Back to Journals » Journal of Pain Research » Volume 13
A Randomized, Open-Label, Bioequivalence Study of Lidocaine Topical System 1.8% and Lidocaine Patch 5% in Healthy Subjects
Authors Gudin J, Argoff C, Fudin J ![]() , Greuber E
, Greuber E ![]() , Vought K, Patel K, Nalamachu S
, Vought K, Patel K, Nalamachu S ![]()
Received 9 November 2019
Accepted for publication 7 March 2020
Published 22 June 2020 Volume 2020:13 Pages 1485—1496
DOI https://doi.org/10.2147/JPR.S237934
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Video abstract presented by Jeffrey Fudin.
Views: 61327
Jeffrey Gudin,1 Charles Argoff,2 Jeffrey Fudin,3 Emileigh Greuber,4 Kip Vought,4 Kalpana Patel,4 Sri Nalamachu5
1Department of Anesthesiology, Englewood Hospital and Medical Center, Englewood, NJ, USA; 2Department of Neurology, Albany Medical Center, Albany, NY, USA; 3Professional Practice, Albany College of Pharmacy and Health Sciences, Albany, NY, USA; 4Scilex Pharmaceuticals Inc., Palo Alto, CA, USA; 5Mid America PolyClinic, Overland Park, KS, USA
Correspondence: Jeffrey Fudin
Professional Practice, Albany College of Pharmacy and Health Sciences, Albany, NY, USA
Tel +1 518-772-4100
Fax +1 518-734-0288
Email [email protected]
Purpose: This study was designed to characterize drug delivery with lidocaine topical system 1.8% vs lidocaine patch 5% through 2 PK studies.
Patients and Methods: Two Phase 1, single-center, open-label, randomized PK studies were performed in healthy adults. In Study 1, 56 subjects received a single intravenous bolus of 0.7 mg/kg of lidocaine as a lead-in to allow for the accurate determination of apparent dose of both products. After a 7-day washout period, subjects were randomized to receive either lidocaine topical system 1.8% or lidocaine patch 5% for 12 hours followed by another 7-day washout period, after which subjects crossed over to receive the other treatment for 12 hours. In Study 2, 54 subjects were randomized to receive either lidocaine topical system 1.8% or lidocaine patch 5% for 12 hours. After a 7-day washout period, subjects crossed over to receive the other treatment. Adhesion and skin irritation assessments were performed after application of the products in Study 2. In both studies, serial blood samples were collected to measure the plasma concentration of lidocaine after product application. Safety assessments and adverse events were monitored in both studies.
Results: The comparative PK analysis demonstrated that the two products, despite their difference in drug load and strength, are bioequivalent. Both products were well tolerated. In Study 2, dermal response scores (skin tolerability after removal) were similar between lidocaine topical system 1.8% and lidocaine patch 5%, with a mean irritation score per patch < 1 (barely perceptible erythema), which is not considered to be clinically significant.
Conclusion: Bioequivalence was demonstrated between lidocaine topical system 1.8% and lidocaine patch 5%. A comparison of the single-time adhesion scores at 12 hours in Study 2 favored lidocaine topical system 1.8% over lidocaine patch 5%. Both products were well tolerated as a single application in healthy adult human subjects.
ClinicalTrials.gov: NCT04144192, NCT04149938.
Keywords: postherpetic neuralgia, topical, lidocaine, pharmacokinetics
Introduction
Each year in the United States, an estimated one million individuals are diagnosed with herpes zoster (HZ), or shingles.1 HZ is caused by reactivation of latent varicella-zoster virus in the dorsal root ganglia and typically presents as a unilateral, painful, blistering rash in the affected dermatome(s).2 It most often occurs in patients ≥50 years old; women are at greater risk than men.3 Approximately 20% of HZ patients will develop postherpetic neuralgia (PHN), a complication characterized by neuropathic pain within the zoster-affected dermatome(s) that is sustained for at least 3 months after the rash has healed.1,4 PHN pain is often associated with allodynia and hyperalgesia and can persist for years.5 Pharmacologic treatment usually includes oral medications, such as antidepressants, anticonvulsants, and opioids, as well as topical analgesics, such as the lidocaine patch.6,7 Advantages of topical drug delivery systems include providing targeted site-specific therapy while minimizing entry and level of drugs in the systemic circulation; this therapy often avoids or reduces side effects or toxicities of commonly used oral medications.8
Lidocaine is an amide local anesthetic that blocks sodium ion channels required for the initiation and conduction of neuronal impulses.9 Blocking sodium channels in the dermal nociceptors of A delta and C fibers reduces the frequency of ectopic discharges that contribute to neuropathic pain. Topical patches and other delivery systems have been developed to deliver lidocaine locally for the treatment of PHN.1,4,10,11 In 1999, the US Food and Drug Administration (FDA) approved lidocaine patch 5% (Lidoderm®, Endo Pharmaceuticals Inc., Malvern, PA) for the relief of pain associated with PHN. The lidocaine patch 5% contains 700 mg of lidocaine (5% = 50 mg per gram adhesive) in an aqueous (“hydrogel”) matrix.12 The analgesic efficacy of topical lidocaine in PHN has been demonstrated in several randomized clinical trials, which show that patients experience pain relief within hours of application of lidocaine patches and that treatment is well tolerated.13,14 Topical lidocaine remains an effective treatment option for PHN over time, with up to 4 years of available safety and efficacy data on long-term treatment outcomes.15,16 Additionally, topical lidocaine has been reported to have analgesic effects in other conditions, such as lower back pain, diabetic peripheral neuropathy, carpal tunnel syndrome, and osteoarthritis pain.11
In 2018, a novel topical system (ie, patches, now referred to as “topical systems” by the FDA) consisting of lidocaine dissolved in an organic acid/polyalcohol solvent system in a polymer-based adhesive matrix was FDA-approved for the relief of PHN pain (ZTlido® [lidocaine topical system 1.8%], Scilex Pharmaceuticals Inc., Mountain View, CA). Each topical system contains 36 mg of lidocaine (1.8% = 18 mg per gram adhesive).9
Similar to lidocaine patch 5%, lidocaine topical system 1.8% was designed with a skin contact area of 140 cm2 and is indicated to be worn for 12 hours per day (maximum of 3 topical systems per day). The nonaqueous formulation of lidocaine topical system 1.8% results in improved biopharmaceutical efficiency and drug delivery compared with lidocaine patch 5%, such that bioequivalent plasma levels are achieved with ~19-fold lower drug load (36 mg vs 700 mg) and lower strength (1.8% vs 5%). The lidocaine topical system 1.8% was developed to have optimal adhesion (ie, maintaining contact with the skin over the 12-hour administration period) and to be thinner and lighter than lidocaine patch 5% so that it can better adhere to contour-challenged areas of the body and maintain contact during normal activities and moderate exercise. A comparison of lidocaine topical system 1.8% and lidocaine patch 5% product attributes is provided in Table 1.
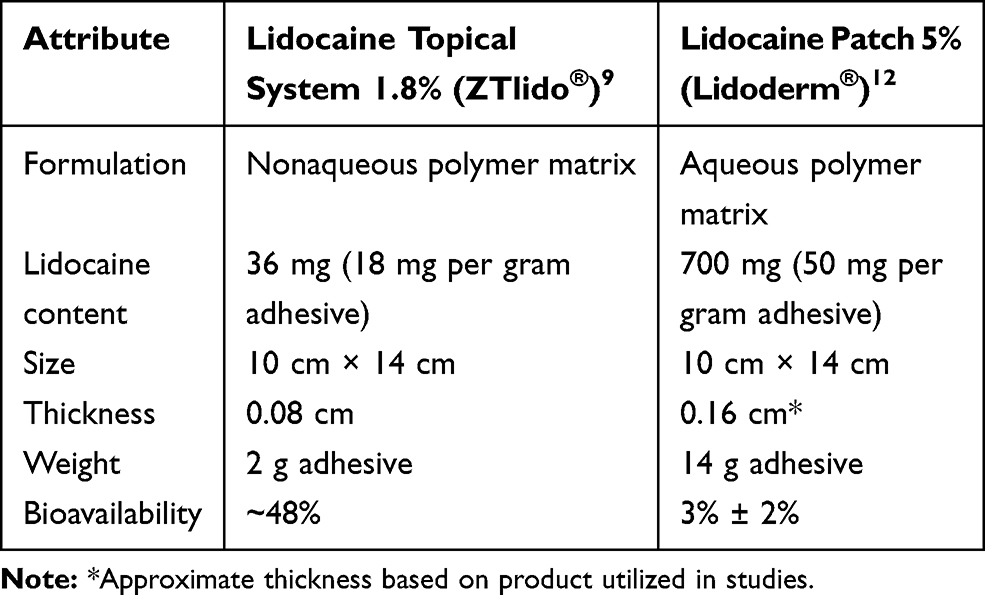 |
Table 1 Characteristics of Lidocaine Topical System 1.8% and Lidocaine Patch 5% |
To characterize drug delivery with lidocaine topical system 1.8% compared with lidocaine patch 5%, 2 separate pharmacokinetic (PK) studies were performed in normal, healthy volunteers. Study 1 was designed to characterize the PK of lidocaine topical system 1.8%, including apparent dose, and Study 2 was designed to confirm comparable PK between lidocaine topical system 1.8% and lidocaine patch 5%. Safety, adhesion, and skin tolerability of both formulations were also assessed.
Materials and Methods
Study Design
Two Phase 1 comparative PK studies were conducted at a single clinical site (TKL Research, Fair Lawn, NJ), approved by an IntegReview ethical review board (IRB; Austin, TX), and conducted in accordance with the ethical principles originating from the Declaration of Helsinki and amendments and the International Conference on Harmonization Good Clinical Practice guidelines and local regulatory requirements. All subjects provided signed informed consent. The studies were not prospectively registered because the studies described were Phase 1 trials, and FDA guidelines state that Phase 1 trials are not applicable drug clinical trials and therefore are not required to be registered. The studies were retrospectively registered at the request of the Editor at ClinicalTrials.gov with the clinical trial registration numbers NCT04144192 and NCT04149938.
Study 1 was a single-center, open-label, randomized study conducted to characterize the comparative single-dose PK and safety of lidocaine topical system 1.8% versus lidocaine patch 5% and included a single-dose PK lead-in period of lidocaine 0.7 mg/kg bolus intravenous (IV) injection. The lidocaine IV injection lead-in period was included to allow for the determination of the apparent dose of both products via IV clearance (IVCL).
Study 2 was a single-center, open-label, randomized, two-period crossover study conducted to characterize the comparative single-dose PK and safety of lidocaine topical system 1.8% and lidocaine patch 5% and to evaluate adhesion performance and dermal irritation. Apparent dose of lidocaine topical system 1.8% was also determined in Study 2 based on the residual drug analysis of skin, used product, product envelopes, and release liners, which allowed for comparison with the apparent dose observed for the product in Study 1 via IVCL. It is noted that Study 1 was performed with tape reinforcement of both products to ensure continuous contact of the products to the skin. Study 2 was performed without tape reinforcement or any other mechanical reinforcement of the products, consistent with the respective prescribing information.
Patients
Inclusion criteria for Study 1 required men and women ≥18 years of age to be healthy, verified by clinical hematology and chemistry laboratory test results and vital signs measurements. Female subjects were not pregnant and used an acceptable form of birth control if they were of childbearing potential. Key exclusion criteria included use of a prescription medication within 14 days or over-the-counter medications within 7 days prior to administration of study medication, known hypersensitivity or allergy to any of the components of the lidocaine topical system formulation, and any serious illness in the 4 weeks preceding the beginning of treatment that resulted in missed work or hospitalization. Study 1 actively recruited 4 geriatric subjects (ie, subjects ≥65 years of age) who were able to meet all the inclusion and exclusion criteria, which was challenging given the exclusion of prescription product use within 14 days of the study.
Key inclusion criteria for Study 2 were similar to those for Study 1, with the inclusion of healthy (confirmed by medical history, laboratory work, and physical exam) men and women between ≥18 and ≤65 years of age. Female subjects were not pregnant and used an acceptable form of birth control if they were of childbearing potential. Subjects were free of any systemic or dermatologic disorder, which, in the opinion of the investigator, would have interfered with the study results or increased the risk of adverse events (AEs). Key exclusion criteria for Study 2 included unwillingness to refrain from using systemic/topical analgesics for 72 hours prior to enrollment and during the study, current use of opioids, planned dental procedures within 30 days of enrollment, and known hypersensitivity or allergy to any of the components of the product formulations. Key exclusion criteria for the PK portion of Study 2 included use of any drug treatment at the time of the study, use of a prescription medication within 14 days prior to administration of study medication or over-the-counter products (including natural food supplements, vitamins, garlic as a supplement) within 7 days prior to enrollment, and during the PK phase, unwillingness to restrict caffeine or grapefruit juice.
Treatments
Study 1 assessed the comparative PK of lidocaine topical system 1.8% and lidocaine patch 5%, with an IV lead-in period. Subjects (N=56) first received a single IV bolus of 0.7 mg/kg of lidocaine administered on Day 1 using prefilled syringes of lidocaine (20 mg/mL or 100 mg/5 mL, 2% solution; Hospira, Inc., Lake Forest, IL). Following a 7-day washout period, subjects were randomized to receive either lidocaine topical system 1.8% or lidocaine patch 5% (Period 1; Day 7). After the 7-day washout period, subjects crossed over and received the other treatment (Period 2; Day 15). For both periods, each treatment consisted of the application of a set of three lidocaine topical systems 1.8% or three lidocaine patches 5% to the skin of each subject’s back with tape reinforcement over a 12-hour administration period. Tape reinforcement consisted of the use of 3M™ Transpore™ surgical tape (3M, St. Paul, MN) on the four corners of the product. If lifting was observed, additional tape reinforcement was allowed along the product edges. Taping of the surface of the product or use of occlusive overlays was not allowed in the study.
In Study 2, subjects were randomized to receive either lidocaine topical system 1.8% or lidocaine patch 5% (Period 1; Day 1). After a 7-day washout period, subjects crossed over and received the other treatment (Period 2; Day 7). Subjects were confined from Day −1 until Day 3 (after PK blood draw), and from Day 7 until Day 10 (after PK blood draw). Following an overnight fast of at least 10 hours, subjects received three lidocaine topical systems 1.8% or three lidocaine patches 5% applied for 12 hours to a defined area of normal skin on the back; all subjects completed a 7-day washout period following Period 1 and Period 2. During Periods 1 and 2, products were evaluated for degree of adhesion by the trained scorer using the FDA-recommended rating scale, and serial PK samples were collected for the determination of lidocaine plasma concentration at predose (time 0) and at scheduled time points up to 48 hours (Day 3 [Period 1] and Day 10 [Period 2]).17
Blood Sampling and Determination of Drug Concentrations
In both Studies 1 and 2, serial blood samples were collected via venipuncture to measure the plasma concentration of lidocaine at predetermined times after application of the product. Blood samples (5 mL at each time point in Study 1 and 6 mL at each time point in Study 2) were taken during each treatment period at preapplication and then after product application at 2, 4, 6, 9, 10, 11, 12, 13, 14, 16, 18, 20, 22, 24, and 48 hours. In Study 1, samples were taken at 5, 10, 20, 30, 45, 60, and 90 minutes and 2, 3, 4, 8, and 12 hours postdose for the lidocaine IV lead-in period. Blood was collected into heparinized or EDTA-containing tubes and centrifuged at 3000 rpm for 10 minutes at 4°C. Plasma was transferred to plastic tubes and frozen until analysis.
Bioanalytical Methods
Plasma concentrations of lidocaine were determined using a validated liquid chromatography with tandem mass spectrometry method. The assay was linear in the range of 0.500 ng/mL to 300 ng/mL for Study 1 and 0.200 ng/mL to 300 ng/mL for Study 2. The lower limit of quantification was 0.500 ng/mL in Study 1 and 0.200 ng/mL in Study 2.
Safety Assessments
Safety assessments included clinical laboratory tests (blood chemistry, hematology, urine analysis), physical examination, vital signs (blood pressure, pulse rate, body temperature), and resting standard 12-lead electrocardiogram at screening, before product administration, and at specified time points throughout Studies 1 and 2. In the Study 1 lidocaine IV lead-in period, subjects had continuous cardiac monitoring during the IV bolus and up to 4 hours post-administration. AEs were monitored throughout both studies.
Adhesion Assessments
In Study 2, adhesion of the products was assessed immediately after application and at 3, 6, 9, and 12 hours, just prior to product removal. Adhesion was scored according to the FDA-recommended scale, where 0: ≥90% adhered (essentially no lift off the skin); 1: ≥75% to <90% adhered (some edges only lifting off the skin); 2: ≥50% to <75% adhered (less than half of the system lifting off the skin); 3: <50% adhered but not detached (more than half the system lifting off the skin without falling off); and 4: 0% (complete detachment).17
Skin Irritation Assessments
In Study 2, dermal response was evaluated after product removal according to the scoring system recommended by FDA guidance.18 The scoring system is based on a scale of 0 to 7 (0=no evidence of irritation; 1=minimal erythema, barely perceptible; 2=definite erythema; 3=erythema and papules/pustules; 4=definite edema; 5=erythema, edema, and papules; 6=vesicular eruption; 7=strong reaction spreading beyond the application site).
Statistical Analyses
In Studies 1 and 2, all statistical processing was performed using the SAS® system (Version 9.2). In Study 1, all PK parameters were summarized by treatment period, using mean, standard deviation (SD), 95% confidence interval (CI), minimum, median, and maximum. Cmax, AUC0-t, and AUC0-∞ were tested for bioequivalence between the two product treatments. The log-transformed quantities were analyzed in a mixed effects analysis of variance model, with treatment, sequence, and period as fixed effects and subject within sequence as a random effect. Estimates of the difference between treatment least square means were obtained from the model with 90% CIs. These estimates and CIs were back-transformed onto the original scale to provide estimates of the geometric means for each treatment and the ratio of the geometric means. By convention, if the 90% CI of the ratio of geometric means was within 80% to 125%, then the two treatments were considered bioequivalent.
Results
Patient Disposition and Baseline Characteristics
In Study 1, a total of 58 subjects were enrolled and 56 (96.6%) completed the study. One subject discontinued because of an AE (urticaria) during the IV lead-in period, and 1 subject withdrew consent. The median age was 39.5 years with four subjects over the age of 65. More than half of subjects were women (62.1%) (Table 2). In Study 2, a total of 54 subjects were randomized; 28 subjects were randomized to Sequence 1 and 26 subjects were randomized to Sequence 2. All subjects (100%) in both Sequence 1 and Sequence 2 completed the study. Among all subjects, the mean age was 41.1 years with an SD of 10.3 years. Thirty-nine subjects (72.2%) were men, and 15 subjects (27.8%) were women (Table 3).
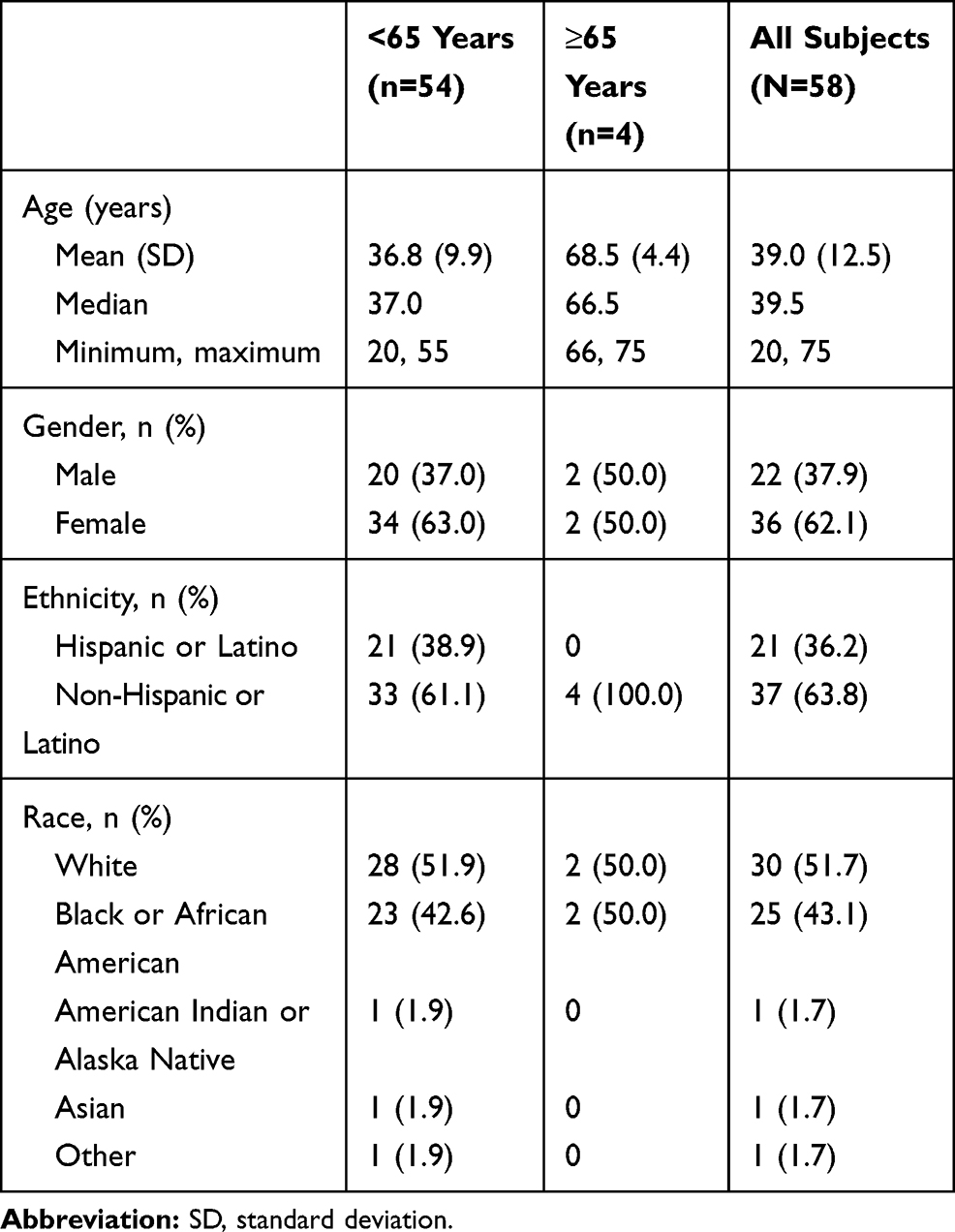 |
Table 2 Demographics and Baseline Characteristics (Study 1) |
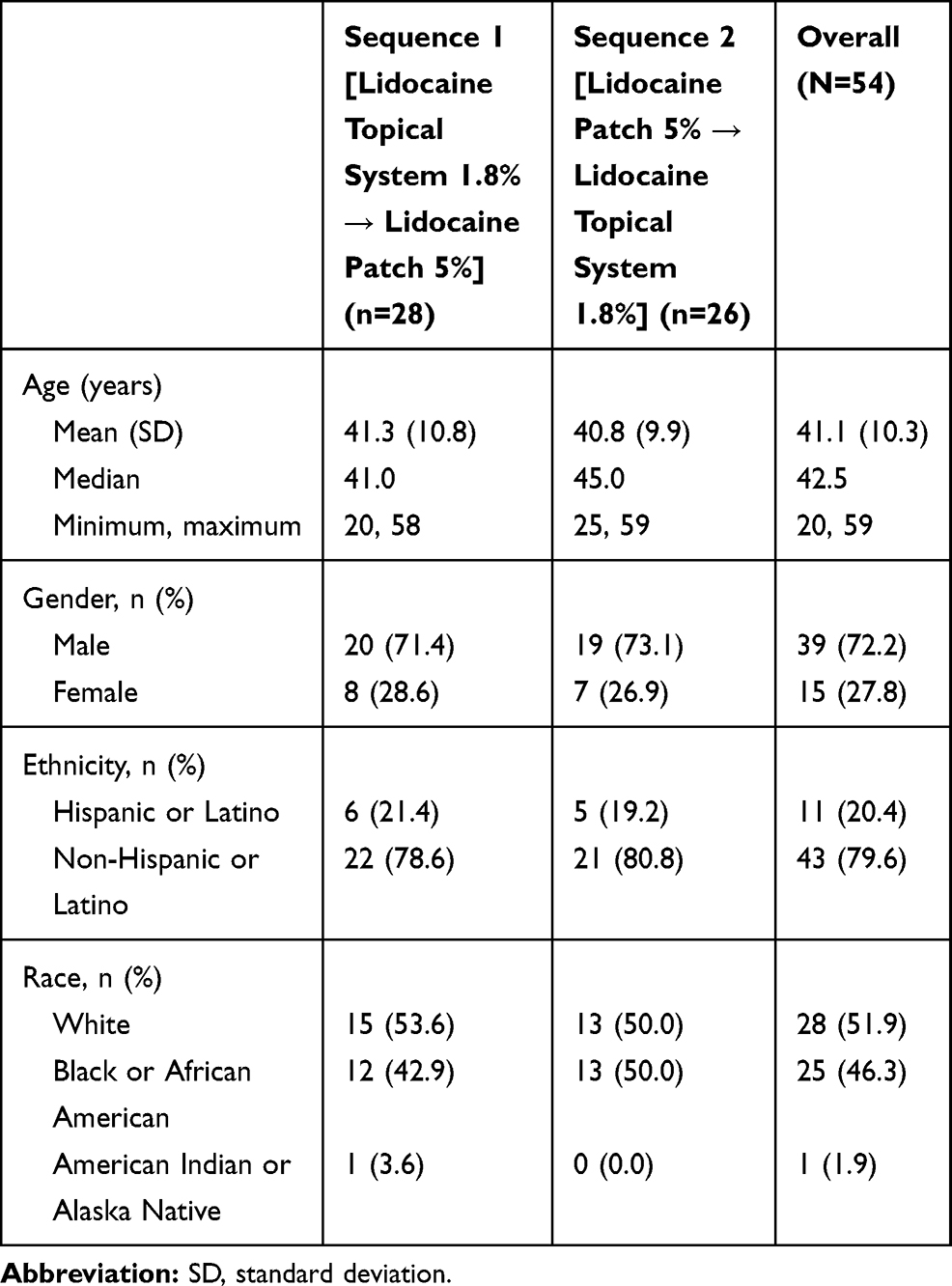 |
Table 3 Demographics and Baseline Characteristics (Study 2) |
PK Assessments
In Study 1, 56 of 58 subjects completed all portions of the study and were included in the PK analysis. The PK of lidocaine delivered as a 0.7 mg/kg IV dose was characterized by a high Cmax (1778.39 ± 2555.09 ng/mL), short Tmax (median of 0.13 hours [0.1–4.0 hours]), and short T1/2 (2.92 ± 0.52 hours) (Table 4, Figure 1). Compared with the IV bolus, the PK profiles of both the lidocaine topical system 1.8% and lidocaine patch 5% were characterized by a more than 20-fold lower Cmax (80.45 ± 25.53 ng/mL for lidocaine topical system 1.8% and 75.38 ± 29.96 ng/mL for lidocaine patch 5%), a longer Tmax (median of 13.95 hours [range 9.0–20.0 hours] for lidocaine topical system 1.8% and 12.69 hours [range 9.1–24.0 hours] for lidocaine patch 5%), and a longer T1/2 (5.56 ± 1.67 hours for lidocaine topical system 1.8% and 6.27 ± 1.77 hours for lidocaine patch 5%). Mean lidocaine plasma concentrations versus time profiles for lidocaine topical system 1.8% and lidocaine patch 5% are shown in Figure 2 and demonstrate a similar PK profile.
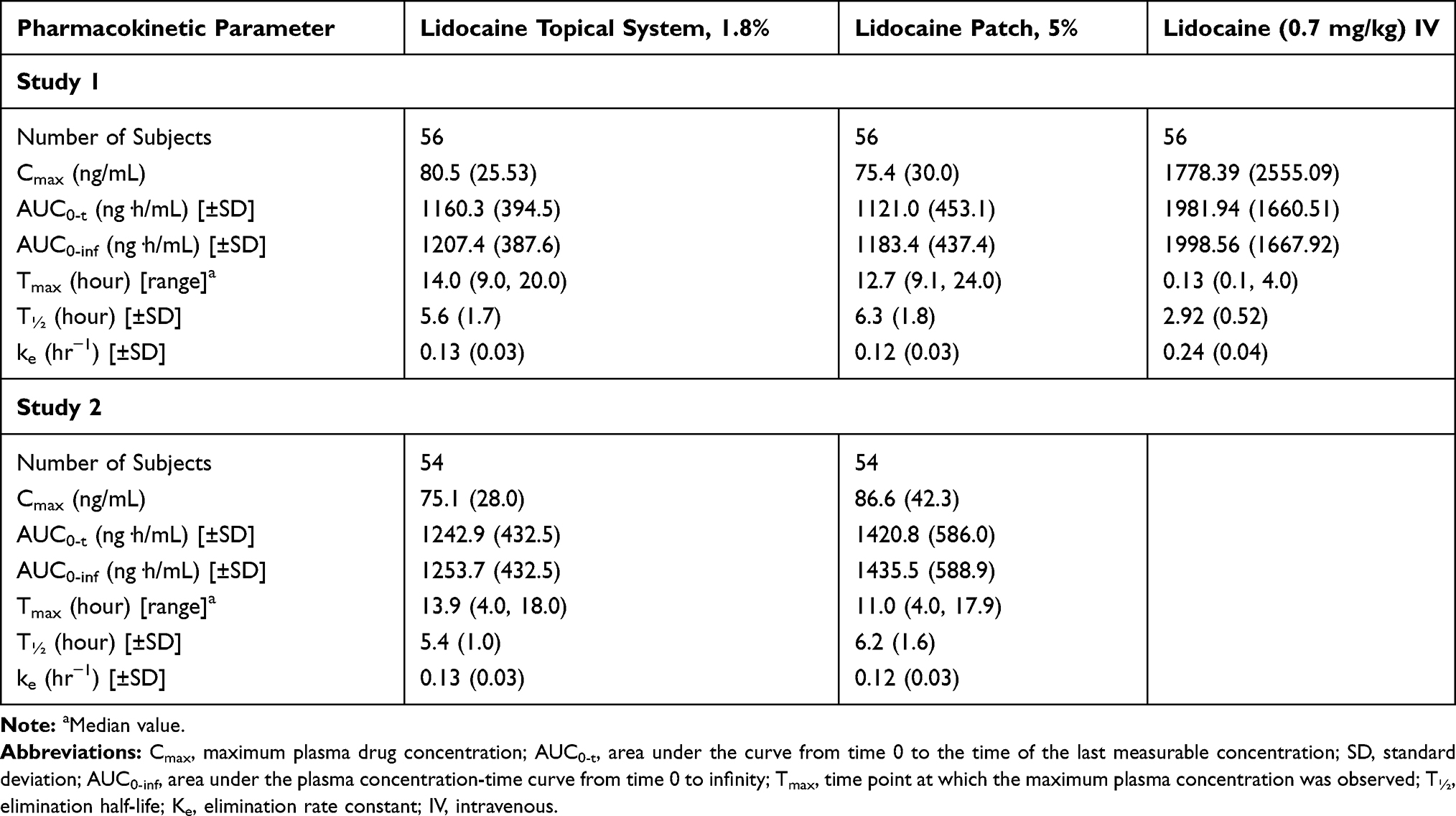 |
Table 4 Pharmacokinetic Parameter Estimates of Lidocaine in Plasma (per Protocol Population) (Studies 1 and 2) |
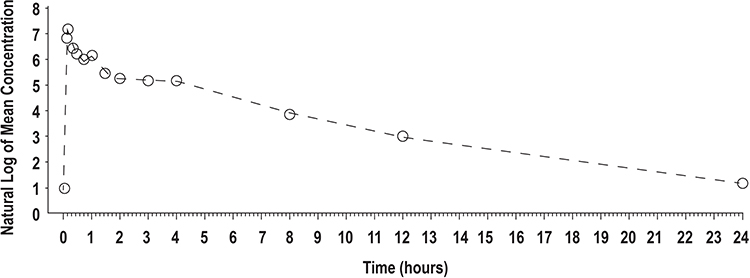 |
Figure 1 Mean lidocaine plasma concentrations versus time profiles for IV lidocaine 0.7 mg/kg, semilog scale (N = 56) (Study 1). Time = 0 is pre-dose measurement. Abbreviation: IV, intravenous. |
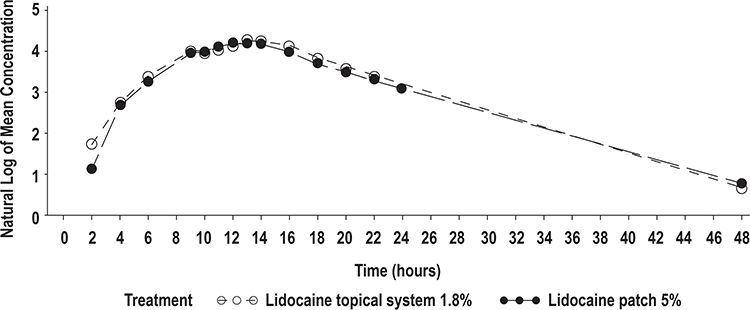 |
Figure 2 Mean lidocaine plasma concentrations versus time profiles for lidocaine topical system 1.8% (N = 56) and lidocaine patch 5%, semilog scale (N = 54) (Study 1). Time = 0 is pre-dose measurement. |
Bioequivalence was assessed by calculating the 90% CI for the geometric least square means ratio of lidocaine topical system 1.8%/lidocaine patch 5% for AUC0-t, AUC0-inf, and Cmax, all of which were entirely within the acceptance range of 0.8 to 1.25, indicating that the products have a very similar rate and extent of absorption. The 90% CIs of all parameters were within the range of 80% to 125%, demonstrating that lidocaine topical system 1.8% and lidocaine patch 5% are bioequivalent (Table 5).
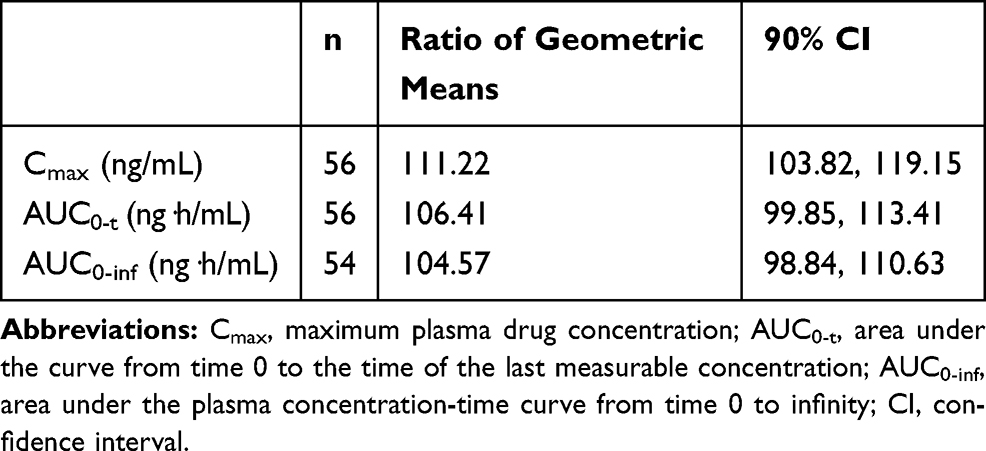 |
Table 5 Analysis of Bioequivalence (Study 1) |
In Study 2, a total of 54 subjects were enrolled and included in the PK analysis. Like in Study 1, the PK parameters of lidocaine from lidocaine topical system 1.8% and lidocaine patch 5% were similar (Table 4). Mean lidocaine plasma concentrations versus time profiles for lidocaine topical system 1.8% and lidocaine patch 5% are shown in Figure 3. The bioequivalence of lidocaine topical system 1.8% compared with lidocaine patch 5% is presented in Table 6. The ratio of geometric least square means and 90% CIs for AUC0-t, AUC0-∞, and Cmax were entirely within the acceptance range of 0.8 to 1.25 for the PK parameter population and by period.
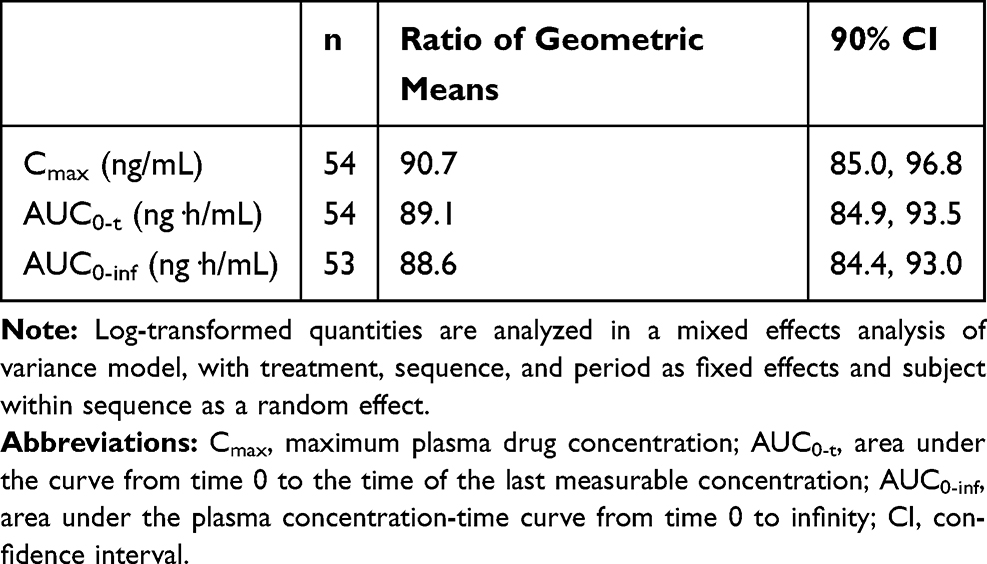 |
Table 6 Analysis of Bioequivalence (Study 2) |
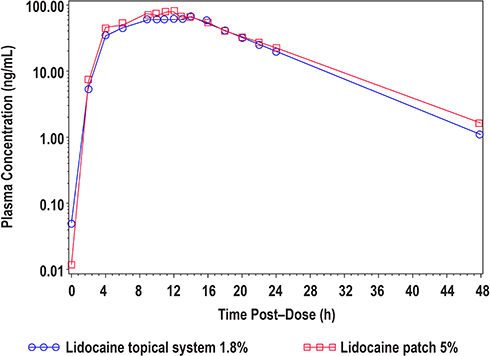 |
Figure 3 Mean lidocaine plasma concentrations versus time profiles for lidocaine topical system 1.8% and lidocaine patch 5% (Study 2). |
Apparent Dose
In Study 1, systemic exposure for both lidocaine topical system 1.8% and lidocaine patch 5% was estimated by applying the clearance rate observed after IV lidocaine bolus by the formula DoseAPP-IV = CLIV × AUCPRODUCT to ascertain the dose absorbed from the products. The results are described in Table 7 and show that 43.99 ± 17.62 mg of lidocaine was absorbed from three lidocaine topical systems 1.8% and 43.74 ± 18.22 of lidocaine was absorbed from three lidocaine patches 5%. While both products deliver the same amount of lidocaine, the bioavailability is very different due to the difference in drug load per system/patch (41% ± 16% for lidocaine topical system 1.8% vs 2% ± 1% for lidocaine patch 5%). These results are consistent with the prescribing information for lidocaine patch 5%, which indicates a bioavailability of 3% ± 2%, albeit on the lower side of the labeled range.12
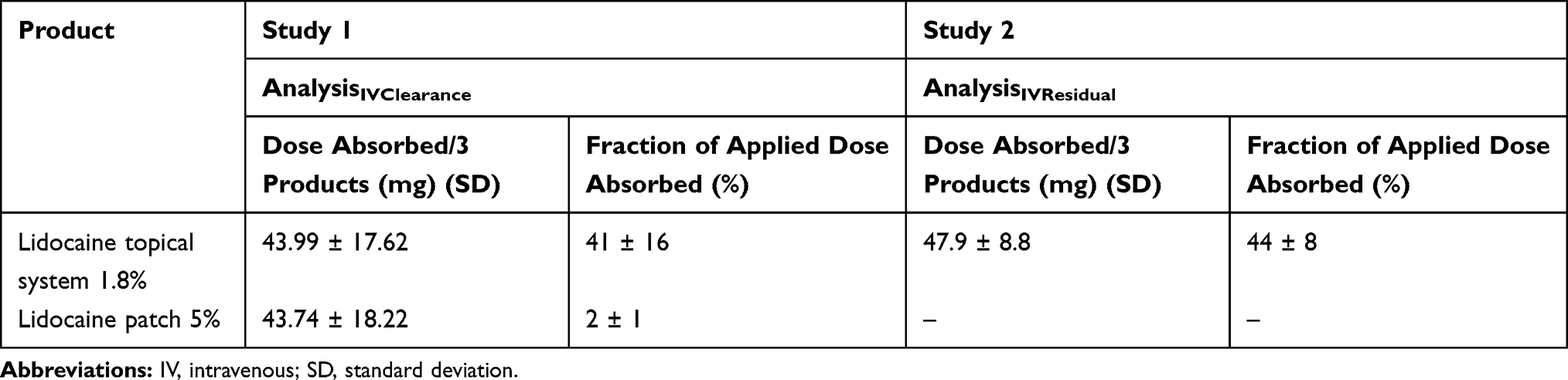 |
Table 7 Summary of Apparent Dose Absorbed (Studies 1 and 2) |
In Study 2, systemic exposure for lidocaine topical system 1.8% was estimated by measuring the amount of residual lidocaine in the used topical system, liner, envelope, and surface of the skin and subtracting the total residual amount from the amount of lidocaine in an unused topical system. The total residual amount of lidocaine recovered was 18.4 ± 2.94 mg/topical system (of the possible 36 mg). As shown in Table 7, 47.9 ± 8.8 mg of lidocaine was absorbed from three lidocaine topical systems 1.8%, with estimated bioavailability of 44% ± 8%. These results are consistent with the apparent dose determined by IVCL observed with lidocaine topical system 1.8% in Study 1.
Adhesion Assessments
In addition to characterizing the PK profile, Study 2 also included assessments of product adhesion. Lidocaine topical system 1.8% had a cumulative mean adhesion score of 0.22 that was statistically superior to lidocaine patch 5%, with a mean score of 0.86 (P<0.001). None of the lidocaine topical systems 1.8% had a score greater than 2. In contrast, 5.0% (8 patches) of the lidocaine patches 5% had a score of 3 (>0% to <50% adhered), and 3.1% (5 patches) became completely detached (score of 4) (Table 8).
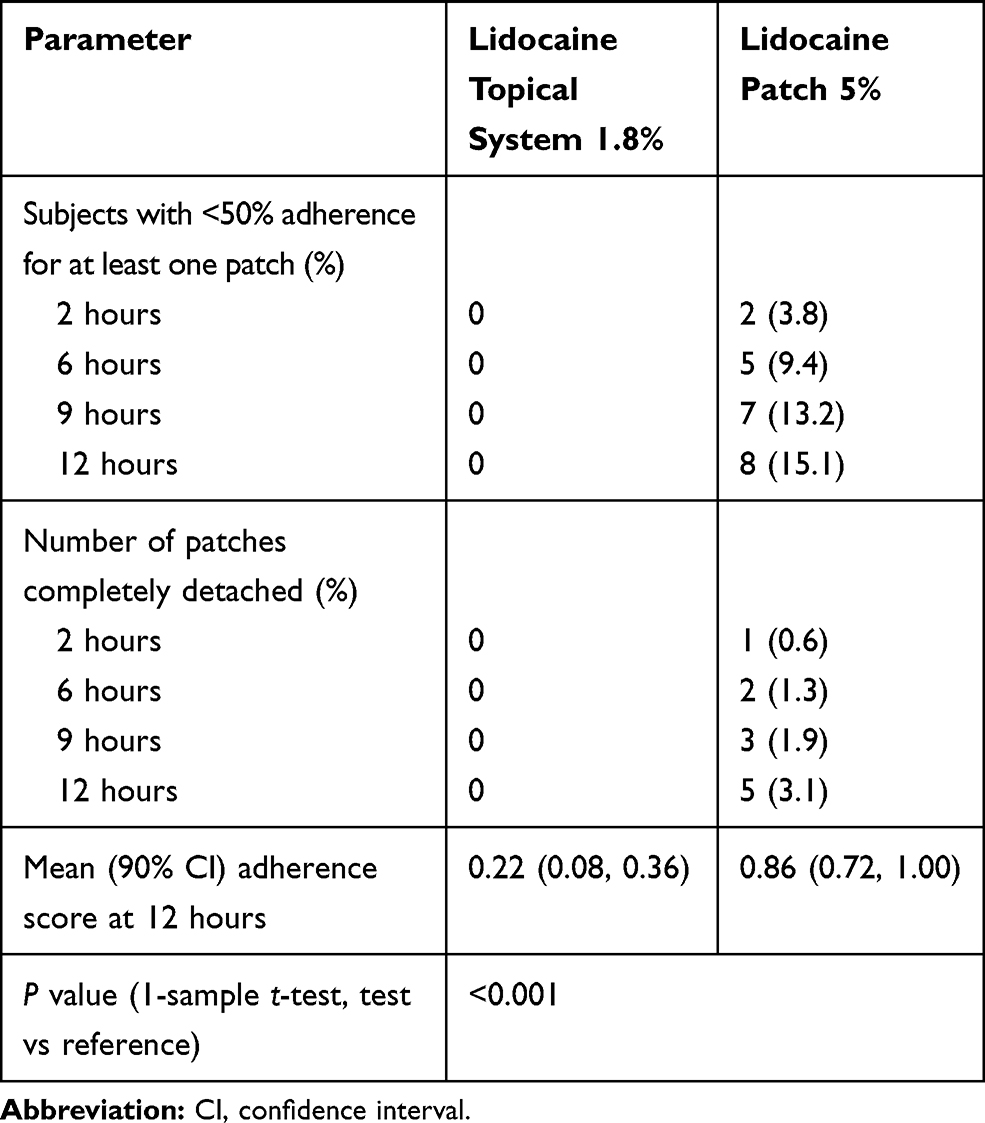 |
Table 8 Summary of Adhesion (Study 2) |
Skin Irritation Assessments
In Study 2, skin irritation was assessed after product removal. The mean sums of irritation scores for the three patches per subject (ie, total score for all three patches) for lidocaine topical system 1.8% and lidocaine patch 5% at 12 hours following application were quite similar. At 12 hours, 63.0% of subjects treated with lidocaine topical system 1.8% and 55.6% of subjects treated with lidocaine patch 5% had an irritation score of 0, indicating no evidence of irritation. Overall, 20 of 54 (37.0%) lidocaine topical system 1.8% subjects compared with 24 of 54 (44.4%) lidocaine patch 5% subjects had at least some evidence of irritation. At a 90% CI, the mean sums of irritation scores were 1.15 and 1.31, respectively, which is not a significant difference (P=0.406). The mean irritation score per patch was <1, which is not considered to be clinically significant (Table 9).
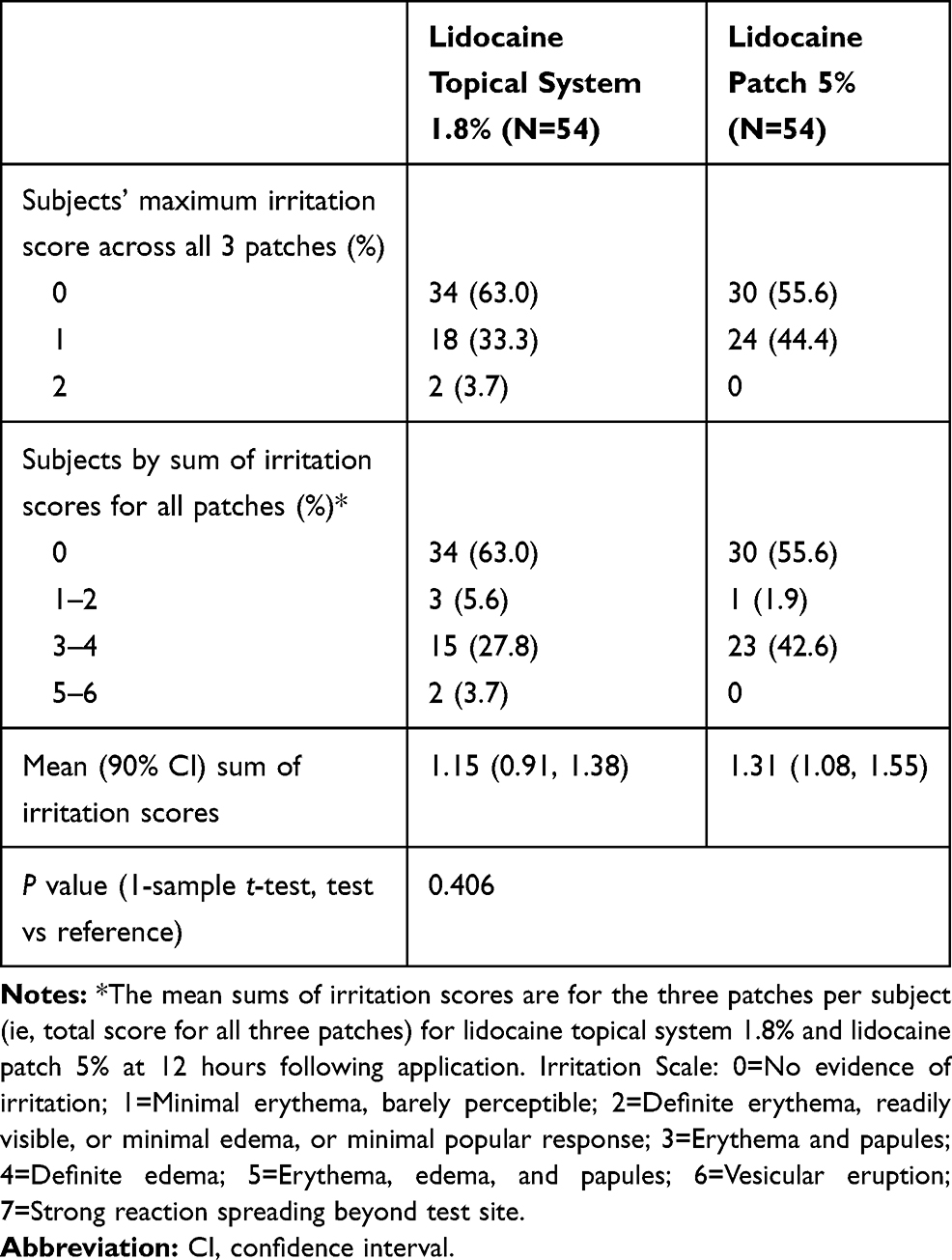 |
Table 9 Summary of Irritation Analysis (Study 2) |
Safety
In Study 1, 12 subjects (20.7%) experienced a total of 17 AEs; one subject experienced an AE that led to study discontinuation, which was not related to the study drug. The overall incidence of AEs was 3.6% in subjects receiving lidocaine topical system 1.8% and 10.5% in subjects receiving lidocaine patch 5% (Table 10). In the group of subjects receiving lidocaine topical system 1.8%, one subject experienced dizziness, headache, and abdominal pain, and a second subject experienced urticaria. All of these AEs were considered possibly related to study medication. In the group of subjects receiving lidocaine patch 5%, one subject experienced anemia and a second subject experienced elevated alanine transaminase levels that were considered possibly related to study medication.
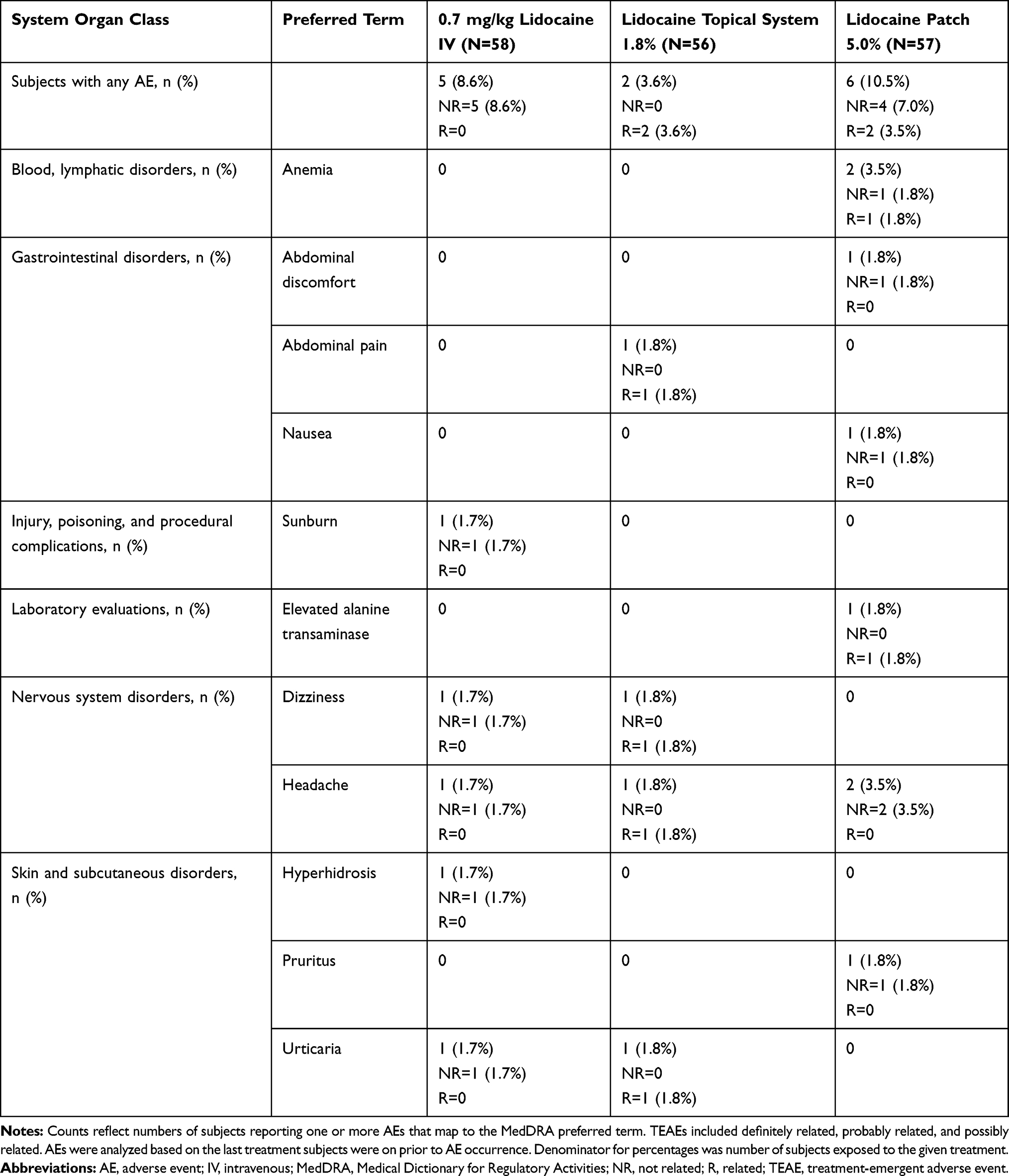 |
Table 10 Overall Summary of Adverse Events (Study 1) |
In Study 2, two AEs were recorded among two subjects (3.7%). One subject (1.9%) receiving lidocaine topical system 1.8% experienced acne and one subject (1.9%) receiving lidocaine patch 5% experienced syncope. Neither AE was judged to be treatment-related. There were no serious or severe AEs and no AEs that led to discontinuation of study medication or the study (Table 11).
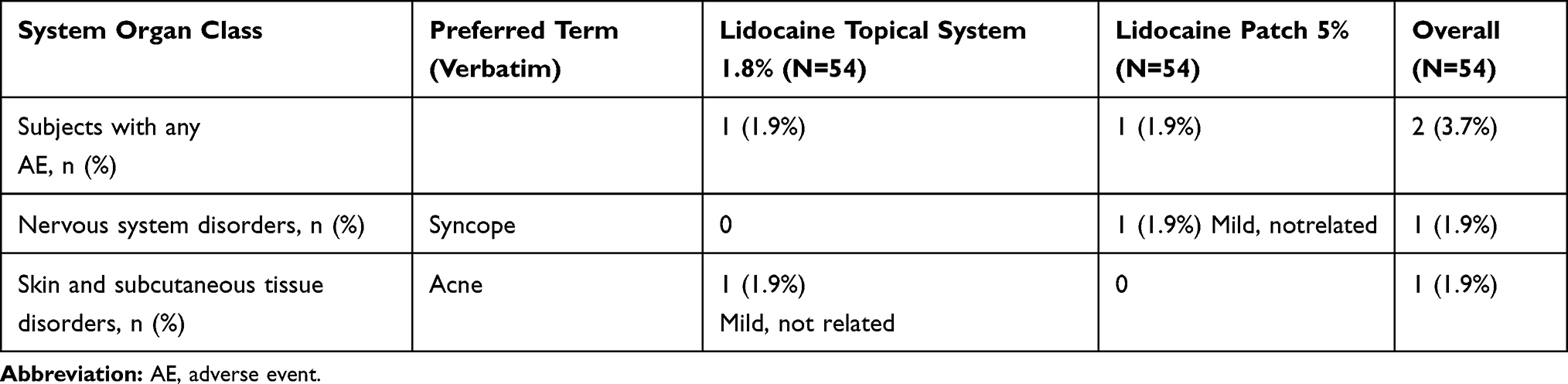 |
Table 11 Overall Summary of Adverse Events (Safety Population) (Study 2) |
Discussion
The PK profiles of two different lidocaine-containing topical systems/patches (ZTlido® 1.8% and Lidoderm® 5%) were compared in two studies in normal, healthy subjects with regard to the systemic rate of exposure (AUC0-t, AUC0-inf) and the extent of absorption (Cmax). The geometric means for these PK parameters were similar, and their 90% CIs were within the predefined bioequivalence range of 80% to 125%.19 Thus, based on these results, it can be concluded that the two products, despite their differences in drug load and strength, are bioequivalent. Both Study 1 and Study 2 were performed to support marker approval in the United States by establishing a pharmaceutical bridge between lidocaine topical system 1.8% and lidocaine patch 5%. In Study 1, the products were reinforced at the corners with tape to ensure that there was no lifting from the skin; however, taping may have the potential to alter drug absorption. Study 2 replicated the design of Study 1 without tape reinforcement. Both studies confirm bioequivalence between lidocaine topical system 1.8% and lidocaine patch 5%. While Study 2 shows that the two products are bioequivalent without tape reinforcement, the disparate adhesion performance noted may be a clinical/PK performance consequence in the real world, as single-dose PK studies inherently limit subject mobility within confinement of the clinic that is not representative of real-life challenges.
Both products were well tolerated, with an overall incidence of treatment-related adverse events treatment-related AEs in the lidocaine topical system 1.8% group and the lidocaine patch 5% group of 3.6% and 3.5%, respectively, in Study 1. No treatment-related AEs were observed in Study 2 for either product. Dermal response scores (skin tolerability after removal) were similar between lidocaine topical system 1.8% and lidocaine patch 5%.
In addition to demonstrating bioequivalence between lidocaine topical system 1.8% and lidocaine patch 5% and showing that the two products have comparable safety and skin tolerability profiles, the data from the present study highlight that lidocaine topical system 1.8% presents several advantages over lidocaine patch 5%, in terms of drug load, residual drug after use, and adhesion. Lidocaine topical system 1.8% delivers the same amount of lidocaine as lidocaine patch 5% with a 19-fold reduction in drug load. It also has low residual drug content after use (~18 mg). Topical delivery systems that minimize drug load and residual drug have an inherently lower risk of inadvertent exposure or systemic safety risks (eg, in the event of dose-dumping). Lidocaine topical system 1.8% is also a lighter, thinner “patch” that demonstrated superior adhesion to lidocaine patch 5% at the end of the labeled wear duration (12 hours).9,12 Adequate adhesion is a critical quality attribute for topical delivery systems; if the product lifts or detaches during wear, dosing may be compromised and there is an increased risk of inadvertent exposure to others.20 This risk is emphasized in the lidocaine patch 5% label with a statement on level of residual drug (665 mg) and bolded warning pertinent to the risk of this level of residual drug. The label for lidocaine topical system 1.8% maintains a warning of risks associated with residual drug, but only states that a used product has “residual drug after use” and has an unbolded warning. This study shows that lidocaine topical system 1.8% is an efficient lidocaine delivery system with improved adhesion relative to lidocaine patch 5% and comparable dermal safety and reduction of residual drug after use.
Importantly, these data demonstrate the discrepancy between product strength and the amount of drug that is delivered to the patient. In this case, a lidocaine topical system with a product strength of 1.8% and 36 mg drug load is bioequivalent (ie, delivers the same level of drug) to a lidocaine patch with a product strength of 5% and 700 mg drug load. This is counterintuitive, as one could reasonably expect that a topical system or patch with a higher strength (2.8-fold) and increased drug load (19-fold) would deliver significantly more drug to the skin; however, topical system delivery technologies have advanced such that product strength and drug load are not necessarily indicators of biopharmaceutical efficiency, and thus, can be misleading and may contribute to physician, pharmacy, and patient confusion over strength and dose.
Conclusion
Overall, bioequivalence was demonstrated between lidocaine topical system 1.8% and lidocaine patch 5%, and thus, these products can be expected to have similar efficacy in managing PHN pain. A statistical comparison of the single-time adhesion scores at 12 hours was statistically significant in favor of lidocaine topical system 1.8% over lidocaine patch 5%. Both products were well tolerated as a single application in healthy adult human subjects. It may be reasonable to anticipate that as topical system/patch technology progresses, the strength presentations as a percentage of drug-to-adhesive will become increasingly meaningless. Likewise, the drug load within these topical products may not be an adequate indication of drug delivery or exposure.
Data Sharing Statement
The authors certify that this manuscript reports original clinical trial data. Individual participant data that underlie the results reported in this article after deidentification (text, tables, figures, and appendices) are available, including the study protocol. Data requests should be submitted in the form of a research proposal to [email protected] for up to 36 months after the publication date.
Acknowledgments
The authors would like to thank study investigator Phillip LaStella, Principal Investigator of TKL Research Inc., and Monil Shah for the design and management of the study, the clinical staff, and the volunteers who made this study possible. Sandi Staros, PhD, of Synergy Medical Education, provided medical writing support.
Disclosure
The abstract of this paper was presented at the PAINWeek 2018 Conference (September 4–8, 2018; Las Vegas, NV) as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in 2018 Postgraduate Medicine: Patel K, Gudin J, Vought K, Shah M, Grimes D, LaStella P, Greuber E. A Randomized, Comparative Pharmacokinetic (PK) Study of ZTlido™ Lidocaine Topical System 1.8% (36 mg) Versus Lidoderm® Lidocaine Patch 5% (700 mg). (2018) PAINWeek Abstract Book 2018, Postgrad Med, 130:sup1, 1–91, DOI: 10.1080/00325481.2018.1512253.
In the past year, Dr. Gudin has been an advisor, consultant, or speaker’s bureau member for Averitas Pharma, BioDelivery Sciences International, Inc., Daiichi Sankyo, Hisamitsu Pharmaceutical Co. Inc., Nektar Therapeutics, Salix Pharmaceuticals, and Scilex Pharmaceuticals Inc. Dr. Fudin has been an advisor, consultant, or speaker’s bureau member for Abbott Laboratories, AcelRx Pharmaceuticals, Acutis Diagnostics Inc., AstraZeneca, BioDelivery Sciences International, Inc., Daiichi Sankyo, Firstox Laboratories, GlaxoSmithKline, Human Half-Cell, Inc., Quest Diagnostics, Salix Pharmaceuticals, and Scilex Pharmaceuticals Inc. Dr. Nalamachu has been a consultant/speaker and has received honorarium and grants from Allergan, AstraZeneca, BioDelivery Sciences International, Inc., Collegium Pharmaceuticals, Daiichi Sankyo, Depomed (Assertio Therapeutics), Eli Lilly and Company, Endo Pharmaceuticals, Ferring Pharmaceuticals, Insys Pharmaceuticals, Pernix Therapeutics, Pfizer Inc., Purdue Pharmaceuticals, and Scilex Pharmaceuticals Inc. Drs. Vought and Greuber have patent application 62/762753 pending to Scilex Pharmaceuticals Inc., and Dr Vought is head of R&D responsible for the design and conduct of the study. Drs. Greuber, Patel, and Vought are employees of Scilex Pharmaceuticals Inc., and they report grants and personal fees from Scilex Pharmaceuticals Inc. The authors report no other conflicts of interest in this work.
References
1. Massengill JS, Kittredge JL. Practical considerations in the pharmacological treatment of postherpetic neuralgia for the primary care provider. J Pain Res. 2014;7:125–132. doi:10.2147/JPR.S57242
2. Shah S, Singaraju S, Einstein A, Sharma A. Herpes zoster: clinicocytopathological insight. J Oral Maxillofac Pathol. 2016;20(3):547. doi:10.4103/0973-029X.190968
3. Le P, Rothberg M. Herpes zoster infection. BMJ. 2019;364:kk5095. doi:10.1136/bmj.k5095
4. Hadley GR, Gayle JA, Ripoll J, et al. Post-herpetic neuralgia: a review. Curr Pain Headache Rep. 2016;20(3):17. doi:10.1007/s11916-016-0548-x
5. Jones J. Postherpetic neuralgia. J Pain Palliat Care Pharmacother. 2015;29(2):180–181. doi:10.3109/15360288.2015.1037520
6. Johnson RW, Rice AS, Solomon CG. Clinical practice. Postherpetic neuralgia. N Engl J Med. 2014;371(16):1526–1533. doi:10.1056/NEJMcp1403062
7. Kondamudi PK, Tirumalasetty PP, Malayandi R, Mutalik S, Pillai R. Lidocaine transdermal patch: pharmacokinetic modeling and in vitro-in vivo correlation (IVIVC). AAPS PharmSciTech. 2016;17(3):588–596. doi:10.1208/s12249-015-0390-1
8. Stanos SP, Galluzzi KE. Topical therapies in the management of chronic pain. Postgrad Med. 2013;125(4 Suppl 1):25–33. doi:10.1080/00325481.2013.1110567111
9. ZTLIDO®. (Lidocaine Topical System): [Prescribing Information]. San Diego, CA, USA: Scilex Pharmaceuticals; 2018.
10. Cohen JI, Solomon CG. Herpes zoster. N Engl J Med. 2013;369(3):255–263. doi:10.1056/NEJMcp1302674
11. Gudin J, Nalamachu S. Utility of lidocaine as a topical analgesic and improvements in patch delivery systems. Postgrad Med. 2020;132(1):28–36. doi:10.1080/00325481.2019.1702296.
12. LIDODERM®. (Lidocaine Patch 5%): [Prescribing Information]. Malvern, PA, USA: Endo Pharmaceuticals; 2018.
13. Rowbotham MC, Davies PS, Verkermpinck C, Galer BS. Lidocaine patch: double-blind controlled study of a new treatment method for post-herpetic neuralgia. Pain. 1996;65(1):39–44. doi:10.1016/0304-3959(95)00146-8
14. Galer B, Rowbotham M, Perander J, Friedman E. Topical lidocaine patch relieves postherpetic neuralgia more effectively than a vehicle topical patch: results of an enriched enrollment study. Pain. 1999;80(3):533–538. doi:10.1016/S0304-3959(98)00244-9
15. Hans G, Sabatwoski R, Binder A, Boesl I, Rogers P, Baron R. Efficacy and tolerability of 5% lidocaine medicated plaster for the topical treatment of post-herpetic neuralgia: results of a long-term study. Curr Med Res Opin. 2009;25(5):1295–1305. doi:10.1185/03007990902901368
16. Sabatwoski R, Hans G, Tacken I, Kapanadze S, Buchheister B, Baron R. Safety and efficacy outcomes of long-term treatment up to 4 years with 5% lidocaine medicated plaster in patients with post-herpetic neuralgia. Curr Med Res Opin. 2012;28(8):1337–1346. doi:10.1185/03007995.2012.707977
17. Guidance for Industry. Assessing adhesion with transdermal and topical delivery systems for ANDAs. Rockville, MD, USA; 2018. Available from: https://www.fda.gov/media/98634/download.
18. Guidance for Industry. Assessing the irritation and sensitization potential of transdermal and topical delivery systems for ANDAs. Rockville, MD, USA; 2018. Available from: https://www.fda.gov/media/117569/download.
19. Guidance for Industry. Statistical approaches to establishing bioequivalence. Rockville, MD, USA; 2001. Available from: https://www.fda.gov/media/70958/download.
20. Wokovich AM, Prodduturi S, Doub WH, Hussain S, Buhse LF. Transdermal drug delivery system (TDDS) adhesion as a critical safety, efficacy and quality attribute. Eur J Pharm Biopharm. 2006;64(1):1–8. doi:10.1016/j.ejpb.2006.03.009
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.