identities of these subpopulations, with each cluster expressing representative subtype-associated genes (Figure 4B).
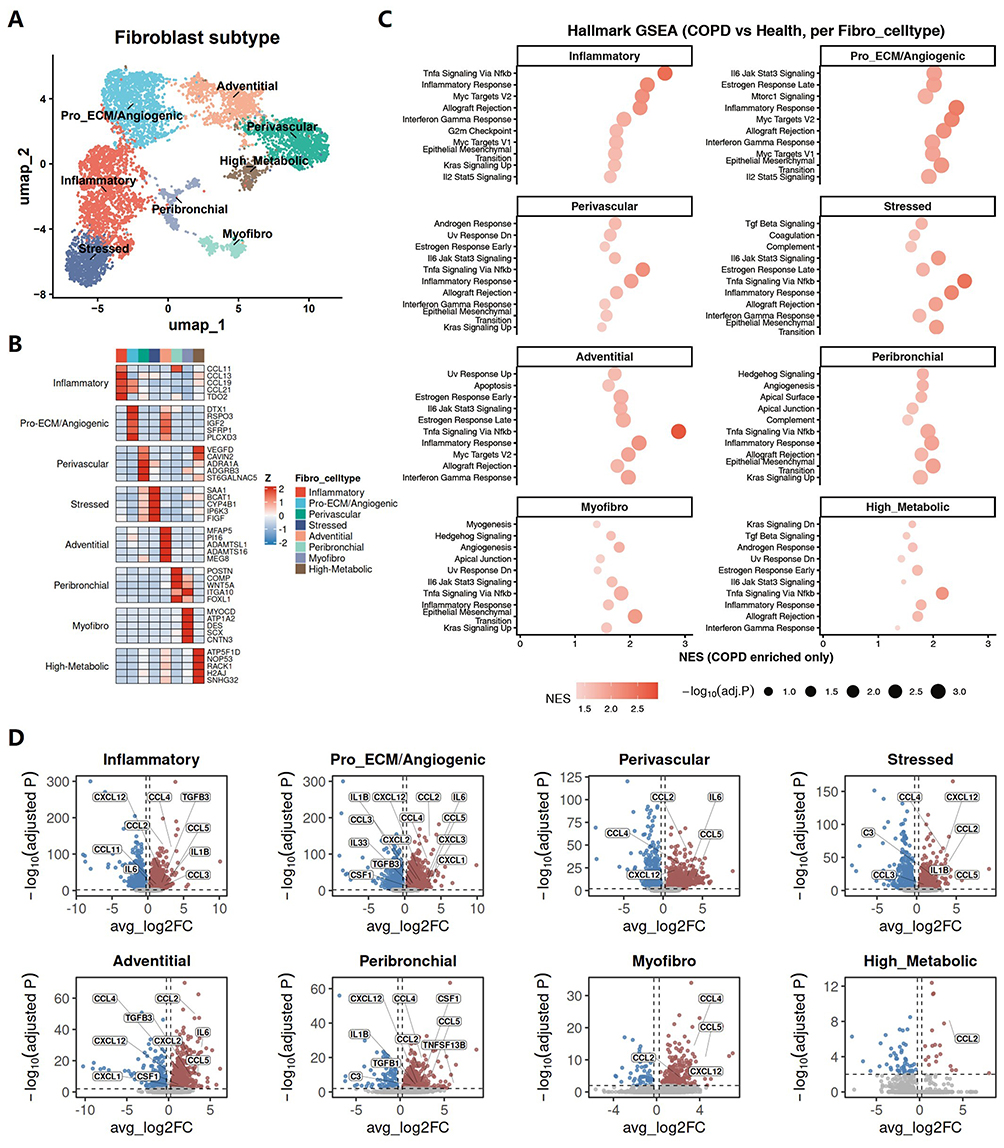 |
Figure 4 Fibroblast subpopulations in COPD uniformly exhibit an inflammatory state. (A) UMAP embedding showing the major fibroblast subpopulations identified in the integrated dataset. (B) Heatmap showing representative marker genes for each fibroblast subpopulation. (C) Hallmark GSEA showing COPD enriched pathways in each fibroblast subpopulation. Dot color indicates normalized enrichment score (NES), and dot size indicates statistical significance. (D) Volcano plots showing the differential expression of selected inflammatory and fibrotic remodeling related molecules, derived from the enhanced communication signals between fibroblasts and other lung cell populations identified in Figure 2E, across fibroblast subpopulations between COPD and healthy lungs. |
To determine whether an inflammatory state was broadly shared across fibroblast subpopulations in COPD, we next performed Hallmark GSEA in each fibroblast subtype. Notably, multiple inflammation related pathways were consistently enriched in COPD across nearly all fibroblast subpopulations, including TNFα signaling via NF-κB, inflammatory response, IL6–JAK–STAT3 signaling, and interferon-γ response (Figure 4C). In addition, several subtypes also showed enrichment of pathways linked to stress adaptation and tissue remodeling, such as epithelial and mesenchymal transition, complement, TGFβ signaling, and angiogenesis, indicating that inflammatory activation was accompanied by broader remodeling related transcriptional changes.
To further substantiate the above findings, we examined the expression of fibroblast derived pro-inflammatory and pro-fibrotic mediators highlighted in Figure 2E across individual fibroblast subpopulations. In agreement with the Hallmark GSEA results, these mediators were broadly elevated across fibroblast subpopulations in COPD (Figure 4D), supporting the presence of a shared inflammatory state across distinct fibroblast populations. Together, these findings indicate that the inflammatory phenotype of fibroblasts in COPD is broadly distributed across subpopulations rather than confined to a single subset.
Defining COPD Associated Fibroblast States and Transcriptional Regulatory Changes
To identify fibroblast subpopulations most closely associated with COPD and explore the core regulatory mechanisms underlying their disease related properties, we characterized fibroblast heterogeneity at both the cellular and transcriptional levels. UMAP visualization showed that all eight fibroblast subpopulations were identifiable in both groups, although their relative distributions differed (Figure 5A). Consistently, compositional analysis revealed altered fibroblast subtype proportions in COPD, indicating substantial remodeling of fibroblast population composition under disease conditions (Figure 5B). Pseudotime analysis further showed that fibroblasts were distributed along a continuous trajectory and could be partitioned into multiple transcriptional states (Figure 5C). When Health and COPD cells were mapped onto the same trajectory, COPD fibroblasts were found to preferentially occupy specific branches and states rather than being evenly distributed across the continuum (Figure 5D and E). Notably, these COPD-enriched states were mainly associated with the Pro_ECM/Angiogenic, Perivascular, and Adventitial fibroblast populations, suggesting that these subpopulations may represent fibroblast programs most relevant to COPD pathogenesis.
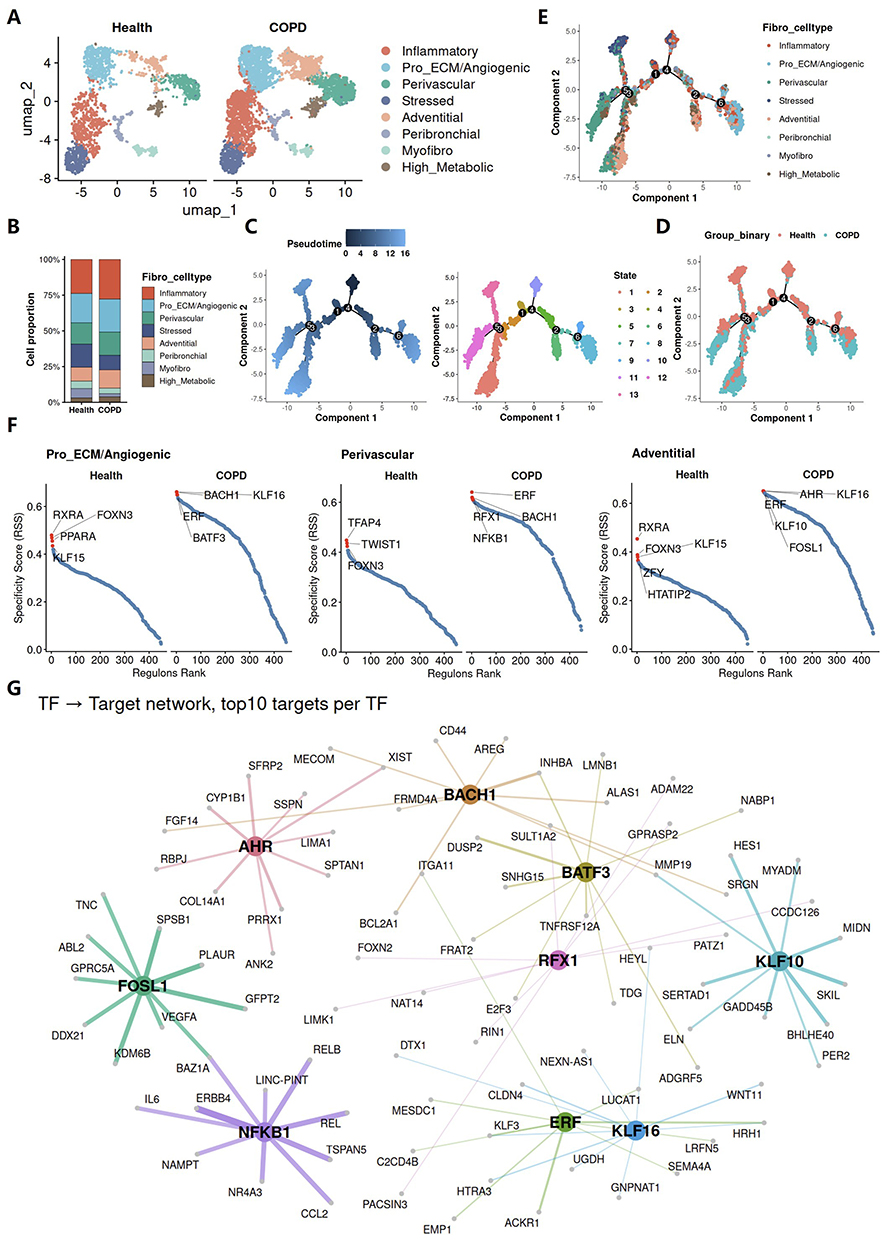 |
Figure 5 Defining COPD associated fibroblast states and transcriptional regulatory changes. (A) UMAP embeddings showing fibroblast subpopulation distributions in the Health and COPD groups. (B) Stacked bar plot showing the relative proportions of fibroblast subpopulations in the Health and COPD groups. (C–E) Monocle2 trajectory analysis of fibroblast subpopulations. (C) Trajectory plots colored by pseudotime and Monocle2-defined state. (D) Trajectory plot colored by condition in the Health and COPD groups. (E) Trajectory plot colored by fibroblast subpopulation. (F and G) SCENIC analysis of transcription factor regulatory networks in fibroblast subpopulations. (F) Regulon specificity score (RSS) ranking plots showing representative regulons in selected fibroblast subpopulations from healthy and COPD lungs. (G) Regulatory network from transcription factors to target genes, showing the top 10 target genes for each highlighted transcription factor. |
To further define the transcriptional basis of these COPD associated fibroblast populations, we compared regulon specificity scores (RSS) between the two groups. All three populations showed marked remodeling of transcriptional regulatory programs in COPD (Figure 5F). Among the top-ranking upregulated regulons were BACH1, KLF16, ERF, and BATF3 in Pro_ECM/Angiogenic fibroblasts; ERF, RFX1, BACH1, and NFKB1 in Perivascular fibroblasts; and AHR, KLF16, ERF, KLF10, and FOSL1 in Adventitial fibroblasts. These top-ranked COPD enriched transcription factors were then included in downstream regulatory network analysis. The resulting transcription factor (TF)–target network revealed extensive interconnections among these regulators (Figure 5G). Among these regulators, NFKB1 occupied a particularly central position in the inferred network, with multiple strong links to downstream targets. As a key component of the NF-κB family, NFKB1 is an important regulator of inflammatory and stress response transcriptional programs.22 Its marked activation in COPD fibroblasts further suggests that fibroblasts undergo pronounced intrinsic inflammatory remodeling in the disease setting. Overall, the transcriptional regulatory network of COPD fibroblasts was profoundly altered, indicating a marked state transition of fibroblasts toward an inflammatory phenotype.
Transcriptional Regulatory Changes in Fibroblasts of COPD are Associated with Activation of Inflammatory Signaling and Fibrotic Remodeling Pathways
Transcriptional regulatory analysis revealed substantial remodeling of the regulatory network in COPD fibroblasts. To further support this finding, we defined each TF together with its downstream target genes as an integrated regulatory module and calculated a module score for each cell to assess its activation status. Consistent with the above findings, the transcriptional regulatory networks activated in COPD fibroblasts also exhibited globally increased module scores (Figure 6A), further supporting their activation in the disease. Expression analysis across fibroblast subpopulations showed that these TFs were also broadly upregulated in COPD, although the magnitude of change varied among fibroblast states (Figure 6B). These results suggest that fibroblasts undergo extensive changes in their transcriptional regulatory networks, which may drive the acquisition of an inflammatory cell state.
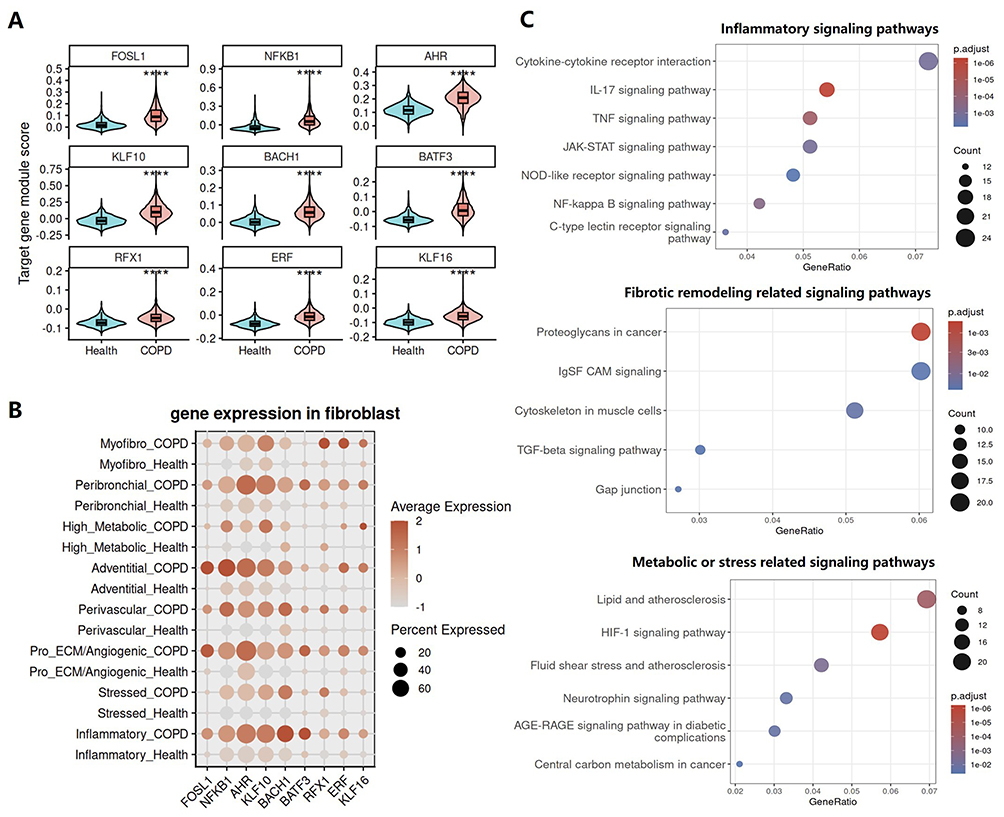 |
Figure 6 Transcriptional regulatory changes in fibroblasts of COPD are associated with activation of inflammatory signaling and fibrotic remodeling pathways. (A) Violin plots showing the target gene module scores of selected transcription factors in fibroblasts from the Health and COPD groups. (B) Dot plot showing the expression of the highlighted transcription factors across fibroblast subpopulations between the Health and COPD groups. (C) Functional enrichment analysis of FOSL1 target genes in fibroblasts, showing pathways related to inflammatory signaling, tissue remodeling, and altered metabolic or stress-response programs. Dot size indicates gene count, color indicates adjusted P value, and the x-axis indicates gene ratio. |
To further examine the association between transcriptional regulatory network remodeling and the broadly observed inflammatory state in COPD fibroblasts, we performed functional enrichment analysis of the target genes downstream of key transcription factors. Because FOSL1 showed particularly prominent activation across the inferred regulatory network, in addition to the canonical inflammatory regulator NFKB1, we selected FOSL1 as a representative example for downstream target gene enrichment analysis. FOSL1 (Fos-like 1, also known as Fra-1) is an important component of the AP-1 transcription factor complex and belongs to the FOS family.23 It typically forms heterodimers with JUN family proteins to bind DNA and thereby regulate the transcription of target genes.24 Current evidence suggests that FOSL1 plays distinct disease specific roles in processes ranging from tissue repair and immune cell differentiation to fibrotic remodeling, oncogenic reprogramming, and smoking related pathogenic responses.25–35 Accordingly, FOSL1 has been implicated in skin and cardiac injury repair, immune inflammatory disorders, fibrotic remodeling diseases, and multiple cancers. Despite these established roles in other disease contexts, its function in COPD remains largely unexplored. Functional enrichment analysis of FOSL1 target genes in COPD fibroblasts showed that their associated functions could be broadly classified into three major categories: inflammatory signaling, fibrotic remodeling, and metabolic or stress related pathways (Figure 6C). Taken together, these results suggest that remodeling of the transcriptional regulatory network in COPD fibroblasts is closely associated with their shift toward an inflammatory state.
Discussion
By integrating single cell RNA sequencing data with publicly available datasets, we generated a relatively comprehensive single cell atlas of the COPD lung. Using this single cell atlas as a framework, we systematically investigated the potential mechanisms underlying the persistence of chronic inflammation in COPD. Our analyses suggest that fibroblasts are not merely passive structural cells responding to fibrotic remodeling, but instead adopt an inflammatory state in COPD lungs that is associated with immune cell recruitment and activation. Regulatory network analysis further indicated that this inflammatory transition was accompanied by extensive remodeling of fibroblast transcriptional programs. Collectively, these findings suggest that chronic inflammation in COPD is not simply a consequence of persistent immune cell activation, but rather a tissue level inflammatory remodeling state that is actively maintained and amplified by structural cells, particularly fibroblasts.
One important question is why fibroblast subpopulations from different anatomical locations exhibited a shared inflammatory state in COPD. This may reflect a convergent response to the common disease microenvironment rather than a loss of anatomical identity. Although these fibroblast subsets occupy distinct niches, they are exposed to overlapping COPD related cues, including chronic inflammatory signaling, oxidative stress and hypoxia. This convergence may also reflect the context dependent plasticity of fibroblasts, whereby chronic injury may drive anatomically distinct fibroblast populations toward similar activated transcriptional programs. However, the specific upstream signals responsible for this shared state remain to be experimentally defined.
Compared with previous studies in COPD, an important strength of our study is that we moved beyond descriptive characterization of disease associated cell states and instead focused on the mechanisms that may sustain chronic inflammation in the COPD lung. Rather than viewing chronic inflammation primarily as a consequence of immune cell activation, our study highlights the inflammatory state of lung tissue as a whole. Within this framework, analysis of intercellular communication and regulatory networks identified fibroblasts as a central coordinating cell population. This finding broadens the role of fibroblasts in COPD, suggesting that they are not merely structural effector cells, but also important immunoregulatory cells within the lung microenvironment. Notably, this regulatory role was not restricted to a single fibroblast subset, but was instead reflected as a broadly shared state across multiple fibroblast subpopulations. Mechanistically, fibroblasts in COPD underwent substantial remodeling of transcriptional regulatory programs, indicating intrinsic reprogramming associated with disease pathology. Taken together, Our data suggests a model in which fibroblast inflammatory reprogramming contributes to sustaining the chronic inflammatory milieu; functional studies are required to test causality.
Although COPD is widely recognized as a chronic inflammatory disease, conventional anti-inflammatory therapies such as corticosteroids provide limited benefit in most patients, except in those with elevated eosinophil counts.36 Several mechanisms have been proposed to explain this corticosteroid insensitivity, including reduced glucocorticoid receptor (GR) expression in COPD neutrophils,37 decreased histone deacetylase 2 (HDAC2) activity that compromises GR-mediated repression of inflammatory gene transcription,38 and the inability of corticosteroids to effectively suppress bacteria induced macrophage IL-8 production and subsequent neutrophil recruitment.39 Current therapies, particularly bronchodilators, primarily provide symptomatic relief rather than interrupting the persistent inflammatory state that drives ongoing disease progression. Our findings offer a new perspective on this therapeutic challenge in COPD. Corticosteroid insensitivity may not be explained solely by abnormalities in immune cells, but may also reflect a central role for inflammatory states in parenchymal cells in disease pathogenesis. In this context, molecular studies targeting parenchymal cells, particularly fibroblasts, may open new avenues for the development of more effective therapies for COPD.
Previous studies have demonstrated heterogeneity among fibroblast subpopulations in COPD and linked distinct fibroblast subsets to disease progression.40,41 Building on these observations, our study further shows that, beyond fibroblast heterogeneity, fibroblast subpopulations in human COPD lung tissue commonly acquire a shared proinflammatory and immunoregulatory state. This shared state was characterized by increased expression of proinflammatory mediators, extensive interactions with both innate and adaptive immune cells, and activation of profibrotic signaling pathways. These findings extend the emerging concept that lung fibroblasts are not merely structural cells but can also function as immunoregulatory cells involved in COPD progression.8 More importantly, our study provides insight into the potential transcriptional regulatory mechanisms underlying this shared fibroblast state, thereby offering concrete directions for subsequent mechanistic studies and potential therapeutic translation.
Several limitations of this study should be acknowledged. First, our conclusions are primarily based on single cell transcriptomic profiling and computational inference, including analyses of cell–cell communication and transcriptional regulatory networks. Although these approaches provide systematic and multidimensional evidence supporting the proposed role of fibroblasts in COPD, the inferred molecular interactions and regulatory mechanisms still require further experimental validation. Second, because single cell RNA sequencing does not directly capture spatial context or protein level activity, the spatial organization and functional relevance of the predicted signaling interactions remain to be confirmed. Third, potential confounding by comorbidities could not be fully excluded. Some patients with COPD had concomitant conditions, including lung tumors, cardiovascular diseases, or cerebrovascular diseases, which may influence disease progression and lung tissue responses. Because comorbidity information was incomplete in public datasets and the number of locally collected samples with detailed clinical information was limited, we could not formally assess or adjust for these effects in the current study. Future studies with larger clinically annotated cohorts are needed to better control for comorbidities and distinguish COPD related fibroblast transcriptional states and phenotypic changes from those driven by comorbid conditions. Nevertheless, our findings provide a comprehensive molecular framework for understanding fibroblast inflammatory reprogramming in COPD and offer specific directions for future mechanistic and functional studies.
Conclusions
In summary, our study suggests that chronic inflammation in COPD is not simply a problem of persistent immune cell activation, but rather a tissue level inflammatory remodeling state that may be actively maintained and amplified by structural cells, particularly fibroblasts. By integrating single cell transcriptomic data and multiple analytical strategies, we identify fibroblasts as central coordinators of the local inflammatory microenvironment in COPD and show that this role is associated with a broadly shared inflammatory state across fibroblast subpopulations and extensive remodeling of transcriptional regulatory programs. These findings not only broaden the current understanding of fibroblast function in COPD, but also provide a conceptual basis for future studies targeting fibroblast associated mechanisms in an effort to better control chronic inflammation and disease progression.
Data Sharing Statement
The datasets generated and/or analyzed during this study are not publicly available due to institutional and patient privacy restrictions. However, the data can be made available from the corresponding author upon reasonable request and with appropriate institutional approvals.
Ethics Approval and Informed Consent
This study was approved by the Institutional Review Board of Zhejiang Provincial People’s Hospital (Approval No. 2025(291)) and was conducted in accordance with the Declaration of Helsinki and all applicable ethical guidelines and regulations governing research involving human participants. Informed consent was obtained from all subjects involved in the study.
Consent for Publication
Not applicable, as this study did not include any identifiable individual person’s data, images, videos, or recordings for publication.
Acknowledgments
The graphical abstract and Figure 1A were created with BioRender.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the sub-project of the National Key Science and Technology Major Project of China (2025ZD0549101 to Yilan Sun) and the Youth Project of National Natural Science Foundation of China (82400006 to Yao Ye).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for Prevention, Diagnosis, and Management of COPD: 2026 Report. Global Initiative for Chronic Obstructive Lung Disease (GOLD); 2026.
2. Chen S, Kuhn M, Prettner K, et al. The global economic burden of chronic obstructive pulmonary disease for 204 countries and territories in 2020–50: a health-augmented macroeconomic modelling study. Lancet Glob Health. 2023;11(8):e1183–19. doi:10.1016/s2214-109x(23)00217-6
3. Kheradmand F, Zhang Y, Corry DB. Contribution of adaptive immunity to human COPD and experimental models of emphysema. Physiol Rev. 2023;103(2):1059–1093. doi:10.1152/physrev.00036.2021
4. Conlon TM, John-Schuster G, Heide D, et al. Inhibition of LTβR signalling activates WNT-induced regeneration in lung. Nature. 2020;588(7836):151–156. doi:10.1038/s41586-020-2882-8
5. Zhang Y, Wei H, Nouws J, et al. Aberrant cellular communities underlying disease heterogeneity in chronic obstructive pulmonary disease. Nat Genet. 2026;58(2):376–391. doi:10.1038/s41588-025-02480-z
6. Hadzic S, Wu CY, Gredic M, et al. Fibroblast growth factor 10 reverses cigarette smoke- and elastase-induced emphysema and pulmonary hypertension in mice. Eur Respir J. 2023;62(5):2201606. doi:10.1183/13993003.01606-2022
7. El Agha E, Thannickal VJ. The lung mesenchyme in development, regeneration, and fibrosis. J Clin Invest. 2023;133(14). doi:10.1172/jci170498
8. Ghonim MA, Boyd DF, Flerlage T, Thomas PG. Pulmonary inflammation and fibroblast immunoregulation: from bench to bedside. J Clin Invest. 2023;133(17). doi:10.1172/jci170499
9. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–2242. doi:10.1016/s0140-6736(22)00470-6
10. Liu G, Philp AM, Corte T, et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Ther. 2021;225:107839. doi:10.1016/j.pharmthera.2021.107839
11. Finney LJ, Mah J, Duvall M, et al. Select airway specialized proresolving mediators are associated with recovery from nonviral chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med. 2025;211(5):803–813. doi:10.1164/rccm.202407-1325OC
12. Winslow S, Odqvist L, Diver S, et al. Multi-omics links IL-6 trans-signalling with neutrophil extracellular trap formation and Haemophilus infection in COPD. Eur Respir J. 2021;58(4):2003312. doi:10.1183/13993003.03312-2020
13. Wu YF, Li ZY, Dong LL, et al. Inactivation of MTOR promotes autophagy-mediated epithelial injury in particulate matter-induced airway inflammation. Autophagy. 2020;16(3):435–450. doi:10.1080/15548627.2019.1628536
14. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
15. Yuan X, Shan M, You R, et al. Activation of C3a receptor is required in cigarette smoke-mediated emphysema. Mucosal Immunol. 2015;8(4):874–885. doi:10.1038/mi.2014.118
16. Calderon AA, Dimond C, Choy DF, et al. Targeting interleukin-33 and thymic stromal lymphopoietin pathways for novel pulmonary therapeutics in asthma and COPD. Eur Respir Rev. 2023;32(167):220144. doi:10.1183/16000617.0144-2022
17. Rabe KF, Celli BR, Wechsler ME, et al. Safety and efficacy of itepekimab in patients with moderate-to-severe COPD: a genetic association study and randomised, double-blind, phase 2a trial. Lancet Respir Med. 2021;9(11):1288–1298. doi:10.1016/s2213-2600(21)00167-3
18. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. doi:10.1056/NEJMoa032158
19. Cambier S, Gouwy M, Proost P. The chemokines CXCL8 and CXCL12: molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell Mol Immunol. 2023;20(3):217–251. doi:10.1038/s41423-023-00974-6
20. Wu X, Qian L, Zhao H, et al. CXCL12/CXCR4: an amazing challenge and opportunity in the fight against fibrosis. Ageing Res Rev. 2023;83:101809. doi:10.1016/j.arr.2022.101809
21. Dupin I, Allard B, Ozier A, et al. Blood fibrocytes are recruited during acute exacerbations of chronic obstructive pulmonary disease through a CXCR4-dependent pathway. J Allergy Clin Immunol. 2016;137(4):1036–1042.e7. doi:10.1016/j.jaci.2015.08.043
22. Song J, Han S, Amaru R, et al. Alternatively spliced NFKB1 transcripts enriched in Andean Aymara modulate inflammation, HIF and hemoglobin. Nat Commun. 2025;16(1):1766. doi:10.1038/s41467-025-56848-0
23. Chen Z, Wang S, Li HL, et al. FOSL1 promotes proneural-to-mesenchymal transition of glioblastoma stem cells via UBC9/CYLD/NF-κB axis. Mol Ther. 2022;30(7):2568–2583. doi:10.1016/j.ymthe.2021.10.028
24. Nimmakayala RK, Seshacharyulu P, Lakshmanan I, et al. Cigarette smoke induces stem cell features of pancreatic cancer cells via PAF1. Gastroenterology. 2018;155(3):892–908.e6. doi:10.1053/j.gastro.2018.05.041
25. Liu Z, Bian X, Luo L, et al. Spatiotemporal single-cell roadmap of human skin wound healing. Cell Stem Cell. 2025;32(3):479–498.e8. doi:10.1016/j.stem.2024.11.013
26. Huang CK, Dai D, Xie H, et al. Lgr4 governs a pro-inflammatory program in macrophages to antagonize post-infarction cardiac repair. Circ Res. 2020;127(8):953–973. doi:10.1161/circresaha.119.315807
27. Benhadou F, Glitzner E, Brisebarre A, et al. Epidermal autonomous VEGFA/Flt1/Nrp1 functions mediate psoriasis-like disease. Sci Adv. 2020;6(2):eaax5849. doi:10.1126/sciadv.aax5849
28. Shetty A, Tripathi SK, Junttila S, et al. A systematic comparison of FOSL1, FOSL2 and BATF-mediated transcriptional regulation during early human Th17 differentiation. Nucleic Acids Res. 2022;50(9):4938–4958. doi:10.1093/nar/gkac256
29. Buquicchio FA, Fonseca R, Yan PK, et al. Distinct epigenomic landscapes underlie tissue-specific memory T cell differentiation. Immunity. 2024;57(9):2202–2215.e6. doi:10.1016/j.immuni.2024.06.014
30. Nayakanti SR, Friedrich A, Sarode P, et al. Targeting Wnt-ß-Catenin-FOSL signaling ameliorates right ventricular remodeling. Circ Res. 2023;132(11):1468–1485. doi:10.1161/circresaha.122.321725
31. Paul S, Hagenbeek TJ, Tremblay J, et al. Cooperation between the Hippo and MAPK pathway activation drives acquired resistance to TEAD inhibition. Nat Commun. 2025;16(1):1743. doi:10.1038/s41467-025-56634-y
32. Dong J, Li J, Li Y, Ma Z, Yu Y, Wang CY. Transcriptional super-enhancers control cancer stemness and metastasis genes in squamous cell carcinoma. Nat Commun. 2021;12(1):3974. doi:10.1038/s41467-021-24137-1
33. Braun S, Bolivar P, Oudenaarden C, et al. Pericytes orchestrate a tumor-restraining microenvironment in glioblastoma. Nat Commun. 2025;16(1):10918. doi:10.1038/s41467-025-66985-1
34. Vallejo A, Erice O, Entrialgo-Cadierno R, et al. FOSL1 promotes cholangiocarcinoma via transcriptional effectors that could be therapeutically targeted. J Hepatol. 2021;75(2):363–376. doi:10.1016/j.jhep.2021.03.028
35. Shi J, Wang X, Tang Y, et al. LY6D identifies persistent stem-like cells driving pancreatic tumourigenesis. Gut. 2026:gutjnl–2025–336460. doi:10.1136/gutjnl-2025-336460
36. Leung C, Park HY, Li X, et al. Transcriptomic profiling of the airway epithelium in COPD links airway eosinophilia to type 2 inflammation and corticosteroid response. Eur Respir J. 2025;65(5):2401875. doi:10.1183/13993003.01875-2024
37. Plumb J, Gaffey K, Kane B, et al. Reduced glucocorticoid receptor expression and function in airway neutrophils. Int Immunopharmacol. 2012;12(1):26–33. doi:10.1016/j.intimp.2011.10.006
38. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131(3):636–645. doi:10.1016/j.jaci.2012.12.1564
39. Lea S, Higham A, Beech A, Singh D. How inhaled corticosteroids target inflammation in COPD. Eur Respir Rev. 2023;32(170):230084. doi:10.1183/16000617.0084-2023
40. Eapen MS, Lu W, Hackett TL, et al. Increased myofibroblasts in the small airways, and relationship to remodelling and functional changes in smokers and COPD patients: potential role of epithelial-mesenchymal transition. ERJ Open Res. 2021;7(2):00876–2020. doi:10.1183/23120541.00876-2020
41. Yun JH, Lee C, Liu T, et al. Hedgehog interacting protein-expressing lung fibroblasts suppress lymphocytic inflammation in mice. JCI Insight. 2021;6(17). doi:10.1172/jci.insight.144575
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Pathogenic Cell in COPD: Mechanisms of Airway Remodeling, Immune Dysregulation, and Therapeutic Implications
Zhou K, Wen Q, Zuo Y, Bai G, Sun R
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:2925-2943
Published Date: 21 August 2025