")
Back to Journals » International Journal of General Medicine » Volume 14
A Review of Three Chinese Cases of Acromicric/Geleophysic Dysplasia with FBN1 Mutations
Authors Shan YC, Yang ZC, Ma L, Ran N, Feng XY, Liu XM, Fu P, Yi MJ
Received 23 February 2021
Accepted for publication 14 April 2021
Published 17 May 2021 Volume 2021:14 Pages 1873—1880
DOI https://doi.org/10.2147/IJGM.S306018
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Yan-Chun Shan, Zhao-Chuan Yang, Liang Ma, Ni Ran, Xue-Ying Feng, Xiao-Mei Liu, Peng Fu, Ming-Ji Yi
Department of Child Health Care, Pediatric Center, Affiliated Hospital of Qingdao University, Qingdao, 266003, People’s Republic of China
Correspondence: Ming-Ji Yi
Department of Child Health Care, Pediatric Center, Affiliated Hospital of Qingdao University, No. 16 of Jiangsu Road, Shinan District, Qingdao, 266003, People’s Republic of China
Tel +86 532 82911223
Email [email protected]
Objective: This study aims to explore the clinical features and molecular diagnosis of FBN1-related acromelic dysplasia in Chinese patients.
Methods: The clinical and genetic features of three FBN1-related acromicric dysplasia (AD)/geleophysic dysplasia (GD) Chinese patients from two families were reviewed, and comprehensive medical evaluations were performed. Targeted next-generation sequencing was used to detect genetic mutations associated with short statures, including FBN1. Sanger sequencing was used to determine the de novo mutation origin.
Results: Patient 1 presented with short stature, short and stubby hands and feet, mild facial dysmorphism, hepatomegaly, delayed bone age and beak-like femoral heads. Patient 2 and this patient’s father merely presented with short stature, wide and short hands, and beak-like femoral heads. One novel mutation, c.5272G>T(p.D1758Y), and one known mutation, c.5183C>T(p.A1728V), were identified in these patients.
Conclusion: The clinical features varied among these patients. The variant c.5272G>T(p.D1758Y) is a novel mutation.
Keywords: FBN1, acromelic dysplasia, acromicric dysplasia, geleophysic dysplasia, short stature
Introduction
Acromelic dysplasia is a heterogeneous group of rare skeletal dysplasias characterized by disproportionate short stature with distal limb shortening. In the 2015 Nosology and Classification of Skeletal Disorders,1 the acromelic dysplasia group consists of 10 conditions, which included, Weill-Marchesani syndrome (WMS), geleophysic dysplasia (GD) and acromicric dysplasia (AD). Specific mutations in the fibrillin-1 (FBN1) gene have been identified in AD, GD and WMS patients.2,3 The FBN1 gene lies on the long arm of chromosome 15 at 15q15-q21.1, and encodes a 2871-aa (350 kDa) structural protein FBN1. This is a cysteine-rich glycoprotein, and the primary component of the 10–12 nm extracellular microfibrils that are broadly distributed in elastic and nonelastic connective tissues.4 Mutations in FBN1 disrupt microfibril formation, result in fibrillin protein malformation, and eventually, weaken connective tissues.5 FBN1 comprises of 47 epidermal growth factor-like (EGF) domains and seven transforming growth factor-β1 binding protein-like (TB) domains.6 Mutations in FBN1 that underlie these acromelic dysplasias are predominantly limited to the hotspots located in exon 41 and exon 42.7 Exons 41 and 42 encode the 5th 8-cysteine domain in FBN1 (the heparin binding TGFβ-binding protein-like domain 5 [TB5] of FBN1).8 FBN1 is the only gene implicated in AD, which is inherited in an autosomal dominant manner.7 In contrast, both GD and WMS are genetically heterogeneous. These three genes, which are known to be associated with GD, are a disintegrin and metalloproteinase with thrombospondin motifs-like 2 (ADAMTSL2) gene,9 FBN1,9 and the recently discovered latent transforming growth factor-beta-binding protein-3 (LTBP3).10,11 For WMS, three genes, including ADAMTS10, ADAMTS17 and LTBP2, cause the autosomal recessive form, while FBN1 causes the autosomal dominant form.3 Acromelic dysplasia is an extremely rare condition, and merely 13 Chinese cases have been reported in literature.3,9,12,13 Among these cases, merely one GD patient was caused by a missense mutation and a novel splicing mutation in ADAMTSL2.12 The present study reports the clinical and genetic findings of another three GD/AD patients from two Chinese families. A retrospective study was conducted by combining the clinical features of these Chinese patients.
Patients and Methods
Patients Information
Family 1
Patient 1 was a nine years and seven months old boy, who was referred to our clinic due to short stature. He was born at 39 weeks of gestation with a birth weight of 3200 g. His parents are non-consanguineous. His father was 163 cm in height, while his mother was 167 cm in height. He presented with marked growth retardation from two years old, while he paralleled his peers in intelligence. He presented with short stature, short limbs, short and stubby hands and feet, and mild facial dysmorphism: round face, flat nose with anteverted nostrils, prominent philtrum, and thick lips (Figure 1). His body height was 108.5 cm (Ht <-4 SD), and his arm span was 100.0 cm. His liver was palpable at 4 cm below the right costal margin. The abdominal ultrasound demonstrated mild hepatomegaly, while cardiac ultrasound revealed no significant findings. In view of his dropping height centile, endocrine investigations, including thyroid function test, insulin-growth-factor 1 and clonidine stimulation test, were performed with normal results. The radiograph of the left hand revealed that his bone age was six years, with cone shaped epiphysis (Figure 1). The pelvis radiograph revealed beak-like femoral heads (Figure 1).
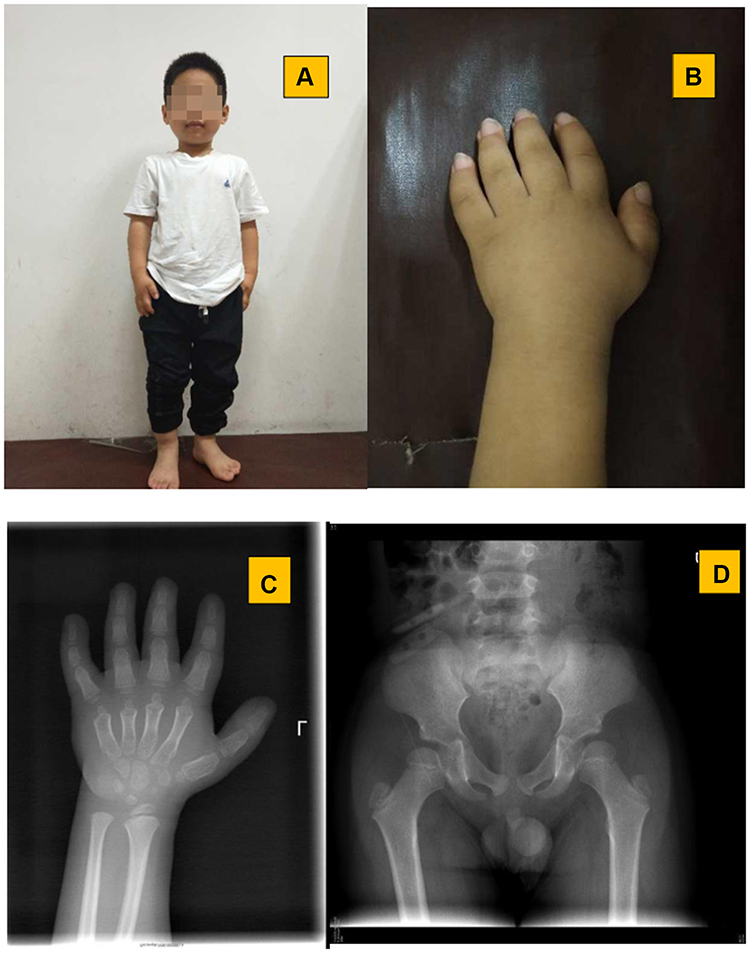 |
Figure 1 Clinical and radiological features of patient 1: (A) short stature and facial profile; (B) small hand and short fingers; (C) delayed bone age and cone shaped epiphysis; (D) beak-like femoral heads. |
Family 2
Patient 2 was a five years and seven months old boy, who was referred to our clinic due to short stature. He was the first child of a non-consanguineous Chinese couple. He was born full term, with a birth weight of 3050 g and a body length of 49 cm. He has a family history of short stature on the paternal side. His mother was 160 cm in height. On examination, his body height was 100.3 cm (Ht <-3 SD), sitting height was 58.0 cm, and arm span was 95.0 cm. His face and head were normal. He had wide and short hands, but there was no joint stiffness (Figure 2). The cardiovascular examination and abdominal examination were unremarkable. Mental and motor development were normal. The endocrinological examination revealed that growth hormone excretion and thyroid function were normal. The radiological examination revealed shortened tubular bones in the hands and a beak-like femur head (Figure 2).
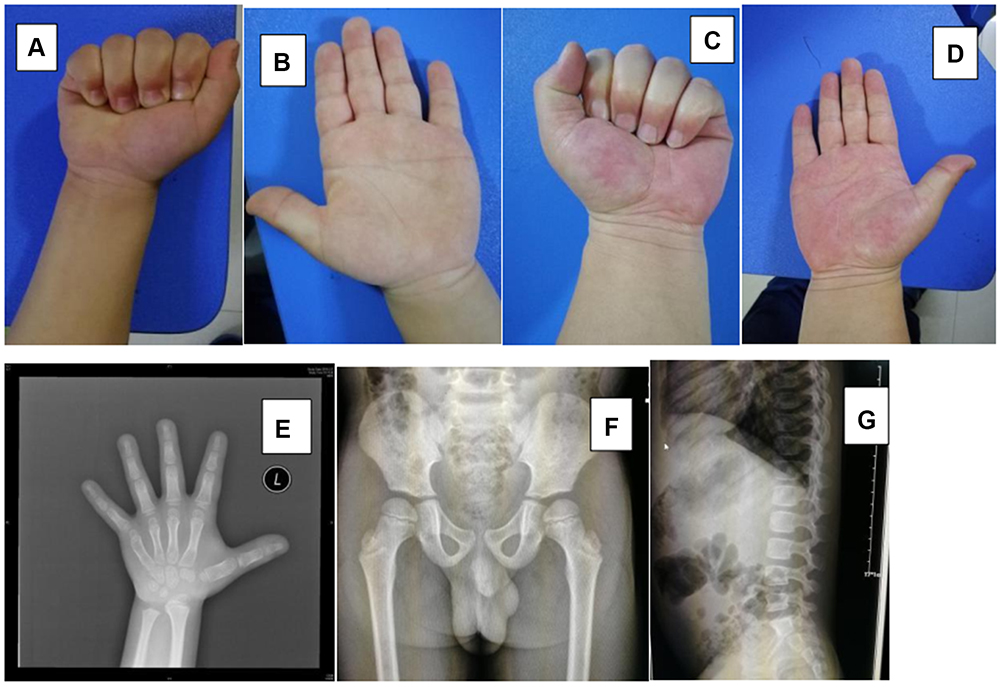 |
Figure 2 Clinical and radiological features of patient 2 and patient 3: (A–D) The presence of small hands, but without joint contracture; (E–G) The images of the left hand, pelvis and vertebral column of patient 2 revealed shortened tubular bones in the hands and beak-like femoral heads. |
Patient 3 was a 35 year-old gentleman, and was the father of patient 2. He had similar short limbs and short stature, with a height of 148.0 cm (Ht <-4 SD). His face and head were normal. He also had wide and short hands (Figure 2). He had no joint stiffness, no organomegaly, no eye problem, and no cardiac respiratory problems. His intelligence was normal.
Genetic Analysis
All procedures performed in the present study were in accordance with the 1964 Helsinki declaration and its later amendments. An informed consent was obtained from the guardians of all patients.
DNA Preparation
A signed informed consent for genetic analysis was provided by the children’s parents. Approximately 2 mL of peripheral blood (EDTA anticoagulant) were collected from the patient and the patient’s parents, and genomic DNA was extracted using a QIAamp Blood Midi Kit (QIAGEN, Germany), according to manufacturer’s instructions. A minimum of 3 ug of DNA was used for the indexed Illumina libraries, according to manufacturer’s protocol (MyGenostics, Beijing). DNA fragments with sizes that ranged from 350 bp to 450 bp, and those that included the adapter sequences were selected for the DNA libraries. The final product was validated using the Nanodrop 2000 (Thermo Fisher, USA) and Agilent Bioanalyzer (Agilent, USA).
Targeted Genes Capture and Sequencing
The coding exons of the 286 genes associated with short stature (including the FBN1 gene) were selected through a gene capture strategy using the GenCap custom enrichment kit (MyGenostics, Beijing), according to manufacturer’s protocol. The enriched libraries were sequenced on an Illumina NextSeq 500 sequencer (Illumina, San Diego, CA, USA) for paired-end reads of 150 bp.
Data Analysis
After the sequencing, low quality variations were filtered using a quality score of ≥20, and the Burrows-Wheeler Alignment tool (BWA) was used to align the clean reads to the reference human genome (hg19). Single-nucleotide polymorphisms (SNPs), and the insertions or deletions (InDels) were determined using the Genome Analysis Toolkit (GATK). Frequencies ≥0.02 of the identified SNPs and InDels were removed in 1000 Genomes, ESP6500, ExAC and Inhouse (MyGenostics). Non-synonymous variants were evaluated using four algorithms (SIFT, Polyphen-2, Mutation-Taster, and GERP++) to predict the pathogenicity.
Validation by Sanger Sequencing
Among all the members of the family, the candidate variable sites were confirmed by Sanger sequencing. The target sequences were sequenced using an ABI 3730 analyzer (Applied Biosystems). The sites of variation were identified by comparing the DNA sequences with the corresponding GenBank reference sequences using the Mutation Surveyor software.
Sequence Analysis
The sequencing depth on the target regions yielded more than 200 on average, and the sample had more than 99.5% targeted regions covered. Meanwhile, the coverage of the targeted exons for >10 reads ranged within 95%.
Results
For patient 1, a novel heterozygous missense mutation in exon 42 of the FBN1 gene, c.5272G>T(p.D1758Y), was identified (Figure 3). The chromosome coordinates is chr15-48752467 (GRCh37/hg19), and transcript ID NM_000138. This variant has not been previously reported in the UMD-FBN1 mutation database (www.umd.be/FBN1/), and is not recorded in the following databases: HapMap, NCBI dbSNP, 1000 human genomes, gnomAD, ExAC and Chinese population database. This novel mutation was also not detected in 100 chromosomes from normal Chinese subjects. Furthermore, the parents did not carry this variant.
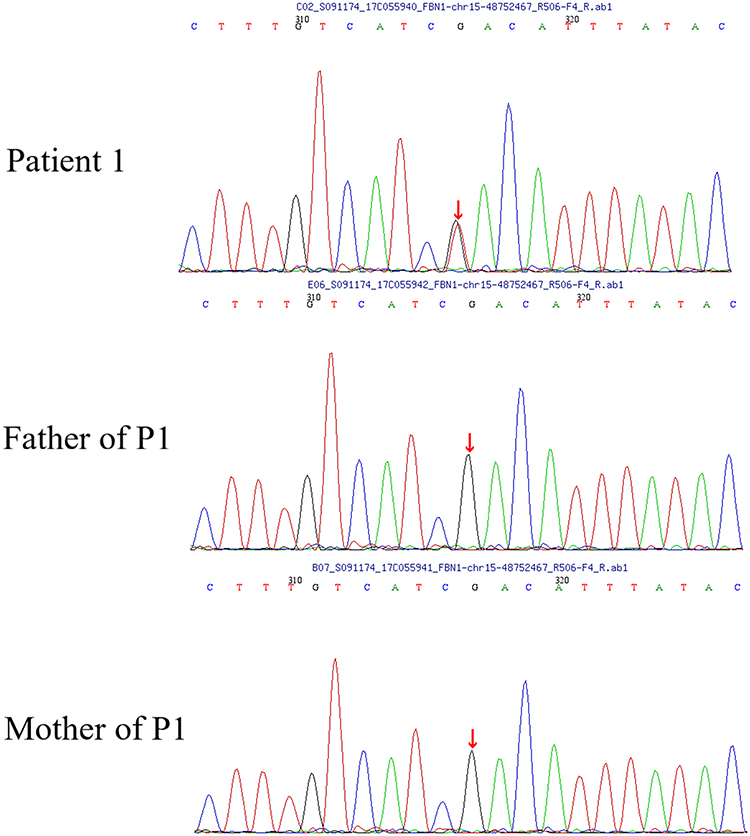 |
Figure 3 A novel heterozygous missense mutation in exon 42 of the FBN1 gene, c.5272G>T (p.D1758Y), was found in patient 1. |
For patient 2 and patient 3, a heterozygous missense mutation, c.5183C>T (p.A1728V), was detected in exon 42 (Figure 4). The chromosome coordinates is chr15-48755320 (GRCh37/hg19), and transcript ID NM_000138. This mutation was previously reported to be associated with GD.6
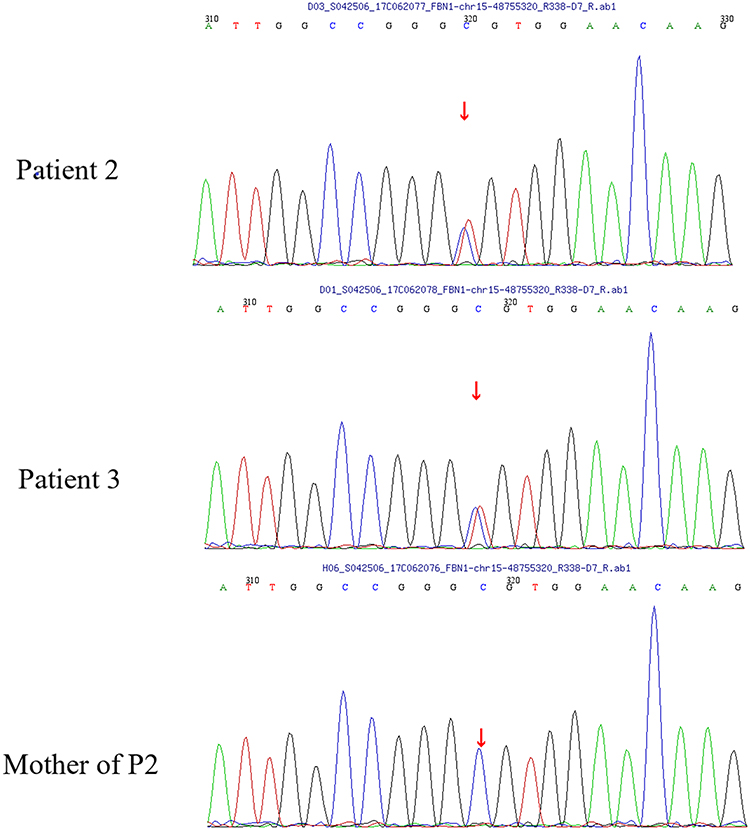 |
Figure 4 A heterozygous missense mutation, c.5183C>T (p.A1728V), was detected in exon 42 in patient 2 and the patient’s father patient 3. |
Discussion
Mutations in the FBN1 gene result in multiple distinct pleiotropic disorders. Most of the more than 3000 mutations known today in FBN1 cause the Marfan syndrome. In contrast, rare mutations in FBN1, which are confined to only certain domains, cause several different types of acromelic dysplasia, including WMS, GD and AD. All three of these acromelic dysplasias share severe short stature, short hands and stiff joints. WMS differs from AD and GD in the presence of microspherophakia,14 while GD has clinical features of progressive cardiac valvular thickening, the characteristic of a “happy” face, hepatomegaly, tracheal stenosis and tip-toeing gait.15 AD only has an autosomal dominant mode of inheritance, and is characterized by distinct facial features, such as a round face, well-defined eyebrows, long eyelashes, a bulbous nose with anteverted nostrils, a long and prominent philtrum, thick lips with a small mouth, a hoarse voice and a pseudomuscular build, and distinct skeleton features, including an internal notch of the femoral head, an internal notch of the second metacarpal, and an external notch of the fifth metacarpal. Mutations of FBN1 cause AD, and the autosomal dominant forms of GD and WMS.
Patient 1 demonstrated short stature, short arm span, special face, stiff joints, skeletal abnormalities and hepatomegaly, supporting the diagnosis of GD. The NGS results identified a heterozygous variant, c.5272G>T(p.D1758Y), in the FBN1 gene of patient 1. Sanger sequencing was used to determine the de novo origin. In 2011, the same amino acid change mutation position p.D1758V, but with a different nucleotide change c.5273A>T, was detected in an Italian patient with AD.9 However, the codon that changed at position 5272 has not been detected in Chinese patients, to date. The novel missense mutation c.5272G>T in exon 42 of FBN1 was identified for the first time in the present Chinese patient, which led to GD. The effect of c.5272G>T was predicted to be deleterious by both SIFT and Polyphen-2, confirming that this variation is an important functional mutation, and has obvious biological effects to this disease.
Patient 2 and the patient’s father demonstrated short stature, and wide and short hands. Most notably, they had no distinguishable faces, and no joint stiffness, cardiac pathology, or organomegaly. A heterozygous missense mutation c.5183C>T(p.A1728V) in exon 42 of FBN1 was detected in patient 2 and the patient’s father. This mutation has been previously reported.9,16 In 2011, this mutation was first detected in a GD patient with cardiac involvement. In 2016, the same heterozygous mutation was found in two patients with isolated, asymptomatic short stature, which was similar to that in patient 1 and patient 3.16 As documented in literature, the same mutations, such as p.Y1699C and p.A1728T, could lead to both GD and AD.9 Similarly, six patients from three families with acromelic short stature were reported in 2018. These patients all carried a heterozygous mutation, c.5284G>A(p.Gly1762Ser), in exon 42 of the FBN1 gene, but they had pleiotropic clinical features.12 Hence, identical FBN1 mutations can give rise to a wide phenotypic spectrum, supporting the unknown epigenetics involved in disease pathogenesis.
To date, a total of 15 FBN1-related acromelic dysplasia Chinese patients had been reported,6,9,12 including the present three patients. Among all these patients, a three year-old girl received a diagnosis of WMS. In all Chinese patients with FBN1-related acromelic dysplasia, the height SDs progressively and negatively deviated from −3 with age. Furthermore, they all presented with short-limbed short stature. No tip-toeing gait was found in all Chinese FBN1-related GD patients. Facial dysmorphism was not always observed in FBN1-related GD/AD Chinese patients. However, the patient 1 reported by us and patient 2 presented in the study conducted by Wang et al3 and the patient 1 presented in the study conducted by Cheng et al12 had a certain degree of phenotypic mimicry. It was particularly noteworthy that some AD/GD patients did not present with stiff joints. The radiological examinations always revealed shortened tubular bones in the hands and beak-like femur heads. Hence, it could be summarized that the clinical features of FBN1- related GD/AD are variable, which range from isolated, asymptomatic short stature to a more classic picture of GD2, with cardiac involvement, facial dysmorphisms and various skeletal anomalies.
Short-limbed short stature is a heterogeneous condition that can result from many diseases. The clinical diagnosis of each disorder that could cause a short-limbed short stature remains challenging. Patients with acromelic dysplasia have similar clinical features to those with lysosomal storage disorders or hypochondroplasia. Normal biochemical results may help to exclude lysosomal storage disorders. Hepatomegaly, which is not usually observed in hypochondroplasia patients, might be used to differentiate mild AD/GD from hypochondroplasia. A genetic analysis was conducted to identify 25 patients who had been clinically diagnosed as having “hypochondroplasia” in Japan. The researcher also found other diseases, including AD/GD, where among, these 25 patients 10 patients had FGFR3-related hypochondroplasia (n=10).17 Targeted next-generation sequencing is an efficient approach to identify the underlying molecular mechanism of genetic bone dysplasia.18 Therefore, genetic diagnosis plays an important role in the diagnosis of acromelic dysplasia. Targeted next-generation sequencing may be clinically adopted to investigate the defective genes of difficult cases of genetic bone dysplasia, which are very critical for the patient’s prognosis prediction and genetic counseling of the family. At present, there is no consensus on the surveillance of AD and GD, and no suitable therapy has been developed for such cases. However, the early definitive diagnosis for these patients can alert clinicians to the possibility of additional complications, and may prevent the use of ineffective growth promoting treatments in this specific patient category.16 Furthermore, an accurate molecular diagnosis is very critical for the patient’s prognosis prediction and genetic counseling of the family.
In summary, by using target next-generation sequencing, three AD/GD patients carrying causative mutations in the TB5 domain of the FBN1 gene were identified. The variant c.5272G>T(p.D1758Y) is a novel mutation. The clinical features were variable among these patients. The present study expands the mutation spectrum in the FBN1 gene, which is crucial for establishing an accurate diagnosis.
Ethics Consent
Patient 3 and a parent or legal guardian of Patients 1 and 2 provided informed consent for the case details and accompanying images to be published. The institutional approval was required to publish the case details. And we have obtained the approval from the Medical Ethics Comments of Affiliated Hospital of Qingdao University.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167A:2869–2892. doi:10.1002/ajmg.a.37365
2. Le Goff C, Cormier-Daire V. From tall to short: the role of TGFβ signaling in growth and its disorders. Am J Med Genet C Semin Med Genet. 2012;160C:145–153. doi:10.1002/ajmg.c.31337
3. Wang Y, Zhang H, Ye J, Han L, Gu X. Three novel mutations of the FBN1 gene in Chinese children with acromelic dysplasia. J Hum Genet. 2014;59:563–567. doi:10.1038/jhg.2014.73
4. Pereira L, D’Alessio M, Ramirez F, et al. Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. Hum Mol Genet. 1993;2:961–968. doi:10.1093/hmg/2.7.961
5. Ramirez F, Caescu C, Wondimu E, Galatioto J. Marfan syndrome; A connective tissue disease at the crossroads of mechanotransduction, TGFβ signaling and cell stemness. Matrix Biol. 2018;71–72:82–89. doi:10.1016/j.matbio.2017.07.004
6. Sakai LY, Keene DR. Fibrillin protein pleiotropy: acromelic dysplasias. Matrix Biol. 2019;80:6–13. doi:10.1016/j.matbio.2018.09.005
7. Sakai LY, Keene DR, Renard M, De Backer J. FBN1: the disease-causing gene for Marfan syndrome and other genetic disorders. Gene. 2016;591:279–291. doi:10.1016/j.gene.2016.07.033
8. Cain SA, McGovern A, Baldwin AK, Baldock C, Kielty CM. Fibrillin-1 mutations causing Weill-Marchesani syndrome and acromicric and geleophysic dysplasias disrupt heparan sulfate interactions. PLoS One. 2012;7:e48634. doi:10.1371/journal.pone.0048634
9. Le Goff C, Mahaut C, Wang LW, et al. Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Genet. 2011;89:7–14. doi:10.1016/j.ajhg.2011.05.012
10. McInerney-Leo AM, Le Goff C, Leo PJ, et al. Mutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia. J Med Genet. 2016;53:457–464. doi:10.1136/jmedgenet-2015-103647
11. Intarak N, Theerapanon T, Thaweesapphithak S, Suphapeetiporn K, Porntaveetus T, Shotelersuk V. Genotype-phenotype correlation and expansion of orodental anomalies in LTBP3-related disorders. Mol Genet Genomics. 2019;294:773–787. doi:10.1007/s00438-019-01547-x
12. Cheng SW, Luk HM, Chu YWY, et al. A report of three families with FBN1-related acromelic dysplasias and review of literature for genotype-phenotype correlation in geleophysic dysplasia. Eur J Med Genet. 2018;61:219–224. doi:10.1016/j.ejmg.2017.11.018
13. Li D, Dong H, Zheng H, et al. A chinese boy with geleophysic dysplasia caused by compound heterozygous mutations in ADAMTSL2. Eur J Med Genet. 2017;60:685–689. doi:10.1016/j.ejmg.2017.09.003
14. Faivre L, Gorlin RJ, Wirtz MK, et al. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003;40:34–36. doi:10.1136/jmg.40.1.34
15. SScott A, Yeung S, Dickinson DF, Karbani G, Crow YJ. Natural history of cardiac involvement in geleophysic dysplasia. Am J Med Genet A. 2005;132A:320–323. doi:10.1002/ajmg.a.30450
16. de Bruin C, Finlayson C, Funari MF, et al. Two patients with severe short stature due to a FBN1 Mutation (p.Ala1728Val) with a mild form of acromicric dysplasia. Horm Res Paediatr. 2016;86:342–348. doi:10.1159/000446476
17. Hasegawa K, Tanaka H. Children with short-limbed short stature in pediatric endocrinological services in Japan. Pediatr Int. 2014;56:809–812. doi:10.1111/ped.12511
18. Zhang H, Yang R, Wang Y, et al. A pilot study of gene testing of genetic bone dysplasia using targeted next-generation sequencing. J Hum Genet. 2015;60:769–776. doi:10.1038/jhg.2015.112
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.