Back to Journals » ImmunoTargets and Therapy » Volume 13
A Self-Activating IL-15 Chimeric Cytokine Receptor to Empower Cancer Immunotherapy
Authors Chen S ![]() , Yang L, Xia B, Zhu H, Piao Z
, Yang L, Xia B, Zhu H, Piao Z ![]() , Jounaidi Y
, Jounaidi Y ![]()
Received 7 August 2024
Accepted for publication 4 October 2024
Published 10 October 2024 Volume 2024:13 Pages 513—524
DOI https://doi.org/10.2147/ITT.S490498
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Sumei Chen,1,* Lingrong Yang,2,* Bing Xia,3 Haitao Zhu,4 Zhenghao Piao,5 Youssef Jounaidi6
1Department of Radiation Oncology, Hangzhou Cancer Hospital, Hangzhou, Zhejiang, 310002, People’s Republic of China; 2Department of Hangzhou Cancer Institution, Hangzhou Cancer Hospital, Hangzhou, Zhejiang, 310002, People’s Republic of China; 3Department of Thoracic Oncology, Hangzhou Cancer Hospital, Hangzhou, Zhejiang, 310002, People’s Republic of China; 4Department of Hepatobiliary Surgery, The Affiliated Hospital of Guizhou Medical University, Guiyang, Guizhou, 550001, People’s Republic of China; 5Department of Basic Medical Science, Hangzhou Normal University, Hangzhou, Zhejiang, 311121, People’s Republic of China; 6Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA, 02114, USA
*These authors contributed equally to this work
Correspondence: Youssef Jounaidi, Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital, Harvard Medical School, 55 Fruit Street, Boston, MA, 02114, USA, Email [email protected]
Background: Enhancing NK cells’ antitumor activity requires sustained cytokine signaling. Interleukin-15 (IL-15) is a potent immunostimulatory cytokine used to armor CAR-NK and CAR-T cell immunotherapies. However, strategies to increase IL-15 expression and antitumor effect may trigger systemic toxicity with the potential to promote oncogenesis and autoimmune diseases.
Methods: To overcome these limitations, we developed a new platform (IL15RB) whereby IL-15 with IL-2 signal peptide is tethered to its receptor, IL2Rβ.
Results: NK92-expressing IL15RB (NK92IL15RB) cells expand indefinitely without exogenous cytokines and have significantly higher anticancer activity than NK-92 stimulated by IL-15, IL-2, or expressing tethered IL-2. NK92IL5RB showed resistance to irradiation and IL-4. However, TGFβ 1 substantially reduced NK92IL5RB killing, suggesting the need to inhibit TGFβ 1 in IL-15-mediated immunotherapies. IL15RB induced strong STAT3 but weaker STAT5 and STAT1 activation compared to IL-2. Chronic exposure of NK92IL15RB cells to cancer cells reduced STAT3 and STAT1 activation irreversibly, suggesting a role in exhaustion. Combination with CAR-CD19 enhanced NK92IL15RB antitumor activity against leukemia and increased its STAT5 activation. NK92IL15RB anti-tumors activity was further enhanced by combination with anti-PD1.
Conclusion: Our data suggest that the tethering of IL-15 to its receptor IL2Rβ empowers NK cell cytolytic activity. Additionally, the tethering of IL-15 will prevent any systemic risk of toxicity.
Keywords: IL-15, IL2RB receptor, natural killer cells, cancer immunotherapy, IL15RB
Introduction
NK cells mediate potent anti-tumor activity without prior exposure to specific antigens.1–3 They attack tumors in an MHC-independent manner, clearing cancer cells that antigen-specific T cells cannot target.4 However, enhancing NK cell cytotoxicity and persistence in vivo is still challenging since NK cells are inhibited and exhausted by multiple mechanisms in the tumor microenvironment (TME). Cytokine-based approaches have been widely used to increase NK cell functions by multiple interleukins that activate NK cells. Among them, IL-15 is emerging as a potent immunostimulatory cytokine to augment the anti-tumor effects of NK cells. IL-15 is a better substitute for IL-2 due to their overlapping functions with the advantage of not stimulating regulatory T cells (Treg). IL-15 directly supports NK cell development in vivo. However, it is poorly translated, processed, or secreted when expressed in its original form and is mostly trans-presented by dendritic cells DC.5 Indeed, the mature NK population collapses in vivo when dendritic cells are depleted.
IL-15 substantially improved CB-NK cells especially when combined with the knockout of the CIS gene expressing soluble IL-15.6 One Phase 1/2 clinical trial using cord blood NK cells engineered to express IL-15 and a chimeric antigen receptor (CAR) against CD19 showed long-term persistence and potent antitumor activity. Secreted IL-15 is currently used to improve CAR-NK and CAR-T immunotherapies. Enhancing NK cells’ antitumor effects requires sustained IL-15 exposure. However, sustaining soluble IL-15 is difficult due to its short serum half-life (less than 1h)7 and the cytokine “sink effect” due to subsequent NK and CD8 T-cell expansion.8 Increasing systemic IL-15 is constrained by its adverse effects, such as systemic toxicity,9 hypotension, thrombocytopenia, and the potential to promote oncogenesis.10 Additionally, reports suggest that secreted IL-15 can help expand primary and CB-NK cells but causes severe9 to lethal toxicity and cytokine release syndrome in animal models.7 This toxicity depends on the dose of secreted IL-15, which will increase with proliferation and persistence. A recent clinical trial shows that activation of primary NK cells with an aAPC trans-presenting IL-15 can unexpectedly lead to graft versus host disease by NK cells.10 Additionally, it is long established that overexpression of IL-15 causes LGL leukemia with either an NK cell or TNK cell phenotype,11 and mice overexpressing mutated HMGI-C express excessive IL-15 that causes NK lymphoma.12 Strategies to increase IL-15 stability and antitumor effect may also trigger autoimmune diseases.13,14 In mice, IL-15-pre-stimulated NK cells were hyporesponsive to a second cycle of IL-15 stimulation.15 To overcome these limitations, we developed a new platform (IL15RB) with contained and sustained IL-15 signaling by tethering IL-15 to receptor IL2Rβ. We investigated whether IL15RB supports NK cell survival, proliferation, and cytotoxicity. Our study suggests that after prolonged exposure to cancer cells, signal transducer and activator of transcription STAT1 and STAT3 activations are reduced, without recovery except for STAT5. NK92IL15RB cytolytic activity is enhanced by the addition of CAR-CD19, but inhibition by immunosuppressive factors such as transforming growth factor TGFβ1 will need to be addressed. Together, our data suggest that tethering IL-15 to its receptor IL2Rβ may empower NK cell-mediated immunotherapies without the potential toxicities associated with soluble IL-15.
Materials and Methods
Reagents
Chloroquine, Matrigel (cat# 126–2.5) (Sigma-Aldrich Co). Horse serum (HS), DMEM/F12, Lipofectamine 2000 and TRIzol (Life Technologies). Fetal bovine serum (FBS) (Atlanta Biologicals). RPMI 1640 (LONZA). Smartscribe and Blueprint Onestep RT-PCR Takara kit (Clontech Laboratories); Platinum SYBR Green qPCR (Invitrogen), PfuUltra DNA polymerase (Stratagene). Human TGFβ1 (Antigenix America Inc). IL-2 was from MGH-DF/HCC Recombinant Protein Core (Boston, MA). Human IL-4, IL-7, and IL-21 (Shenandoah Biotechnology Inc), Human IL-15 (STEMCELL Technologies Inc), CyQUANT™ LDH Cytotoxicity Assay Kit (Invitrogen Biotechnology Inc).
Cells
HEK-293T, NK-92, PC-3, MDA-MB-231, BT474, KYSE-150, NCI-H1975, TE-1, A2780, cells were from ATCC. Tumor cell lines were cultured in complete RPMI-1640 medium and HEK-293T in complete DMEM/F12. NK-92 and derived cell lines were grown in RPMI-1640 as described earlier16 with 100 IU/mL IL-2. All cell lines and assay cultures were maintained at 37°C and 5% CO2. Cell lines were used for a maximum of twenty passages. Monitoring for mycoplasma contamination was done using MycoFluor mycoplasma detection kit (Molecular Probes).
Chimera IL15RB construction
IL-15 transcript variant 2, mRNA NM_172175.3 was cloned from a human brain mRNA library using primers: IL-15 sense BamHI ON1052, TGCAGGATCCACCTAATGCCTTCATGGTATTGGG; and IL-15 reverse XhoI ON1053, TGCACTCGAGGAATCAATTGCAATCAAGAAGTG.
IL15RB cloning adopted the same design we previously reported for tethered IL-2 to IL2Rβ17 by the tethering of IL-15 to C-myc tag (EQKLISEEDL), followed by IL2Rα extracellular shaft (EMETSQFPGEEKPQASPEGRPESETSC), and IL2Rβ mature protein. IL-15 was fused to IL2Rβ by PCR extension using bridge oligo between IL-15 and IL2Rβ (c-myc-shaft of IL-2 Rα): included in oligos ON1066 and ON1067.IL-15 fragment was amplified using forward ON1052, TGCAGGATCCACCTAATGCCTTCATGGTATTGGG, and reverse ON 1067, GGTCTTCTTCCGAAATGAGCTTCTGCTCAGAAGTGTTGATGAACATTTGGAC.
To amplify Cmyc-IL2Rα-IL2Rβ fragment, we used clone CIRB17 as a template using ON 1066 sense (at the junction between Il-15 and c-myc), GTCCAAATGTTCATCAACACTTCT GAGCAGAAGCTCATTTCGGAAGAAGACC, and reverse ON854 (3’end of IL2Rβ), AGCTTCTAGACTCGAGTTATCACACCAAGTGAGTTGGGTCCTGACCCTGG. However, neither IL-15 cloned from the brain nor its derivative IL15RB did provide survival to NK-92 stably expressing these forms. The reason for this lack of expression is the non-cleavage of IL-15 signal peptide. To allow correct processing of IL-15, the signal peptide of IL-15 was replaced by the signal peptide from interleukin-2 (IL2sp), which includes the cleavage site TNSAP. IL2sp: (MYRMQLLSCIALSLALVTNSAP) was added by PCR to the IL-15 mature sequence. NWVNVISDLKKIEDLIQSMHIDATLYTESDVHPSCKVTAMKCFLLELQVISLESGDASIHDTVENLIILANNSLSSNGNVTESGCKECEELEEKNIKEFLQSFVHIVQMFINTS.
The addition of IL2sp was done using IL15RB as a template in two PCR steps using in the first step IL2sp/IL-15 junction oligo #1 CATTGCACTAAGTCTTGCACTTGTCACAAACAG TGCACCTAACTGGGTGAATGTAATAAG and vector reverse primer CTCACATTGCCAAAAGACGGC. In the second step, IL2sp/IL-15 junction was added using oligo #2 CATTGCACTAAGTCTTGCACTTGTCACAAACAGTGCACCTAACTGGGTGAATGTAATAAG, and vector reverse CTCACATTGCCAAAAGACGGC, then cloned in lentiviral vector CSCW mcherry at NheI-XhoI. All constructs were verified by sequencing.
Lentivirus production and NK cell lines transduction
HEK-293T cells were transfected using Lipofectamine 2000 with 2.4 μg total DNA of pVSV, (Clontech Laboratories), pCMVdr8.2dvrp Addgene (plasmid # 8455) and the lentiviral construct CSCW-GFP or CSCW-mCherry vectors MGH Vector Core (Boston, MA) to express either CIRB or IL15RB, using the ratios 1: 0.4: 1, respectively. Six hours post-transfection, the media was changed, and lentiviral supernatant was collected 3 days later and filtered through a 0.45um PES filter syringe. NK-92 cells were infected by spinoculation at 1800g for 45 minutes at room temperature using a multiplicity of infection (MOI) of 46 lentiviral particles per cell in a 2mL-Eppendorf tube containing 2×105 cells. Infected cells were plated with IL-2 (100IU/mL) for 2 days and weaned of exogenous IL-2 after that.
Western Blot
Phosphorylated STAT1, 3, and 5 were detected using rabbit anti-Stat5 Phospho (Tyr694 used at 1:500) antibody, purified mouse anti-Stat3 Phospho (Tyr705 used at 1:1000), and purified mouse anti-Stat1 Phospho (Ser727 used at 1:1000), all from BioLegend. IL-15 was detected using polyclonal anti-human IL-15 (PeproTech, Rocky Hill, NJ). Secondary antibodies used are donkey anti-Mouse IRDye 800CW and goat anti-Rabbit IRDye 680RD antibodies obtained from Li-Cor, Biosciences. All antibodies were used as specified by the manufacturer.
Flow cytometry
NK cell markers expression was verified using mouse anti-human antibodies to CD25-APC, CD56- PE-CY7 and CD122-PE, NKP30-PE, NKP44-PE-CY7, NKP46-FITC, Granzyme B-FITC, Perforin-PE, Interferon-γ-APC, TNF-α- APC-CY7 and CD107- PE-CY7 were from BioLegend, DAPI from Invitrogen. Cells were sorted at the MGH Flow Cytometry Core facility using a BD 5 laser SORP FACS Vantage SE Diva system (BD Biosciences) and Hangzhou cancer hospital CytoFLEX benchtop flow cytometer 4 lasers for 9-color research flow cytometry (Beckman Coulter Life Sciences). FACS data and ∑Median statistics were analyzed using the FlowJo 10.8.1 software (Tree Star, Inc).
Cytotoxicity of NK cells
NK-92 cell lines’ anti-cancer effect was evaluated against prostate cancer (PC-3), breast cancer (MDA-MB-231 and BT474), esophageal squamous cell carcinoma (KYSE-150 and TE-1), ovarian cancer (A2780) and non-small cell lung cancer cells (NCI-H1975). PC-3 was first plated 24 hours prior to adding NK-92 (pre-stimulated with IL-15 for 48 hours, 10ng/ml), NK92IL2sp15 expressing untethered IL2sp15 or NK92IL15RB and co-cultured NK cells, and cancer cells at 0.5 E/T ratio for two or 4 days. PC-3, MDA-MB-231, KYSE-150, and NCI-H1975 cells (32x103), BT474 (64x103), TE-1, and A2780 cells (50x103) were first plated in triplicate in 24-well plate, 24 hours prior to adding NK-92 (pre-stimulated with IL-2 for 48 hours, 100IU/ml), NK92CIRB and NK92IL15RB at the indicated E/T ratio. Co-cultured cells were then incubated for 2 or 4 days. The viability of cancer cells was determined using a 0.2% crystal violet in a 2% alcohol solution, followed by extraction using 70% ethanol and an absorbance reading at 562nm. Alternatively, cell viability and growth were determined with Trypan blue exclusion using a Luna II automated cell counter. Leukemia cell viability was determined using CyQUANT™ LDH Kit.
Impact of Pre-Exposure to Immunosuppressors on NK Cells Cytotoxicity and Viability
To determine the impact of immunosuppressors TGFβ1 on the growth of NK92IL15RB (75x103 cells), cells were plated in triplicate in a 6-well plate, grown under TGFβ1 (10ng/mL) for 3 days, NK92IL15RB cells (32x103 cells) were plated in triplicate in a 12-well plate, grown under IL-4 (10ng/mL, 20ng/mL, and 30ng/mL) for 4 days, Cell viability and growth were determined with Trypan blue exclusion using a Bio-Rad TC20TM automated cell counter.
Separately, NK92IL15RB (75x103 cells) were plated and exposed for 24 hours to IL-4 (10ng/mL). PC-3 cells (32x103 cells) were then added to IL-4 treated NK cells at an E/T ratio of 2:1. Co-cultured cells were incubated for 4 days, and cell viability of cancer cells was determined using crystal violet staining.
Survival and cytotoxicity of irradiated NK92IL15RB and NK92 cells pre-exposed to IL-15
To compare the viability of NK-92 stimulated by IL-15 and NK92IL15RB after irradiation, NK-92 cells were pretreated with IL-15 (10ng/mL) for 2 days then irradiated 10Gy (0.83Gy for 12 min), NK cells were then cultured, and their survival was determined using Trypan Blue exclusion every 24 hours for 3 days. Irradiated NK cell lines’ anti-cancer effect was evaluated against PC-3. Cancer cells (32x103) were first plated in triplicate in 24-well plate 24 hours prior to adding irradiated NK92IL15RB or NK-92 cells (pre-exposed to IL-15) after 8Gy X-ray irradiation at the indicated E/T ratio. Co-cultured cells were then incubated for 2 days. The killing of cancer cells was determined using crystal violet, as described earlier.
IL15RB Synergy with CAR-CD19 Against CD19-Positive Leukemia Cells
CD19-negative erythroleukemia cell-line K562 cells or CD19-positive B cell precursor acute leukemia cell line Nalm-6 (104 cells/well in 100μL of medium) were plated in triplicate wells in a 96-well plate overnight. NK92IL15RB and CD19-CAR-NK92IL15RB cells (2x104) were then added and co-cultured at 37°C with 5% CO2 for 5 hours. Cytotoxicity was determined using CyQUANT™ LDH kit, as per manufacturer instructions.
Tumor Growth Delay Experiments
All animal care and procedures were performed according to protocols reviewed and approved by Zhejiang Chinese Medical University Institutional Animal Care and Use Committee (IACUC), from which ethical and legal approval was obtained prior to the commencement of the study (approval number 20231030–04). All experiments were performed following Zhejiang Chinese Medical University institutional and national guidelines and regulations. Hangzhou Cancer Hospital does not have an animal center that meets the requirements for NCG mice. Therefore, we placed the animals in the Animal Experiment Center of Zhejiang Chinese Medical University, Hangzhou, from which the ethical and legal approval was obtained and which provided mouse accommodation, food, water, and mouse intravenous injection services. PC-3 cells were suspended in serum-free RPMI-1640 containing 15% Matrigel and injected subcutaneously (s.c). as 4×106 PC-3 cells in a volume of 0.2 mL using a 0.5-inch 29-gauge needle and a 1 mL insulin syringe in 7-week-old (24–25g) male NCG (NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt) mice (GemPharmatech, Jiangsu, China). Tumor areas (length x width) were measured twice a week using Vernier calipers (Manostat Corp., Switzerland), and tumor volumes were calculated based on: Volume = π/6 (length x width)3/2. Treatment with NK-92 or NK92IL15RB alone or in combination with anti-PD1 (Nivolumab) was initiated when the average tumor volume reached ~150 mm3. NK cells were irradiated with 1000cGy, suspended in PBS and administered via the tail vein as four-weekly injections (5x106 cells in 200ul per mouse). Anti-PD1 (150ug/25g/mouse) from Selleck chemicals LLC, was injected i.p. immediately after NK cells injection. The same treatment was repeated a second time 6 days later after the first NK cell injection.
Statistical Analysis
Statistical significance of differences was determined by a two-tailed Student’s test and one-way ANOVA, paired Tukey’s Multiple Comparison test. All tests included comparisons to untreated samples or as indicated in the text. Statistical significance is indicated by *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Analyses were performed using Prism software version 8 (GraphPad Software).
Results
Design, Construction, and Cell Surface Expression of the IL15RB Chimera
We previously reported the CIRB platform tethering IL-2 to its receptor IL2Rβ and its ability to enhance NK cell activation and proliferation without the cytotoxic effect of exogenous IL-2.17 We sought to further improve this platform by novel signaling from other potent cytokines. We first tested the impact of multiple cytokines on the viability of NK-92. In a two-day period, IL-2 at 50IU/mL increased NK-92 proliferation by 3.6 folds followed by IL-15 (10IU/mL) ~2.8 folds (Figure 1A). IL-21 (10IU/mL) marginally increased NK-92 proliferation, while IL-7 (10IU/mL) did not. However, IL-15 enhanced NK-92 cytotoxicity against PC-3 more than when stimulated by IL-2 (Figure 1B). Therefore, we chose IL-15 to build the chimera IL15RB by tethering IL-15 to receptor IL2Rβ with an intervening C-myc tag and IL2Rα (CD25) extracellular domain linker (Figure 1C). However, native IL-15 variant 2, cloned from human brain RNA and introduced via lentivirus was not able to sustain NK-92 proliferation suggesting improper processing/translation of IL-15 in NK-92 cells. We resolved this issue by the introduction of IL-2 signal peptide (IL2sp: MYRMQLLSCIALSLALVTNSAP) instead of IL-15 native signal peptide. This allowed NK-92 survival and proliferation in the absence of cytokines. We also introduced IL2sp in IL2sp15RB fusion proteins (hereafter called IL15RB). As expected, NK92IL15RB cells express the chimera IL15RB at the cells surface, with an estimated size of ~95 kDa, detected in cell membrane preparation using an anti-IL-15 antibody (Figure 1D). The expression of chimeric IL15RB at the cell surface was also confirmed by flow cytometry using anti-IL2Rβ, which can detect both the endogenous IL2Rβ and the chimeric IL15RB form (Figure 1E). Due to IL15RB expression, NK92IL15RB express higher IL2Rβ than NK-92. NK92IL15RB and NK-92 have comparable CD56 expression. However, NK92IL15RB showed higher CD25 expression than NK-92 (Figure 1E). This result is in clear contrast with our prior observation that NK-92 expressing tethered IL-2 to receptor IL2Rβ showed downregulated CD25.17
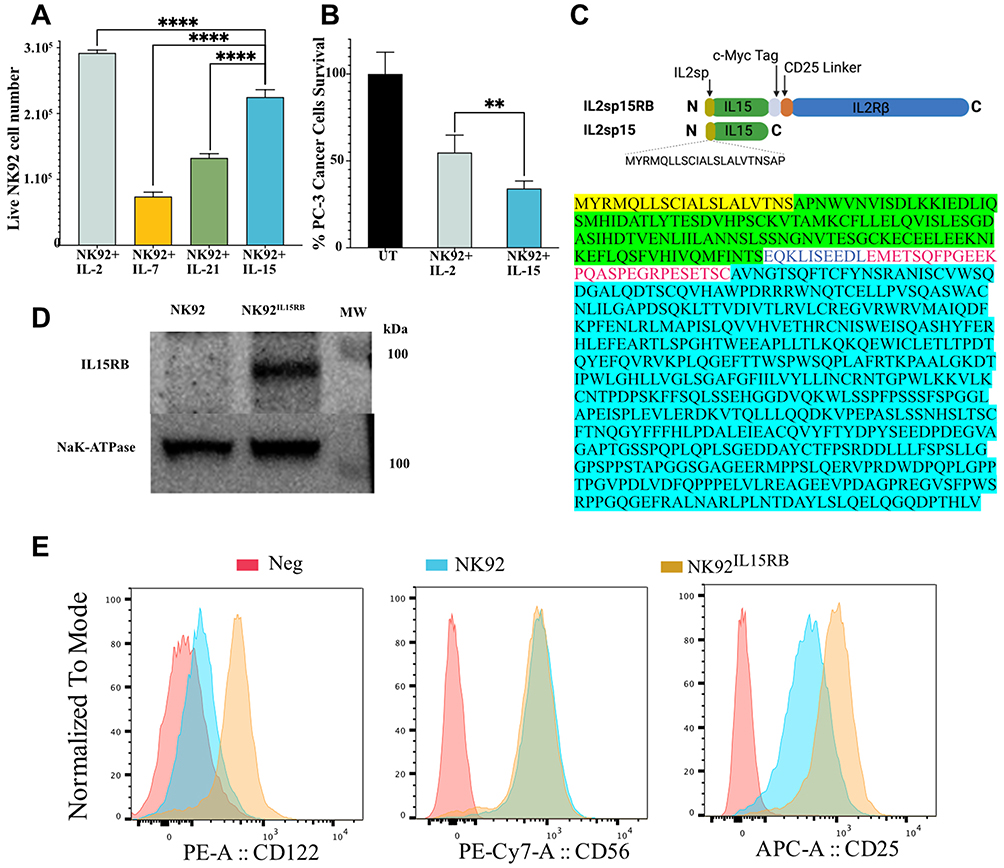 |
Figure 1 Tethering IL-15 to IL2Rβ. (A), IL-2 (50IU/mL) drives stronger proliferation of NK-92 cell within 48h, followed by IL-15 (10ng/mL), IL-21 (10ng/mL) and IL7 (10ng/mL) (n=3). (B), NK-92 cells pre-stimulated by soluble IL-15 (10ng/mL) for 48h are more cytotoxic to PC-3 cells than when stimulated by IL-2 (100IU/mL) (E/T:0.5, n=3). (C), Schematics and sequence of IL2sp15Rβ fusion proteins with the joining c-myc tag and IL2Rα linker. (D), IL2sp15Rβ fusion proteins detection by anti-IL-15 antibody in cell membrane extracts. (E), NK92IL15RB express classical NK markers CD56 and higher CD25 with higher surface expression of IL2Rβ than parental NK92. Data are shown as the mean ± SD. ns represents no significant difference. **p<0.01, ****p<0.0001. |
IL15RB enhances NK cell cytotoxicity
We next evaluated NK92IL15RB cytotoxicity compared to NK-92 cells prestimulated by soluble IL-15 for 2 days against PC-3 cancer cells for another 2 days. The continuous signaling in NK92IL15RB exerted significantly higher cytotoxicity than soluble IL-15 (Figure 2A). To determine whether this higher cytotoxicity is due to IL15RB continuous signaling, we compared NK92IL15RB cytotoxicity to NK-92IL2sp15 expressing untethered IL2sp15 (Figure 1C), which allows cytokine-independent expansion and continuous IL-15 signaling. Figure 2B shows that IL15RB induced significantly better cytotoxicity than the IL2sp15 form in NK92IL2sp15, suggesting that the signaling from IL15RB is more potent than soluble IL-15. Similarly, IL15RB signaling induced better cytotoxicity against human lung cancer PC-9, compared to IL-2 tethered to IL2Rβ CIRB in NK92CIRB or to soluble IL2sp15 in NK92IL2sp15 (Figure 2C), despite the faster cell growth of NK-92 under the signaling of tethered IL-2 in CIRB compared to IL15RB (Figure 2D). Moreover, NK92IL15RB killed, more efficiently than NK-92 stimulated by IL-2, several human cancer cell lines: KYSE-150 TE-1, A2780, PC-3, and NCI-H1975, at different ratios (Figure 2E). Similarly, NK92IL15RB performed slightly better than NK92CIRB against MDA-MB-231, PC-3, and BT474.
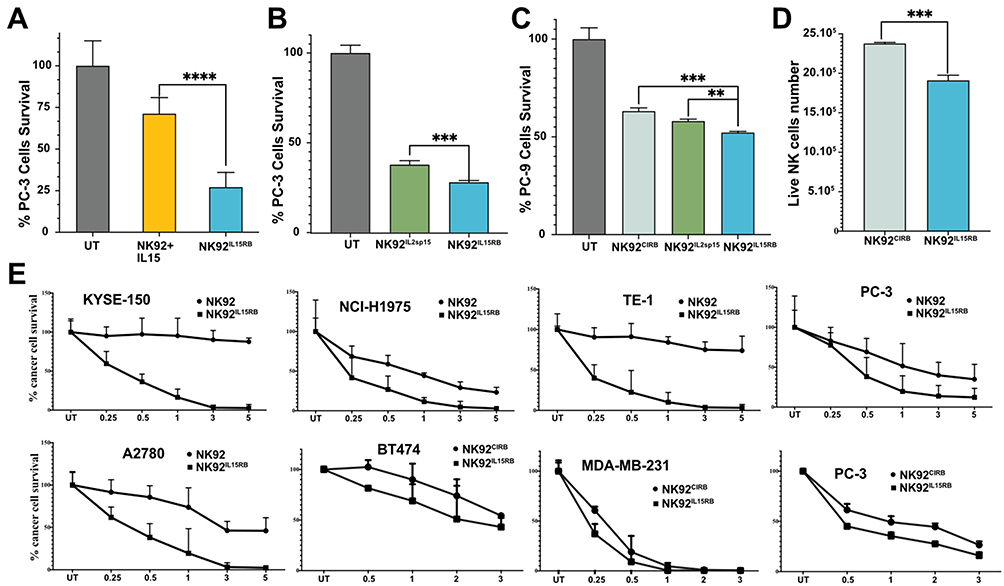 |
Figure 2 IL15RB augments NK-92 cytotoxicity. (A) NK92IL15RB outperforms the anti-cancer activity of NK-92 stimulated by soluble IL-15 in PC-3 cells (E/T=0.5:1, n=3). (B) Comparison of the anticancer effect of NK92IL15RB and NK92IL2spIL15 against PC-3 cells at E/T 0.5:1 (n=3). (C) Anticancer effect of NK92IL15RB, NK92IL2spIL15, and NK92CIRB against lung cancer cells PC-9 at E/T 0.5:1 (n=3). (D) IL15RB fusion protein induces a slower growth of NK92IL15RB compared to NK92CIRB like the impact of soluble IL-15 versus IL2 in NK92 cells. (E) the cytotoxicity of NK92IL15RB at different effector-to-target ratios (E/T) is better than the anti-cancer activity of parental NK92 and NK92CIRB in multiple cancer cell lines (n=3). Data are shown as the mean ± SD. ns represents no significant difference. **p<0.01 ***p<0.001, ****p<0.0001. |
NK92IL15RB resistance to immunosuppression and synergy with CAR-CD19
We previously reported that the growth of NK-92 cells constitutively expressing IL-2 was significantly inhibited by TGFβ1 and IL-4, while NK92CIRB cells expressing tethered IL-2 were more resistant to IL-4, TGFβ1, and dexamethasone.17 The growth of NK-92 treated with the soluble IL-15 was not affected by TGFβ1, while NK-92 cells expressing untethered IL2sp15 were marginally affected (Figure 3A). NK92IL15RB growth was counterintuitively increased by TGFβ1 treatment. IL-4 treatment marginally inhibited NK92IL15RB growth between 10 and 20ng/mL with no inhibition at 30ng/mL, suggesting a biphasic effect (Figure 3B). PC-3 cancer cell killing was reduced when NK92IL15RB cells were pretreated with TGFβ1, or Dex and this inhibition was more pronounced than in NK92CIRB which harbor signaling from tethered IL-2 (Figure 3C). However, IL-4 did not affect the cytotoxicity of both cell lines and even slightly improved it. Overall, these results suggest that IL15RB enhanced cytotoxicity over CIRB might be curtailed by TGFβ1.
 |
Figure 3 Impact of TGFβ1, IL4, Dexamethasone, irradiation, and CAR-CD19 on NK cells. (A) NK92 stimulated with soluble IL-15 (10ng/mL), NK92IL15RB and NK92IL2spIL15 cells were plated with TGFβ1 (10ng/mL) for 3 days of growth (n=3). Growth of viable cells was counted using Trypan blue exclusion assay, and all NK cell viability was higher than 90%. Data are presented as live cell count. (B) Compare the effect of different doses of IL-4 on NK92IL15RB growth for 4 days. (C) NK92IL15RB, and NK92CIRB cell lines were exposed for 24 hours to IL-4 (10ng/mL), TGFβ1 (10ng/mL), Dex (0.5uM), or no drug (UT). Cancer cells (32 ×103 cells) were then added to NK cells at E/T ratio of 0.5:1, then incubated for 4 days. Cancer cell viability was determined using a crystal violet extraction assay. Data are presented as mean percentage cell number relative to NK cells-free controls (UT). (D), Cell survival of NK cells irradiated in vitro at 10Gy and then plated in complete NK92 media was determined using Trypan Blue exclusion every 24 hours. The survival advantage of NK92IL15RB cells was statistically significant on days 1 and 2 (One-way Anova test). (E), Anticancer effect of NK92IL15RB and NK92 stimulated with soluble IL-15 (10ng/mL) for 48h against PC-3 cells at E/T 0.5:1 (n=3) after 8Gy irradiation. (F), CAR-CD19 using CD28 domain enhances NK92IL15RB killing in a CD19+ B cell precursor leukemia, not in CD19- chronic myelogenous leukemia. Data are shown as the mean ± SD. ns represents no significant difference. **p<0.01, ***p<0.001, ****p<0.0001. |
NK-92 has been approved for use in clinical trials with the requirement for irradiation to eliminate any proliferation risk. We determined the impact of irradiation (10Gy) on the survival of NK-92 expressing the chimeric IL15RB compared to NK-92 pre-stimulated by IL-15 for 2 days. IL15RB continuous signaling allowed a significantly better survival of NK92IL15RB than NK-92 at days 1 and 2 post-irradiation (Figure 3D). Similarly, irradiated NK92IL15RB killed PC-3 cells significantly more than NK-92 pre-stimulated by IL-15 (Figure 3E) suggesting IL15RB signaling enhances the survival of irradiated NK cells to mediate prolonged killing of cancer cells. Previous studies reported the advantage of using soluble IL-15 to armor CD19-CAR-NK cells. Therefore, we examined whether IL15RB signaling could synergize with a chimeric antigen receptor against CD19. CD19-CAR-NK92IL15RB killing of CD19-negative K562 did not differ significantly from the killing mediated by NK92IL15RB. However, CD19-positive NALM6 were killed more efficiently by CD19-CAR-NK92IL15RB, suggesting specificity and synergy (Figure 3F).
Activation and expression profiles of cytotoxicity effectors in NK92IL15RB
IL-2 and IL-15 signaling are largely mediated by STAT protein activation pathways. Examination of the impact of IL15RB and soluble IL-15 (10ng/mL) on STAT1, 3 and 5 activations revealed comparable effects in NK-92 (Figure 4A). However, compared to soluble IL-2 signaling or tethered IL-2 in CIRB, IL15RB showed relatively, reduced STAT1 and 5 but increased STAT3 activation (Figure 4B). Interestingly, the introduction of CD28 containing CD19-CAR in NK92IL15RB led to an increase in the activation of STAT5 and a reduction in STAT3 with no change in STAT1. We next evaluated the impact of NK92IL15RB exhaustion stemming from long exposure (6 days) to PC-3 cells at E/T 0.5:1. NK cells were then allowed to recover for 1 week, after which we examined the recovery of STAT protein activation. Chronic exposure to PC-3 cells led to a substantial reduction of activation for all STATs (Figure 4C and D), with STAT3 being the most significantly reduced (~59%). After 1 week of recovery, STAT5 activation under IL15RB signaling was restored to pre-exposure status, while STAT1 did not recover and remained at the post-exposure level. A partial recovery was observed for STAT3 but remained substantially below the pre-exposure level. This data suggests that STAT signaling and TGFβ1 inhibition may be critical factors in the exhaustion countering IL-15-mediated signaling.
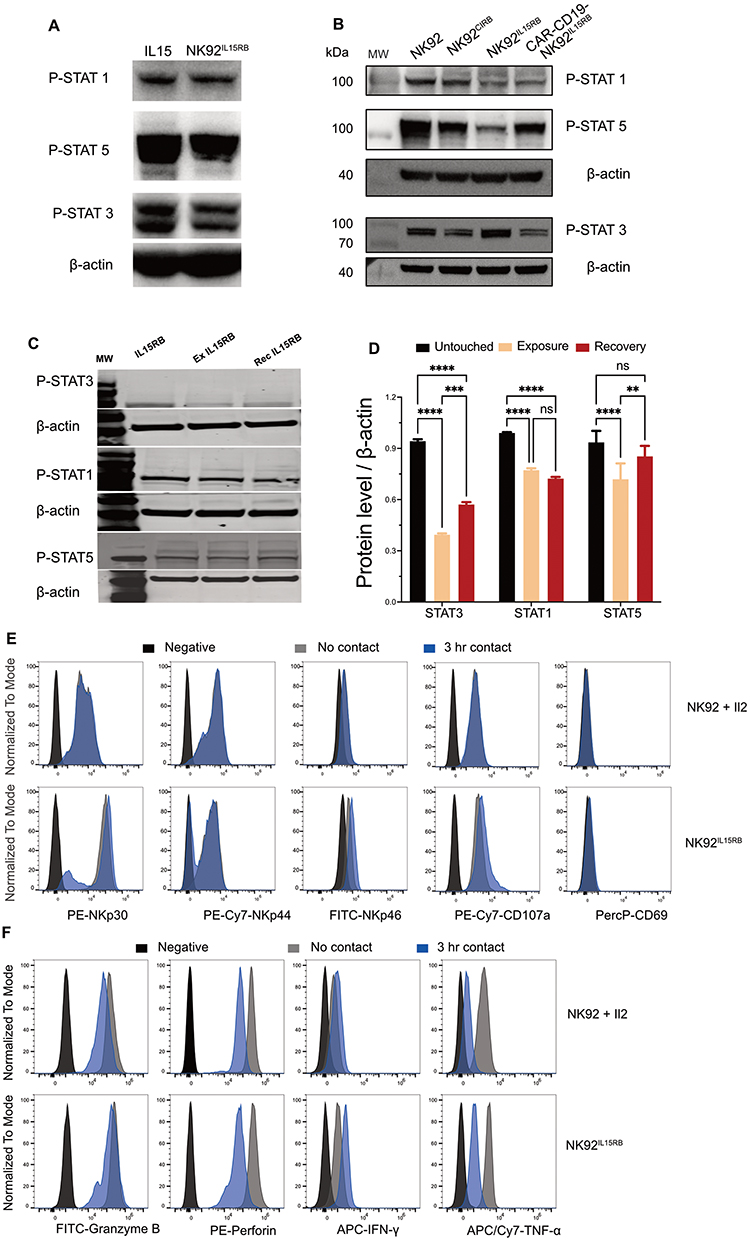 |
Figure 4 Comparison of IL-2 and IL-15 signaling controls STATs activation in NK92. (A) Soluble IL-15 (10ng/mL) for 48h and IL15RB fusion protein impact on phosphorylated STAT1, 3 and 5 detected using rabbit anti-Stat5 Phospho (Tyr694) antibody, purified rabbit anti-Stat3 Phospho (Tyr705), Purified mouse anti-Stat1 Phospho (Ser727). (B) phosphorylated STAT1, 3 and 5 expressions in NK92 stimulated by IL2 (100IU/mL), NK92CIRB, NK92IL15RB and CD19 CAR-NK92IL15RB. (C) phosphorylated STAT1, 3, and 5 in fresh, exhausted, and recovered NK92IL15RB cells. Exhausted NK92IL15RB by exposure to PC-3 cells at E/T 0.5:1 for 6 days, then recovered one week to get recovered NK92IL15RB cells (rec IL15RB) after removing from PC-3 cells. (D) Gel quantification of Western blot bands in (C). (E) Effectors IFN, GzmB, Perf-1, and TNF-α1 change in fresh NK-92 stimulated by IL-2 (100IU/mL) and NK92IL15RB compared to after 3h contact with PC-3 cells. (F) Natural cytotoxicity receptors and active markers expression in NK-92 and NK92IL15RB with or without contact with PC-3 cells for 3hr. Data are shown as the mean ± SD. ns represents no significant difference. **p<0.01, ***p<0.001, ****p<0.0001. |
Natural cytotoxicity receptors (NCRs) NKp30, NKp44, and NKp46 are important activators of NK cells. We examined the impact of tethered IL15RB expression on NCR levels at a resting state, but also after a short (3 hr) exposure to cancer cells (Figure 4E). IL15RB induced a substantially higher basal expression of NKp30, which with NKp46 increased marginally, following exposure to PC-3 cells, compared to IL-2. IL15RB more than IL-2, induced upon contact with PC-3 cells activation marker CD107a which correlates with both cytokine secretion and NK cell-mediated lysis of target cells. Effector enzymes were reduced after exposure to PC-3 cancer cells suggesting delivery via immunological synapses (Figure 4F). However, IFN production was increased by IL15RB during contact, while it was reduced under IL-2. Similarly, TNF-α and Granzyme B were less depleted compared to IL-2 after contact with cancer cells.
IL15RB enhances NK cell antitumor activity
Solid tumors constitute a significant challenge to immunotherapies and require multiple-treatment modalities to achieve meaningful responses. We reported improved antitumor activity of NK-92 cells expressing tethered IL-2 compared to soluble IL-2. We next evaluated the antitumor activity of irradiated NK92IL15RB compared to irradiated K-92 in NCG mice implanted subcutaneously with PC-3 tumors. We also assessed if the blockade of PD-1 by anti-PD-1 (Nivolumab) antibody could enhance NK92IL15RB antitumor activity. Tumors were treated at an initial volume approaching 150mm3 (Figure 5A). The first injection of NK92IL15RB induced an immediate stabilization within 24 hours and a reduction in tumor size by 13% when combined with anti-PD-1. The following injections sustained a tumor delay of 8 days at the end of the treatment combining NK92IL15RB and anti-PD-1. NK92IL15RB cells alone affected a tumor growth delay of 5.5 days. However, a tumor growth delay of only 2.5 days was obtained when using irradiated NK-92 cells. Anti-PD-1 injections alone did not cause any tumor delay. Overall, the treatment with NK92IL15RB alone or combined with anti-PD-1 did not cause any toxicity as determined by animal body weights (Figure 5B).
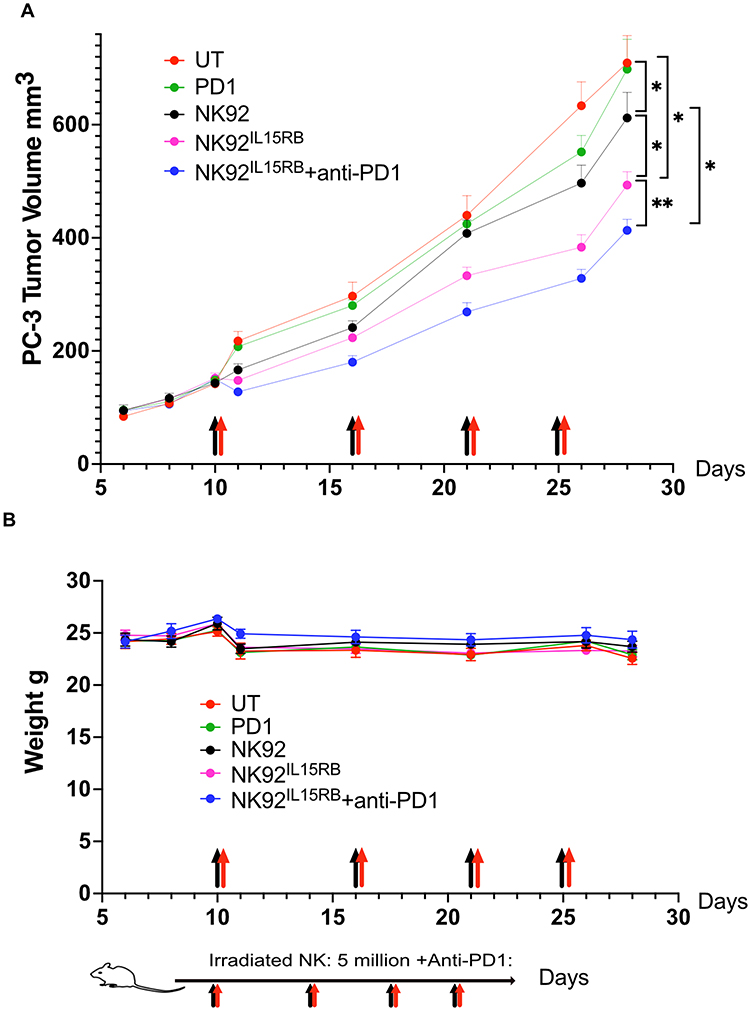 |
Figure 5 NK92IL15RB anti-tumor activity is enhanced by immune checkpoint inhibitors. (A) When PC-3 tumor volume reached ~150mm3, irradiated NK92 and NK92IL15RB cells (4x106 cells in 200ul per mouse) were injected into mice (n=5, black arrows) via the tail vein and the IC inhibitors (anti-PD1 (150ug i.p, red arrows) was injected immediately after NK cells. A second NK cell injection was carried out 6 days later, a third 5 days later, and a fourth 4 days later. Tumor sizes were monitored until 29 days post-tumor implantation. (n=5 tumors). (B) mice body weight remained stable during treatment, suggesting the lack of toxicity from combination treatment. Data are shown as the mean ± SD. (n=5 animals), ns represents no significant difference. * p<0.05, **p<0.01. |
Discussion
NK cells are constantly released from the bone marrow and distributed into organs to become tissue residents or circulate in the blood. During their short lifespan, they depend on other immune cells for survival by competing with T-regulatory cells for IL-2 secreted by T-cells and benefit from IL-15 trans presentation by dendritic cells and macrophages. Most NK stimulation in vivo occurs through IL-15 trans-presentation by DCs, as demonstrated by mature NK population collapse in vivo when DCs are depleted.5 IL-15 can extend the telomeres by enhancing the telomerase activity in NK, NKT, and CD8 T-cells.18 Telomeres erosion with age at a rate of 50 bp/year in human T-cells,19 implies that old individuals at risk of cancer have immune cells, including NK cells, with shorter telomeres.20 Unlike T-cells, this problem may be resolved by using allogenic NK cells from young donors or cord blood. In vitro, the viability of CB-NK cells expressing soluble IL-15 and CAR-CD19 declined precipitously within a few days after plating,9 suggesting telomeres loss due to insufficient activation from CARs or inadequate IL-15 signaling. IL-15 substantially improved CB-NK cells, especially when combined with the knockout of the CIS gene.6 However, secreted IL-15 by genetically modified primary and CB-NK cells can causes severe9 to lethal toxicity and cytokine release syndrome in animal models7 and may lead to NK cells exhaustion by a metabolic defect.21 Therefore, infusing self-sustaining NK cells with IL-15 signaling to safely ensure NK cell survival, enhance cytolytic activity and provide resistance to exhaustion is critical. In this study, we sought to improve NK cell cytotoxicity by a self-contained IL-15 signaling by tethering IL-15 to its receptor IL2Rβ via a linker. In this configuration, IL-15 is self-presented to its receptor without the need for IL-15Rα. Our previous model, tethering IL-2 to IL2Rβ, was superior to the soluble IL-2 produced and secreted by NK-92 cells.17 Similarly, this study shows an advantage in tethering IL-15 to its receptor IL2Rβ, which enhanced the cytolytic activity of NK cells more than free IL-15 (Figure 2A, B). The chimeric IL15RB also showed better NK activation compared to tethered IL-2 (Figure 2C) despite the high cell proliferation induced by Tethered IL-2 (Figure 2D). Similarly, IL15RB enabled higher cytolytic activity against multiple cancer cell lines (Figure 2E). We previously reported NK-92 expressing tethered IL-2, but not secreted IL-2, relative resistance to immunosuppression by TGFβ1, Dexamethasone and IL-4.17 NK92IL15RB cell growth was not inhibited by TGFβ1 and was even enhanced comparatively to NK-92-producing free IL2sp15, whose growth was reduced. This might be explained by the need to produce sufficient IL2sp15 to counter TGFβ1 inhibition. Indeed, soluble IL-15 added directly into the media was able to prevent this growth inhibition by TGFβ1. NK92IL15RB cell growth was reduced by doses of IL-4 ranging from 10–20ng/mL with no effect at higher doses (30ng/mL), suggesting a biphasic response in NK-92 cells. A report of biphasic response to IL-4 was also reported for its ability to stimulate angiogenesis at lower doses and inhibit it at higher doses in a serum-dependent manner.22 Despite IL-4 growth inhibition, NK92IL15RB cytotoxicity against PC-3 cells was not affected. However, Dexamethasone and TGFβ1 were able to significantly reduce NK92IL15RB cytolytic activity.
Irradiated NK92IL15RB cells showed significantly higher survival than NK-92 stimulated by soluble IL-15 (Figure 3D). This translated into higher cytotoxicity of irradiated cells against PC-3 cancer cells (Figure 3E). NK92IL15RB armed with a CAR against CD19 was able to exert higher and specific killing of CD19 positive cells NALM-6 cells compared to CD19-negative K562 cells (Figure 3F). This suggests the potential of IL15RB signaling to synergize with CARs. Surprisingly, our data show that the addition of CD28-CD3 containing CD19-CAR increases STAT5 but reduces STAT3 activation in NK92IL15RB cells (Figure 4B). A recent report demonstrated that CD28-CD3-based CARs recruit kinases such as LCK and ZAP70 with their signaling cascades to enhance signaling in NK cells.23 LCK overexpression and activation, as well as constitutive activation, were reported to activate STAT5.24–26 Therefore, STAT5 activation by IL15RB signaling may be further enhanced by CAR-based CD28-LCK activation. Exposure of NK92IL15RB cells to PC-3 cancer cells for 6 days led to profound changes in STAT activation that remained after 7 days of recovery, suggesting exhaustion. Notably, STAT3 activation was reduced by 59% and only recovered to ~40% of the pre-exposure level. STAT1 and STAT5 activation were reduced by ~22%. Although STAT5 activation recovered 7 days later, STAT1 did not. Therefore, IL15RB signaling may be hindered by reduced STAT1 and 3 deactivation and TGFβ1 inhibition.
Overall, our studies show the benefit of tethering IL-15 to its receptor IL2Rβ. Although soluble IL-15 and IL15RB produced comparable STAT activations. IL15RB signaling provides sustained activation and enhanced killing compared to the soluble IL-2 or soluble IL-15. This strategy reduces the potential toxicity from soluble IL-15 and allows synergy with CARs. Future studies will evaluate IL15RB in human primary or cord blood NK cells. These studies will evaluate whether our results are comparable to NK-92 and will determine if blocking TGFβ1 inhibition augments NK cell’s anticancer activity.
Acknowledgments
Supported by DAPPCM Innovation Grant 232129 and Myles Shores Fellowship 241317 (Y.J).
Supported by the Scientific Research Funds of Zhejiang Province Health Department 2024ky205 (SC) and Hangzhou Agricultural and Social Development Key Scientific Research Project 202004A19 (BX).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Massachusetts General Hospital has filed a provisional patent on the technologies described in this paper: provisional patent MGH 2023-168, listing the authors YJ. and SC as inventors. The authors report no other conflicts of interest in this work.
References
1. Caligiuri MA. Human natural killer cells. Blood. 2008;112(3):461–469. doi:10.1182/blood-2007-09-077438
2. Vidal SM, Khakoo SI, Biron CA. Natural killer cell responses during viral infections: flexibility and conditioning of innate immunity by experience. Curr Opin Virol. 2011;1(6):497–512. doi:10.1016/j.coviro.2011.10.017
3. Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. 2010;142(6):847–856. doi:10.1016/j.cell.2010.08.031
4. Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol. 2009;9(8):568–580. doi:10.1038/nri2604
5. Guimond M, Freud AG, Mao HC, et al. In vivo role of Flt3 ligand and dendritic cells in NK cell homeostasis. J Immunol. 2010;184(6):2769–2775. doi:10.4049/jimmunol.0900685
6. Daher M, Basar R, Gokdemir E, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137(5):624–636. doi:10.1182/blood.2020007748
7. Christodoulou I, Ho WJ, Marple A, et al. Engineering CAR-NK cells to secrete IL-15 sustains their anti-AML functionality but is associated with systemic toxicities. J Immunother Cancer. 2021;9(12). doi:10.1136/jitc-2021-003894
8. Hangasky JA, Waldmann TA, Santi DV. Interleukin 15 Pharmacokinetics and Consumption by a Dynamic Cytokine Sink. Front Immunol. 2020;11:1813. doi:10.3389/fimmu.2020.01813
9. Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–531. doi:10.1038/leu.2017.226
10. Shah NN, Baird K, Delbrook CP, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015;125(5):784–792. doi:10.1182/blood-2014-07-592881
11. Fehniger TA, Suzuki K, Ponnappan A, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med. 2001;193(2):219–231. doi:10.1006/bcmd.2001.0379
12. Baldassarre G, Fedele M, Battista S, et al. Onset of natural killer cell lymphomas in transgenic mice carrying a truncated HMGI-C gene by the chronic stimulation of the IL-2 and IL-15 pathway. Proc Natl Acad Sci U S A. 2001;98(14):7970–7975. doi:10.1073/pnas.141224998
13. Yokoyama S, Watanabe N, Sato N, et al. Antibody-mediated blockade of IL-15 reverses the autoimmune intestinal damage in transgenic mice that overexpress IL-15 in enterocytes. Proc Natl Acad Sci U S A. 2009;106(37):15849–15854. doi:10.1073/pnas.0908834106
14. Waldmann TA. The biology of IL-15: implications for cancer therapy and the treatment of autoimmune disorders. J Investig Dermatol Symp Proc. 2013;16(1):S28–30. doi:10.1038/jidsymp.2013.8
15. Frutoso M, Morisseau S, Tamzalit F, et al. Emergence of NK Cell Hyporesponsiveness after Two IL-15 Stimulation Cycles. J Immunol. 2018;201(2):493–506. doi:10.4049/jimmunol.1800086
16. Tam YK, Maki G, Miyagawa B, Hennemann B, Tonn T, Klingemann HG. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum Gene Ther. 1999;10(8):1359–1373. doi:10.1089/10430349950018030
17. Jounaidi Y, Cotten JF, Miller KW, Forman SA. Tethering IL2 to Its Receptor IL2Rbeta Enhances Antitumor Activity and Expansion of Natural Killer NK92 Cells. Cancer Res. 2017;77(21):5938–5951. doi:10.1158/0008-5472.CAN-17-1007
18. Watkinson F, Nayar SK, Rani A, et al. IL-15 Upregulates Telomerase Expression and Potently Increases Proliferative Capacity of NK, NKT-Like, and CD8 T Cells. Front Immunol. 2020;11:594620. doi:10.3389/fimmu.2020.594620
19. Rufer N, Brummendorf TH, Kolvraa S, et al. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J Exp Med. 1999;190(2):157–167. doi:10.1084/jem.190.2.157
20. Akbar AN, Beverley PC, Salmon M. Will telomere erosion lead to a loss of T-cell memory? Nat Rev Immunol. 2004;4(9):737–743. doi:10.1038/nri1440
21. Felices M, Lenvik AJ, McElmurry R, et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight. 2018;3(3). doi:10.1172/jci.insight.96219
22. Volpert OV, Fong T, Koch AE, et al. Inhibition of angiogenesis by interleukin 4. J Exp Med. 1998;188(6):1039–1046. doi:10.1084/jem.188.6.1039
23. Acharya S, Basar R, Daher M, et al. CD28 costimulation augments CAR signaling in NK cells via the LCK/CD3Z/ZAP70 signaling axis. Cancer Discov. 2024. doi:10.1158/2159-8290.CD-24-0096
24. Cazzaniga V, Bugarin C, Bardini M, et al. LCK over-expression drives STAT5 oncogenic signaling in PAX5 translocated BCP-ALL patients. Oncotarget. 2015;6(3):1569–1581. doi:10.18632/oncotarget.2807
25. Shi M, Cooper JC, Yu CL. A constitutively active Lck kinase promotes cell proliferation and resistance to apoptosis through signal transducer and activator of transcription 5b activation. Mol Cancer Res. 2006;4(1):39–45. doi:10.1158/1541-7786.MCR-05-0202
26. Welte T, Leitenberg D, Dittel BN, et al. STAT5 interaction with the T cell receptor complex and stimulation of T cell proliferation. Science. 1999;283:5399):222–5. doi:10.1126/science.283.5399.222
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.