Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
A randomized, double-blind study of the efficacy and tolerability of extended-release quetiapine fumarate (quetiapine XR) monotherapy in patients with major depressive disorder
Authors Wang G, McIntyre A, Earley W, Raines S, Eriksson H
Received 20 June 2013
Accepted for publication 2 October 2013
Published 30 January 2014 Volume 2014:10 Pages 201—216
DOI https://doi.org/10.2147/NDT.S50248
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Gang Wang,1 Alexander McIntyre,2 Willie R Earley,3 Shane R Raines,3 Hans Eriksson4
1Beijing Anding Hospital, Capital Medical University, Beijing, People's Republic of China; 2Department of Psychiatry, Penticton Regional Hospital, Penticton, BC, Canada; 3AstraZeneca Pharmaceuticals, Wilmington, DE, USA; 4AstraZeneca R&D, Södertälje, Sweden
Objectives: To evaluate the efficacy and tolerability of once-daily extended release quetiapine fumarate (quetiapine XR) monotherapy in patients with major depressive disorder (MDD).
Patients and methods: This was a 10-week (8-week active treatment/2-week post-treatment) randomized, double-blind, placebo- and active-controlled study (D1448C00004). Patients received quetiapine XR 150 mg/day, escitalopram 10 mg/day, or placebo; patients with an inadequate response (<20% improvement in Montgomery–Åsberg Depression Rating Scale [MADRS] total score) at week two received double-dose treatment. The primary end point was week eight change from randomization in MADRS total score. Secondary end points included MADRS response (≥50% improvement) and remission (score ≤8); Hamilton Rating Scale for Depression total and item 1; Hamilton Rating Scale for Anxiety total, psychic, and somatic; Clinical Global Impressions – Severity of Illness total; Pittsburgh Sleep Quality Index (PSQI) global; and Quality of Life Enjoyment and Satisfaction Questionnaire – Short Form percentage maximum total scores. Tolerability was assessed throughout.
Results: A total of 471 patients was randomized. No significant improvements in MADRS total score were observed at week eight (last observation carried forward) with either active treatment (quetiapine XR, -17.21 [P=0.174]; escitalopram, -16.73 [P=0.346]) versus placebo (-15.61). There were no significant differences in secondary end points versus placebo, with the exception of week-eight change in PSQI global score (quetiapine XR, -4.96 [P<0.01] versus placebo, -3.37). Mixed-model repeated-measures analysis of observed-case data suggested that the primary analysis may not be robust. Most commonly reported adverse events included dry mouth, somnolence, and dizziness for quetiapine XR, and headache and nausea for escitalopram.
Conclusion: In this study, neither quetiapine XR (150/300 mg/day) nor escitalopram (10/20 mg/day) showed significant separation from placebo. Both compounds have been shown previously to be effective in the treatment of MDD; possible reasons for this failed study are discussed. Quetiapine XR was generally well tolerated, with a profile similar to that reported previously.
Keywords: antidepressive agents (pharmacological action), antipsychotic agents, sustained-release preparations, treatment efficacy, clinical trial, Phase III
Introduction
Despite the plethora of available antidepressants (>25 agents are currently approved for major depressive disorder [MDD]), many patients discontinue treatment due to side effects.1 Furthermore, a considerable proportion of patients fail to achieve remission following initial treatment, eg, only 28% of patients in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study achieved remission following treatment with citalopram.2 Those patients who do not respond to their treatment or are unable to tolerate it may receive a number of different pharmacotherapies until the optimum one is identified. This suggests a need for new treatment options for patients with MDD.
Once-daily extended release quetiapine fumarate (quetiapine XR) is approved in the US and Europe for the treatment of schizophrenia, bipolar disorder (both bipolar mania and bipolar depression), and more recently as adjunctive treatment for patients with MDD who have had suboptimal response to antidepressant monotherapy.3,4 It is also licensed as a monotherapy for the treatment of MDD in some countries, including Australia and Canada.5,6 The present randomized, placebo-controlled study is part of the clinical development program investigating quetiapine XR in patients with MDD. To date, three acute monotherapy studies,7–9 two acute adjunct studies,10,11 one maintenance study,12 and one acute monotherapy study in the elderly13 have reported positive efficacy and acceptable tolerability of quetiapine XR in patients with MDD.
The design of the current study (D1448C00004) was identical to study D1448C000037: a modified fixed-dose design consisting of a fixed initial dose for 2 weeks followed by a doubling of the dose of randomized treatment for those patients not responding to therapy at week two. The modified fixed-dose design was intended to reflect both clinical practice and the recommendation that nonresponsive patients receive an increase in their medication dose.14
The primary hypothesis of the current study was that quetiapine XR would be more effective than placebo in reducing Montgomery–Åsberg Depression Rating Scale (MADRS) total score from randomization to week eight in adult patients with MDD. However, in the primary analysis, neither quetiapine XR nor the active control escitalopram separated from placebo, which is an unexpected result, as both agents have demonstrated efficacy in this indication. In addition to presenting the results of the study, this article also discusses the most likely explanations for this failed study.
Patients and methods
Study design
This 10-week, multicenter, parallel-group, placebo- and active-controlled, double-blind, randomized, Phase III study (D1448C00004, Amber);15 consisted of a 1- to 4-week enrollment/washout period, an 8-week randomized treatment period, and a 2-week drug-discontinuation/tapering follow-up period.
The study was performed in accordance with the Declaration of Helsinki and International Conference on Harmonisation/Good Clinical Practice guidelines. All patients provided written informed consent.
Patients
Male or female outpatients (aged 18–65 years) with a documented diagnosis meeting the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV) criteria for MDD (single episode/recurrent) and confirmed by Mini-International Neuropsychiatric Interview16 were eligible for inclusion in the study. Patients were required to have a Hamilton Rating Scale for Depression (HAM-D)17 17-item total score ≥22 and HAM-D item 1 (depressed mood) score ≥2 at both enrollment and randomization.
Exclusion criteria included: diagnosis of any DSM-IV Axis I disorder other than MDD within 6 months prior to enrollment or any DSM-IV Axis II disorder impacting on the patient’s current psychiatric status; a current depressive episode lasting >12 months or <4 weeks in duration; a history of inadequate response to treatment (two or more classes of antidepressants each for ≥6 weeks) during the current depressive episode; a DSM-IV diagnosis of substance or alcohol abuse within 6 months prior to enrollment; a current serious suicidal or homicidal risk; a HAM-D item 3 (suicide) score ≥3 or a suicide attempt within the past 6 months; or a clinically relevant medical illness or clinically relevant findings (including laboratory tests or electrocardiogram [ECG]).
Randomization
Randomization was neither site- nor country-specific, and was generated using a computer-based system. Randomization numbers were allocated via a computer-based randomization system in a strictly sequential manner to assign patients to either quetiapine XR, escitalopram, or placebo in a ratio of 1:1:1. To ensure blinding, placebo tablets/capsules were identical in appearance, smell, and taste to their respective active-treatment (quetiapine XR or escitalopram) tablets/capsules. A double-dummy method was used, and the number of tablets/capsules dispensed was the same across all treatment groups. All study medication was administered orally once daily in the evening.
Study medication and dosing schedule
Dose titration for quetiapine XR was 50 mg on days one and two, increasing to 150 mg on days three to 14. Escitalopram was dosed at 10 mg/day on days one to 14. At day 15, (week two), all patients with an inadequate response (<20% reduction from randomization in MADRS18 total score) had their dose doubled (quetiapine XR 300 mg/day; escitalopram 20 mg/day). The daily dose of escitalopram was based on prescribing information and the European Medicines Agency guidelines that recommend that an adequate dose of antidepressant is used in clinical trials.19,20
Investigators were not informed of the criteria for dose increase and an interactive voice-response system (IVRS) was used to blind dose increases. After week two, patients continued to receive their assigned doses for the remaining randomized treatment period. The initial dosage and up-titration of escitalopram was in accordance with the prescribing information.20
At the end of week eight (day 57), patients receiving quetiapine XR 150 mg/day or escitalopram 10 mg/day discontinued active treatment and took placebo until day 63 (post-treatment day six) to maintain the study blinding. Patients receiving quetiapine XR 300 mg/day or escitalopram 20 mg/day had their dose down-titrated to 150 mg/day or 10 mg/day, respectively, from day 57 until day 63. From day 64, patients received no treatment.
Prior and concomitant treatment
Prior to randomization, patients were not permitted to receive: antipsychotics, mood stabilizers, or antidepressants within 7 days; monoamine oxidase inhibitors, anxiolytics, drugs that induce or inhibit the hepatic metabolizing cytochrome P450 3A4 enzymes, or hypnotics within 14 days; fluoxetine within 28 days; a depot antipsychotic injection within two dosing intervals prior to randomization; or electroconvulsive therapy within 90 days.
During the study, lorazepam (2 mg/day or equivalent), zolpidem tartrate (10 mg/day), zaleplon (20 mg/day), zopiclone (7.5 mg/day), or chloral hydrate (1 g/day) were permitted for insomnia if treatment had been ongoing for 28 days prior to enrollment. Anticholinergics could be used to treat emergent extrapyramidal symptoms (EPS), but not prophylactically. Patients were permitted to receive psychotherapy during the study if it had been ongoing for ≥3 months prior to randomization. During the active-treatment period of the study, other psychoactive medications were not permitted. During the second week of the follow-up period (days 64–71), physicians could prescribe other medications, including alternative antidepressants, if clinically warranted.
Efficacy evaluations
The primary efficacy variable was the change from randomization to week eight in MADRS total score. To reduce the likelihood of rater-associated inflation of primary efficacy scale scores, HAM-D assessments comprised part of the inclusion criteria, but the primary efficacy end point utilized the MADRS scale.
Additional efficacy evaluations included change from randomization to each assessment in MADRS total score, change in individual MADRS items at week eight, MADRS response (≥50% reduction from randomization in MADRS total score) at weeks one and eight, the proportion of patients with a Clinical Global Impressions – Improvement (CGI-I)21 score of 1 (“very much improved”) or 2 (“much improved”), and MADRS remission (total score ≤8) rates at week eight. Additional definitions of remission were MADRS total scores of ≤10 and ≤12 (analyzed post hoc). Changes from randomization at week 8 in HAM-D total and item 1 (depressed mood) scores, Hamilton Rating Scale for Anxiety (HAM-A)22 total, and psychic and somatic cluster scores and CGI – Severity of Illness (CGI-S) score21 were also evaluated.
Clinical assessments of MADRS, HAM-A, and CGI-S total scores were conducted on day one (randomization), and at weeks one, two, four, six, and eight. In addition, CGI-S total score was assessed at enrollment (baseline). HAM-D scores were determined at enrollment, day one, and week eight. CGI-I scores were recorded at weeks one, two, four, six, and eight. Where possible, the same trained rater conducted all assessments for a given patient for a specific scale, to reduce scoring variability among raters. Raters received computer-based training (provided by PharmaStar/UBC) and needed to be certified as a qualified rater by the sponsor. High levels of interrater agreement were demonstrated for MADRS baseline (κ=0.826) and follow-up (κ=0.861) assessments.
Patient-reported outcomes
Quality of Life Enjoyment and Satisfaction Questionnaire (Q-LES-Q)23 – short form (SF) and Pittsburgh Sleep Quality Index (PSQI)24 were assessed at randomization and weeks four and eight. Change from randomization to week eight in Q-LES-Q (items 1–14) percentage maximum total, item 15 (satisfaction with medication), and item 16 (overall quality of life) scores were reported.
Changes in PSQI were used to assess several dimensions of sleep, including quality, latency, duration, efficiency, sleep disturbances, use of medication, and daytime dysfunction.
Analysis of primary efficacy variable by age, sex, ethnicity, disease severity, and continent/country
To examine whether the outcome of the study was affected by various factors, analyses of the primary end point were carried out for patient subgroups, including age, sex, ethnicity, disease severity, and continent/country. Response rates at week eight were also analyzed by continent/country (Asia, Europe, North America, and South Africa).
Safety and tolerability
The incidence, severity, and withdrawal due to adverse events (AEs) were recorded throughout the study. Assessment of serum glucose (fasting), lipid (fasting), and prolactin levels, and twelve-lead ECG recordings were performed at enrollment and week eight (fasting serum glucose was also assessed at week four). Body weight and vital signs were measured at enrollment and all subsequent visits up to week eight.
The Simpson–Angus Scale (SAS)25 and Barnes Akathisia Rating Scale (BARS)26 were used to assess EPS at randomization, week four, and week eight. The self-administered, 14-item Changes in Sexual Functioning Questionnaire (CSFQ) was assessed at randomization, week four, and week eight, and was used to measure illness- and medication-related changes in sexual functioning, with males and females completing separate versions.27
During the 2-week, post-treatment, drug-discontinuation follow-up phase, treatment discontinuation signs and symptoms (TDSS) were assessed using an 18-item TDSS scale, which was developed by AstraZeneca as a hybrid of the 17-item discontinuation AE scale28 and the 43-item discontinuation emergent signs and symptoms scale.29 Patients completing the randomized period were asked to rate discontinuation symptoms using the TDSS scale. Patients completed the TDSS by IVRS at the study center during the final randomized treatment period visit (day 57) and on post-treatment days seven and 14, and by telephone at home on post-treatment days one, three, and five. Patients were asked whether the symptom was “present” or “absent.” TDSS total scores were calculated for each visit by summing the number of patient-reported treatment-emergent symptoms (TDSS items) present.
Statistical analyses
The modified intent-to-treat (MITT) population (randomized patients who received one or more doses of study treatment and had randomization and one or more postrandomization MADRS total score assessments) was used for the analysis of primary and secondary efficacy variables. The safety population included patients who received one or more doses of study treatment. The TDSS population included patients who completed 8 weeks of double-blind treatment and had baseline (week eight) and one or more postbaseline TDSS assessments.
Sample size was calculated to allow demonstration of a significant difference between quetiapine XR and placebo for the primary efficacy variable, and was achieved by assuming an anticipated difference of 3.5 units from placebo and a standard deviation (SD) of 9 for the change in MADRS total score from randomization at week eight. For a two-sided hypothesis test with a 5% significance level (α=0.05), a sample size of 140 evaluable patients per treatment group was required to ensure 90% power. Escitalopram was included as an active control for the purpose of assay sensitivity (ie, for comparison with placebo); the study was not powered to make any direct comparisons between quetiapine XR and escitalopram.
Analysis of the change from randomization at week eight in MADRS total score (primary efficacy variable) and Q-LES-Q-SF% maximum total score (secondary efficacy variable of particular interest) was conducted using an analysis of covariance (ANCOVA) model, including treatment (fixed effect), center (random effect), and baseline MADRS total score (covariate) as explanatory variables. A last observation carried forward (LOCF) approach was used for imputation of missing data. To assess the robustness of the primary analysis results, point estimates for the changes in MADRS total score were calculated at each time point for observed-case (OC) data using a mixed-model repeated-measures (MMRM) analysis that included center, treatment, baseline MADRS total score, visit, and treatment-by-visit interaction terms.
Type I error (α=0.05) was controlled using a sequential testing procedure for the two comparisons of primary interest. If the reduction in MADRS total score from randomization at week eight with quetiapine XR was significantly greater than with placebo, then the hypothesis relating to the change in Q-LES-Q-SF% maximum total score from randomization to week eight was to be tested. Any comparisons between escitalopram and placebo were not adjusted for multiplicity.
All other continuous efficacy variables were analyzed using the same ANCOVA model as the primary efficacy variable. MADRS response and remission rates were analyzed using logistic regression models, which included terms for center, treatment, and baseline MADRS total score. Model-based point estimates of odds ratios and 95% confidence intervals were reported. The number needed to treat (NNT) for MADRS responders was calculated using the formula:

Changes in Q-LES-Q overall quality of life (item 16) and satisfaction with medication (item 15) scores from randomization to week eight were presented by descriptive statistics. All statistical tests were two-sided with a significance level of 5% (unless otherwise specified), and with the exception of the primary efficacy variable and secondary efficacy variable of particular interest, were reported as nominal P-values.
Results
Patient population
This study was conducted at 54 centers in Canada, People’s Republic of China, Finland, South Korea, Malaysia, Mexico, The Philippines, South Africa, and Spain between May 2006 and June 2007. In total, 660 patients were screened, and 471 eligible patients with MDD were randomized to receive quetiapine XR, escitalopram, or placebo. Of those randomized, 468 patients received treatment and were included in the safety-analysis set; the MITT population comprised 459 patients (nine patients were excluded due to missing randomization or postrandomization MADRS scores) (Figure 1).
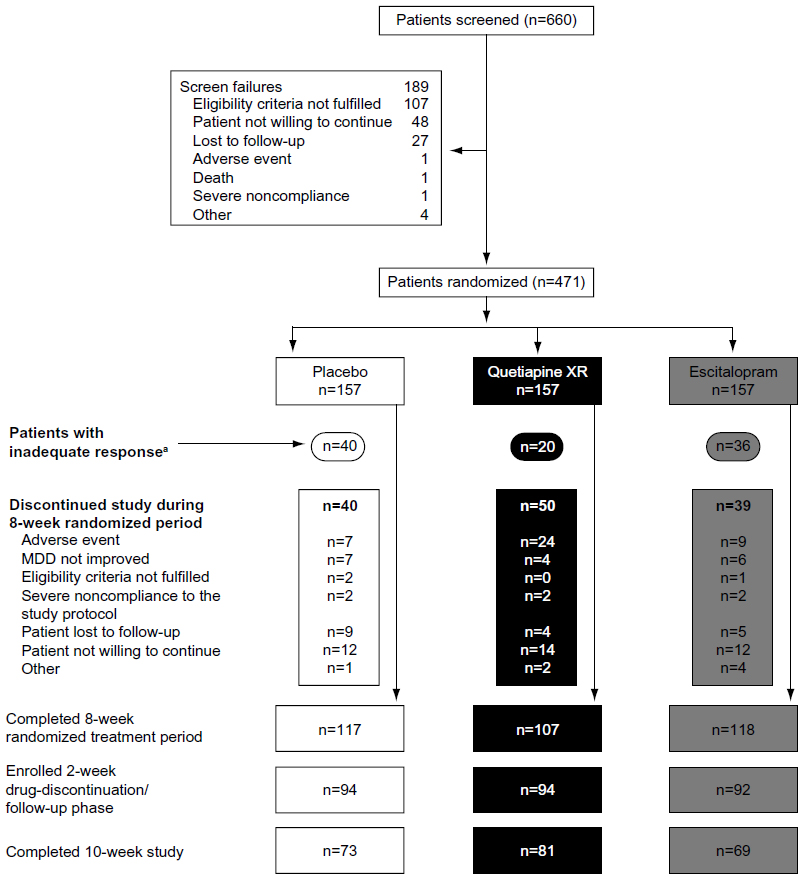 | Figure 1 Patient disposition. |
The treatment groups were generally well balanced with respect to demographic and clinical characteristics (Table 1). Overall, 68.2%, 75.2%, and 74.5%, respectively, of patients in the quetiapine XR, escitalopram, and placebo groups completed the randomized treatment phase; of these, 75.7%, 58.5%, and 62.4%, respectively, completed the 2-week follow-up period. The most common reasons for withdrawal during the randomized treatment period were an AE in the quetiapine XR group and “patient not willing to continue” in the escitalopram and placebo groups.
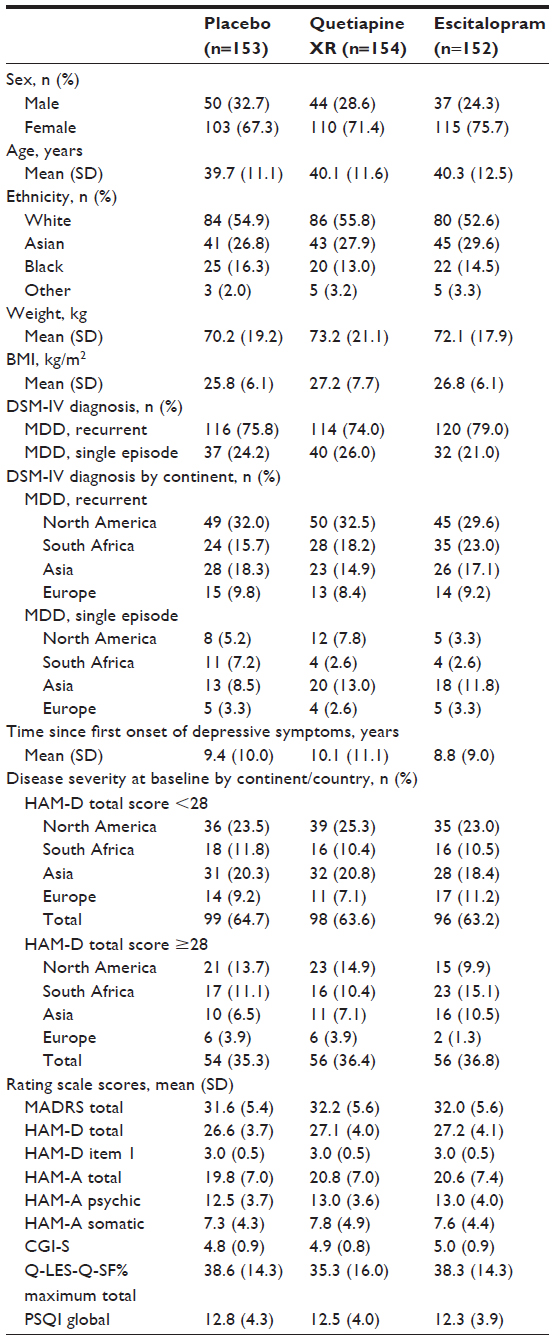 | Table 1 Demographic and baseline clinical characteristics (MITT population) |
In the quetiapine XR (150 mg/day), escitalopram (10 mg/day), and placebo groups, 13.0% (20 of 154), 23.7% (36 of 152), and 26.1% (40 of 153) of patients, respectively, met the criteria for inadequate response at week two and were up-titrated to double their initial randomized dose (MITT population). The mean (SD) daily dose was 139.8 (44.0) mg/day for quetiapine XR and 10.7 (3.0) mg/day for escitalopram during the randomized treatment period (safety population).
High levels of adherence to study medication (based on tablet counts consistent with ≥80% and ≤120% consumption of doses) were observed in each treatment cohort (97.4%, 96.7%, and 99.7% in the quetiapine XR, escitalopram, and placebo groups, respectively [MITT population]).
Prior and concomitant medication
Prior to randomization, 21.6% of patients were receiving benzodiazepines, 11.3% were receiving selective serotonin-reuptake inhibitors, and 4.9% were receiving nonselective monoamine-reuptake inhibitors.
The proportions of patients who received concomitant sleep medication (hypnotics/sedatives) at some time during the randomized phase were 5.4%, 7.4%, and 7.3% in the quetiapine XR, escitalopram, and placebo groups, respectively; benzodiazepines were received at some time during the randomized phase by 15.3%, 22.2%, and 22.7% of patients, respectively. Anticholinergic use at some time during randomized treatment occurred in 5.4%, 2.8%, and 3.6% of patients in the quetiapine XR, escitalopram, and placebo groups, respectively.
Efficacy
At week eight, neither quetiapine XR nor escitalopram significantly reduced MADRS total scores from randomization (LOCF): least-squares mean (LSM) of −17.21 (P=0.174) and −16.73 (P=0.346), respectively, versus −15.61 for placebo (Figure 2A), although at weeks two and four there was a significant effect of treatment (P<0.05 and P<0.01, respectively).
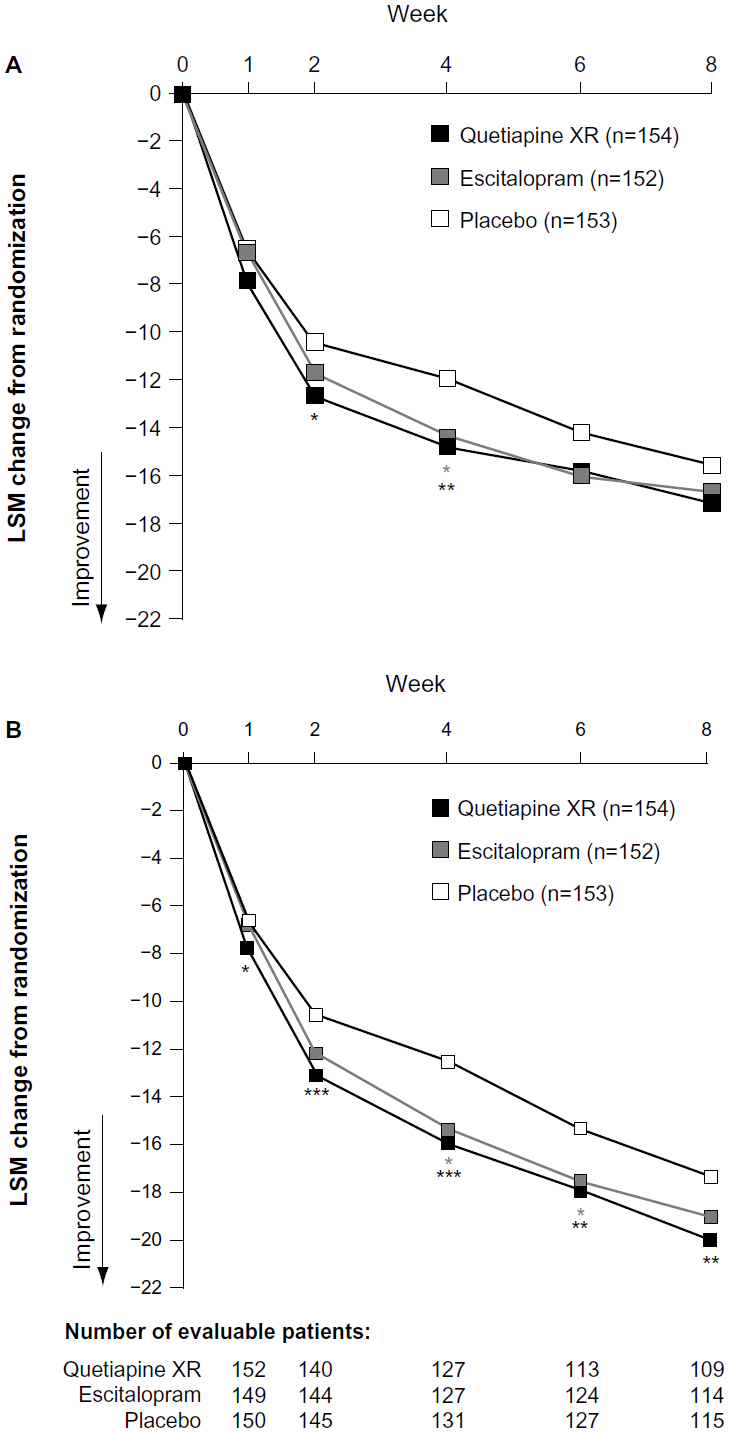 | Figure 2 (A and B) Change from randomization to week eight in MADRS total score. (A) LOCF approach; (B) MMRM analysis of observed-case data (MITT population). |
The mean changes from randomization to week eight in MADRS total score for quetiapine XR, escitalopram, and placebo, respectively, were −19.4, −19.8, and −18.3 for patients with an adequate response at week two, and −13.1, −8.3, and −10.3 for patients with an inadequate response at week two (Figure 3).
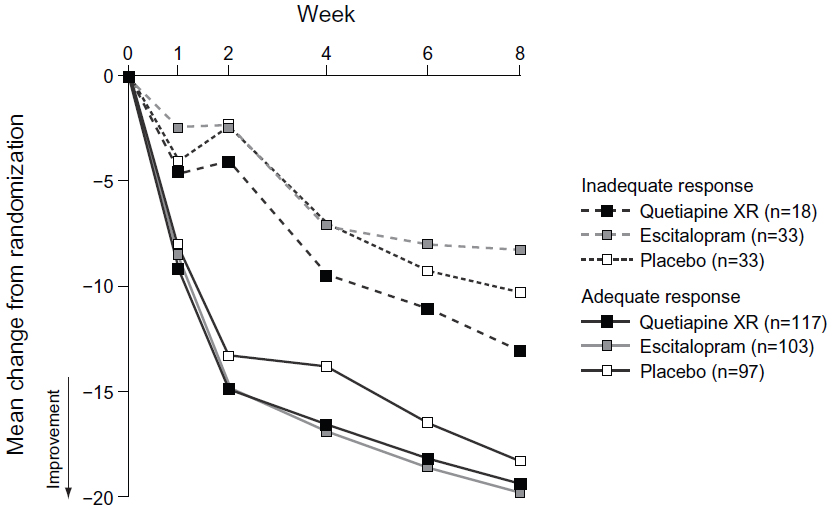 | Figure 3 Change in MADRS total score over time in patients with an adequate and inadequate responsea,b at week two (LOCF; MITT population). |
Of the individual MADRS items, significant improvement at week eight was seen only in MADRS item 4 (reduced sleep) in the quetiapine XR group (LSM change −2.77; P<0.001) compared with placebo (−1.94) (Figure 4).
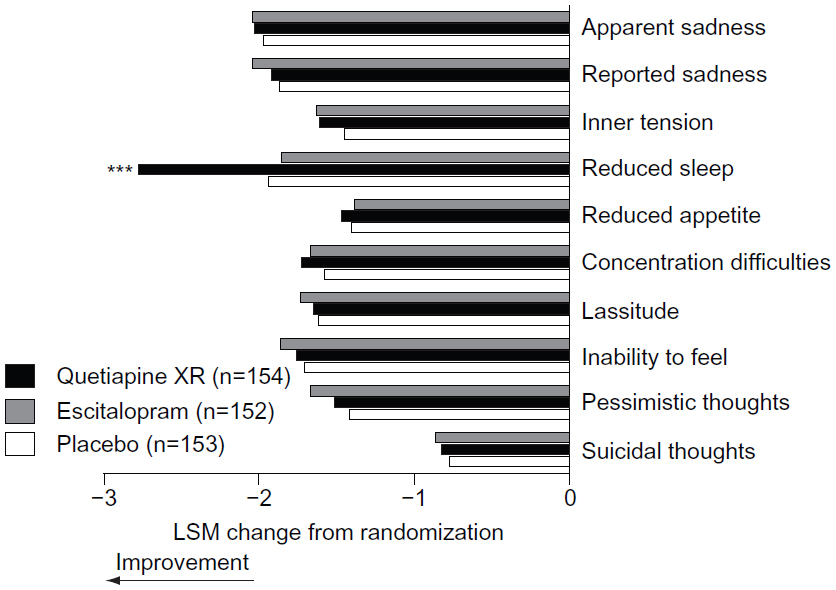 | Figure 4 Change in individual MADRS item scores from randomization to week 8 (MITT population; LOCF). |
With the exception of the PSQI, there were no statistically significant differences for quetiapine XR or escitalopram compared with placebo in any of the secondary end points (Table 2). The NNT using MADRS response at week 8 was 10.6 for quetiapine XR and 11.3 for escitalopram compared with placebo. Post hoc analyses of remission rates at week eight using the criterion of MADRS total score ≤10 were 44.8% (P=0.376) for quetiapine XR, 48.0% (P=0.157) for escitalopram, and 40.5% for placebo; for MADRS total score ≤12, remission rates were 55.2% (P<0.05), 52.0% (P=0.146), and 44.4%, respectively.
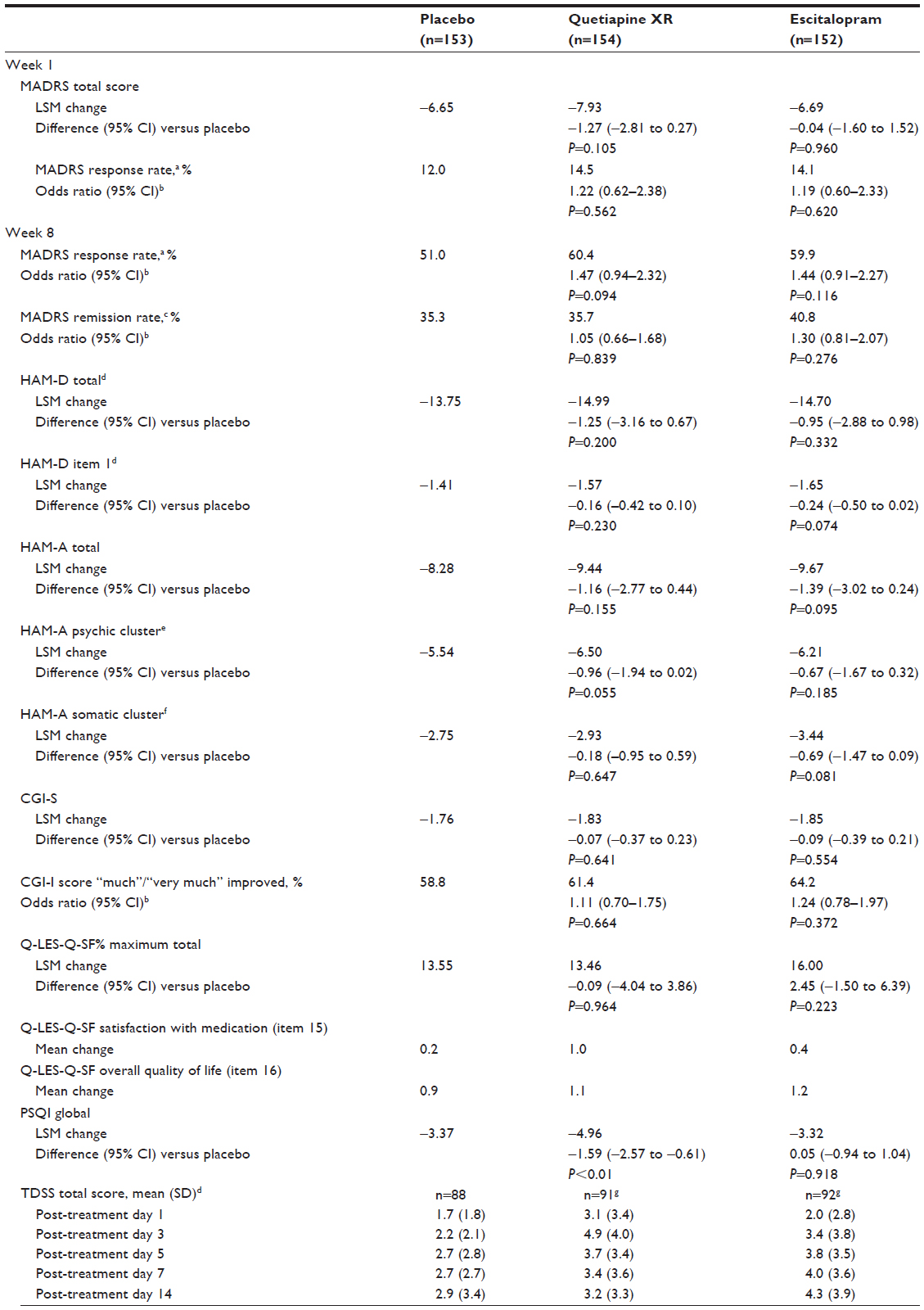 | Table 2 Change from randomization at week one and at week eight in secondary efficacy variables (MITT population, LOCF unless otherwise stated) and TDSS total scores over time during the drug discontinuation/tapering follow-up phase (TDSS population; OC) |
Analysis of primary efficacy variable by MMRM analysis, patient subgroups, and continent/country
Using the MMRM analysis of OC data, LSM reductions in MADRS total score at week eight were quetiapine XR −20.00 (P<0.01) and escitalopram −19.03 (P=0.189) versus −17.34 (placebo) (Figure 2B).
Subgroup analyses of the primary efficacy variable did not reveal any discernible effects for age, sex, or disease severity (Table 3); the only statistically significant change in MADRS total score at week eight was for patients from Asia receiving escitalopram (−20.68; P<0.05 versus placebo [−15.57]). Analysis of the response rate at week eight by continent/country provided similar results to those for the change in the primary efficacy variable by continent/country at week 8 (Table 3).
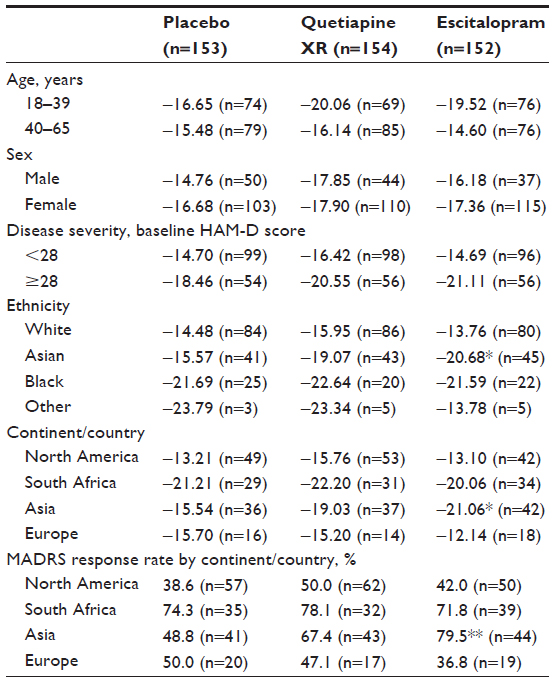 | Table 3 LSM change in MADRS total score from randomization to week eight by age, sex, ethnicity, disease severity, and continent, and MADRS response rates by continent (LOCF; MITT population)a |
Safety and tolerability
The overall incidence of AEs was 86.6% (quetiapine XR), 81.4% (escitalopram), and 73.5% (placebo); serious AEs were reported by four (2.5%), three (1.9%), and one (0.6%) patients, respectively. Two serious AEs were considered treatment related by the study investigator (depression and suicide attempt) and occurred in one patient (who had not received a dose increase) in the quetiapine XR group, and led to the withdrawal of the patient from the study. Treatment-related AEs were reported by 79.6%, 67.9%, and 52.3% of patients in the quetiapine XR, escitalopram, and placebo treatment groups, respectively. There were no deaths in this study.
The proportion of patients who discontinued due to an AE was 15.9%, 7.1%, and 4.5% for quetiapine XR, escitalopram, and placebo, respectively. The most frequently reported AEs leading to discontinuation were: sedation and dizziness (each n=5) with quetiapine XR; nausea (n=3), dizziness, and depressed mood (each n=2) with escitalopram; and palpitations and insomnia (each n=3) with placebo. The most common AEs (>5% in any group) and AEs of special interest (EPS, sexual dysfunction, suicidality, somnolence, and nausea and vomiting) are shown in Table 4.
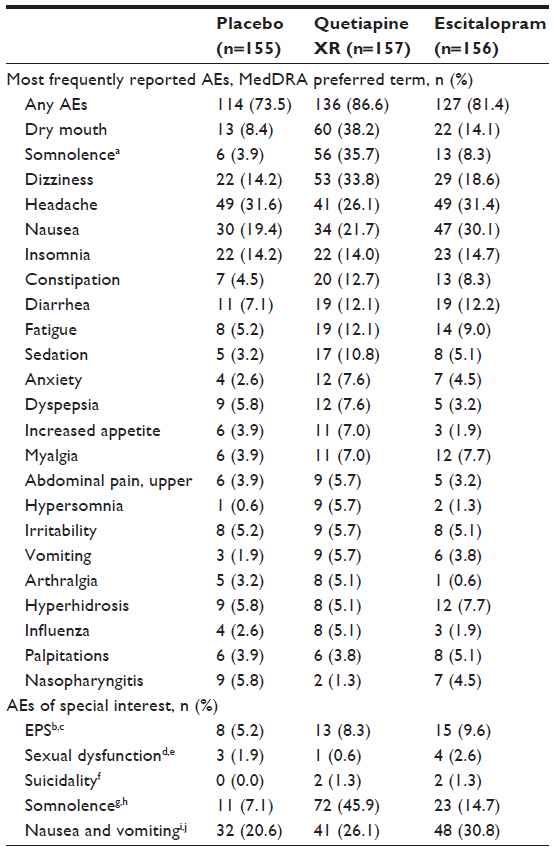 | Table 4 Most frequently reported AEAEs (occurring at an incidence of >5% in any group) and AE s of special interest (safety population) |
EPS
At the end of treatment, 87.6%, 91.7%, and 91.1% of patients in the quetiapine XR, escitalopram, and placebo groups, respectively, experienced an improvement/no change in SAS total scores; 96.6%, 94.5%, and 95.9% of patients, respectively, experienced an improvement/no change in BARS global scores.
Sexual dysfunction
For males, mean improvement from randomization in CSFQ total score at end of treatment was 2.5 with quetiapine XR, 2.4 with escitalopram, and 0.6 with placebo; for females, these changes were 2.4, 1.4, and 1.6, respectively.
Suicidality
AEs potentially related to suicidality were reported in four patients: two in the quetiapine XR group (suicide attempt and suicide ideation), which were considered treatment related, and two in the escitalopram group (suicidal behavior and suicide ideation), which were considered not to be treatment related. All four patients were withdrawn from the study. The proportions of patients with an MADRS item 10 (suicidal thought) score ≥4 at any time following randomization were 2.7%, 4.8%, and 1.4% for quetiapine XR, escitalopram, and placebo, respectively (OC data).
Somnolence
The majority of AEs potentially related to somnolence occurred within the first 4 days of treatment and were generally mild or moderate in intensity (87.0%–90.9%). Of the total number of patients reporting somnolence, 52.1%, 50.0%, and 40.0% reported this as an ongoing AE on the last day of treatment in the quetiapine XR, escitalopram, and placebo groups, respectively. The incidence of hypersomnia was higher in the quetiapine XR (5.7%) and escitalopram groups (1.3%) compared with placebo (0.6%) (Table 4).
Vital signs
Two patients each in the quetiapine XR and placebo groups had AEs of syncope; all AEs, except one in the quetiapine XR group, were considered treatment related. There were no notable differences in the mean changes from randomization to end of treatment in vital signs (including orthostatic changes) or ECG results between treatment groups. Mean changes at end of treatment in supine pulse were +1.7, −3.0, and −2.0 bpm in the quetiapine XR, escitalopram, and placebo groups; mean changes in the QTc (Fridericia correction) interval at week eight were −0.5, +4.0, and −1.1 milliseconds, respectively.
Clinical laboratory parameters
One patient in the escitalopram group experienced an AE of decreased neutrophil count (<0.8 × 109 cells/L), which was neither serious nor considered related to study treatment. Eight AEs potentially related to diabetes mellitus (Medical Dictionary for Regulatory Affairs preferred terms: thirst, polyuria, and blood glucose increased) were reported during the study: thirst (four [2.5%]) and polyuria (one [0.6%]) in the quetiapine XR group; thirst and polyuria (one [0.6%] each) in the escitalopram group; and blood glucose increased (one [0.6%]) in the placebo group; of these, one AE (polyuria) in the escitalopram group was severe in intensity.
Table 5 shows mean changes and clinically important shifts from normal to clinically important values (randomization to end of treatment) for clinical laboratory parameters, including glucose, lipid, and prolactin data. Mean changes (randomization to end of treatment) in confirmed fasting glucose were +0.4, +0.3, and +1.2 mg/dL in the quetiapine XR, escitalopram, and placebo groups, respectively; one (0.8%), three (2.4%), and three (2.5%) patients, respectively, had a clinically relevant increase in fasting glucose at treatment end. At treatment end, clinically relevant increases in total cholesterol were reported for four (3.7%), five (5.0%), and three (3.1%) patients, respectively, and clinically relevant increases in low-density lipoprotein cholesterol were reported for three (2.8%), four (3.9%), and four (3.9%) patients, respectively. Clinically relevant decreases in high-density lipoprotein cholesterol at treatment end occurred in eight (8.5%), six (6.3%), and three (3.2%) patients, respectively. The mean changes in triglyceride levels (randomization to treatment end) were +8.4, +3.6, and +3.6 mg/dL in the quetiapine XR, escitalopram, and placebo groups, respectively (Table 5); clinically important increases occurred in 14 (13.1%), eight (7.8%), and five (5.0%) patients, respectively.
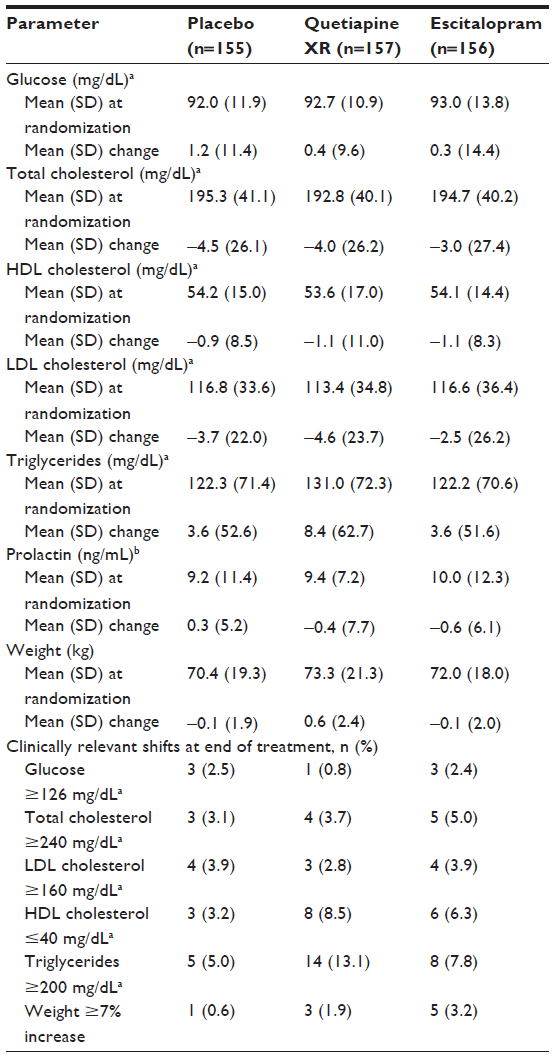 | Table 5 Changes in clinical laboratory parameters and body weight and proportion of patients with clinically relevant shifts (defined within the table) from randomization to end of treatment (safety population; LOCF) |
Body weight
At the end of treatment, patients in the quetiapine XR, escitalopram, and placebo groups experienced a mean (SD) weight change of +0.6 (2.4), −0.1 (2.0), and −0.1 (1.9) kg (Table 5); the proportion of patients experiencing a ≥7% increase in weight was 1.9%, 3.2%, and 0.6%, respectively.
Two-week drug discontinuation/ tapering follow-up phase
The most common AEs (more than two patients) during the drug discontinuation/tapering follow-up phase were headache (n=6) in the quetiapine XR group and insomnia and nausea (each n=4), headache, dizziness, and irritability (each n=3) in the escitalopram group. No AEs were reported by more than two patients in the placebo group.
Mean TDSS total scores are shown in Table 2. The most pronounced signs and symptoms following quetiapine XR discontinuation related to insomnia, nausea, chills, headache, and muscle aches; other signs and symptoms noted for quetiapine XR were crying, agitation, sweating, muscle tension, fatigue, and vomiting. After discontinuation of escitalopram, signs and symptoms included crying, agitation, mood swings, vivid and unusual dreams, sweating, muscle aches, muscle tension, fatigue, and diarrhea.
Discussion
This study assessed the efficacy and tolerability of quetiapine XR monotherapy in patients with MDD. Although quetiapine XR (150/300 mg/day) monotherapy and escitalopram (10/20 mg/day) monotherapy both reduced depressive symptoms, significantly superior efficacy over placebo was not established for either agent for the primary efficacy analysis or for most secondary efficacy end points. Lack of superior efficacy over placebo with escitalopram at clinically efficacious doses indicates a lack of assay sensitivity in this study, and coupled with lack of separation from placebo with quetiapine XR, means that this is a failed study rather than a negative study (the latter being characterized by the study drug not separating from placebo while the other active treatment does). In addition, the discrepancy between the primary LOCF analysis and the OC data suggests that the primary analysis may not have been robust.
Failure to demonstrate statistical difference from placebo in antidepressant trials is not uncommon, with failure rates of up to 60% being reported.30–33 However, it is of note that of eight studies from a clinical development program evaluating the effectiveness of quetiapine XR in patients with MDD, this is the only failed study.7–13 It is important that findings from negative/failed studies are reported and that the reasons underlying such outcomes are considered, as this may aid the design of future clinical studies.
The results from this failed international study are inconsistent with a previously reported US study of quetiapine XR (150/300 mg/day; D1448C00003) that used an identical modified fixed-dose design, but had no active control.7 Bortnick et al7 reported that quetiapine XR (150/300 mg/day) significantly improved depressive symptoms from week one onwards. The improvement in MADRS total score at week eight in quetiapine XR-treated patients was similar across the two studies (−17.21 and −16.49). However, the improvement in placebo-treated patients was larger in our study (−15.61) than in study D1448C00003 (−13.10), suggesting that placebo response may have accounted for the difference in outcomes. Similar improvements were observed in placebo-treated patients at weeks one to four across the two studies, including similar rates of placebo-treated patients requiring dose increases (approximately 26%). This observation suggests that any increases in placebo response for the current study occurred after the week four assessment. Failure to demonstrate efficacy may be affected by cultural and geographic factors, which have been reported to influence signal detection in mood-disorder studies,34 as well as the number and characteristics of study sites.35–37 The present study was undertaken in 54 centers in Finland, Spain, South Korea, Malaysia, People’s Republic of China, The Philippines, Canada, Mexico, and South Africa, whereas study D1448C00003 was performed in 35 centers in the US, and thus results may have been affected by this variation in countries and study centers.
Literature suggests that study design and disease characteristics may play a part in increased placebo response. While the study designs were nearly identical, the probability of receiving placebo was lower (33%) in the current study compared to D1448C00003 (50%). Trials with a lower probability of receiving placebo have been reported to have higher placebo response rates.38 Disease characteristics in the present study signaled a lack of depressive chronicity that (given shorter illness duration) has been associated with an increased placebo response in patients with MDD.39,40 Compared with study D1448C00003, more patients in the present study were experiencing a single (first) MDD episode (23.7% versus 10.0%, respectively), and patients had more recently experienced their first depressive episode (9.4 versus 13.6 years). Analyses of the primary end point by age, sex, baseline disease severity, and ethnicity did not reveal any discernible effects for these variables.
Quetiapine XR monotherapy was generally well tolerated in this study; the pattern of AEs was consistent with the known pharmacological profile of quetiapine.3,4 Although the tolerability profile of quetiapine XR was similar to that of previously published reports, the overall incidence of AEs in the placebo group (73.5%) appeared higher than that reported by Bortnick et al in study D1448C000037 for the placebo group (61.9%). Similarly, treatment-related AEs in the placebo group (52.3%) of the present study were higher than those reported in study D1448C000037 for the placebo group (28.4%), and the proportion of patients who discontinued the treatment was higher in the present study (4.5%) compared to that reported in study D1448C000037 for the placebo group (2.6%). These higher rates of AEs and discontinuations are consistent with the higher placebo efficacy response observed in this study, possibly indicative of greater levels of expectation for patients in this study.
No new safety findings were noted for escitalopram.20 Quetiapine XR was not associated with an increased incidence of AEs related to sexual dysfunction, QT prolongation, syncope, or neutropenia compared with placebo, nor with any notable changes in vital signs. A relationship between either quetiapine XR or escitalopram and increased suicidality could not be established in this study; black-box warnings about suicidality are required on package inserts for antidepressants.41
There were no notable differences between treatment groups in mean changes from randomization for any clinical laboratory parameters in this study; however, there was a greater incidence in shifts to clinically important high triglyceride levels in the quetiapine XR group compared with placebo and escitalopram. In the quetiapine XR group, body weight changes were consistent with those reported previously in patients with MDD receiving quetiapine XR as acute monotherapy.7–9 Clinical recommendations advise serum glucose, lipid, and insulin levels and body weight/body mass index are assessed during antipsychotic treatment.42
Study limitations include the short study duration and exclusion of patients with comorbidities. Furthermore, the study design precluded comparison of quetiapine XR 150 versus 300 mg/day, as it mimicked medication titration used in clinical practice when a patient does not respond to the initial treatment dose. However, patients in the study may have required different dose adjustments than those allowed by the modified fixed-dose study design. Furthermore, restrictions on concomitant medications are not reflective of clinical practice. The discrepancy between LOCF and OC analyses may be due to a possible bias of the LOCF analysis towards initial values at randomization; however, an analysis to investigate this further was beyond the scope of this report.
In summary, neither quetiapine XR (150/300 mg/day) nor the active comparator escitalopram (10/20 mg/day) demonstrated statistically significant separation from placebo in the primary efficacy outcome variable in this study. The placebo response observed here may have contributed to the lack of significant differences for quetiapine XR and the active control escitalopram compared with placebo.
Acknowledgments
This paper was previously presented at the 8th International Forum on Mood and Anxiety Disorders, Vienna, Austria, November 12–14, 2008 (poster presentation). This study (Amber: D1448C00004) was supported by AstraZeneca Pharmaceuticals; Clinical Trials Registry number NCT00351169. We thank Jocelyn Woodcock, MPhil from Complete Medical Communications Limited, who provided medical writing support funded by AstraZeneca. The following investigators were involved in the study: Richard Bergeron, Paul Latimer, Claire O’Donovan, Paul Lesperance, Sidney Kennedy, Shaila Misri, Alexander McIntyre, Satpal Girgla, Jean-Guy Gagnon, Autar Munshi, Smadar Tourjman, Andree Daigneault, Nizar Ladha, and Pierre Blier (Canada); Niufan Gu, Gang Wang, Jian Hu, Shiping Xie, and Xiaoping Wang (People’s Republic of China); Grigori Joffe, Liisa Lahdelma, Jussi Turtonen, Sanna Blanco Sequeiros, Tarja Ruotsalainen, Olli Piirtola, Marko Sorvaniemi, and Markku Timonen (Finland); Won-Myong Bahk, Min-Soo Lee, Se Joo Kim, and Young Chul Shin (South Korea); Ahmad Sulaiman, Benjamin Chan, and Suarn Singh (Malaysia); Vasavan Agambaram, Shlomo Brook, Yao Mfodwo, Karen Vukovic, and Marius Steyn (South Africa); Celso Iglesias García, Raúl Vázquez-Noguerol, and Manuel Franco Martín (Spain); Efren Reyes, Constantine Della, Jacqueline Sy, Agnes Padilla, and Ruby Manalastas (Philippines); and Isaac De La Parra, Sergio Villaseñor, Susana Garcia, Rogelio Apiquian, Ricardo Secin, Felipe Ortega, and Juan Bautista Corral (Mexico).
Disclosure
Gang Wang has received research grants from AstraZeneca and Janssen, has participated on advisory boards for AstraZeneca, Eli Lilly, Janssen, Merck Sharp and Dohme, and Pfizer, and conducted research/studies for AstraZeneca, Eli Lilly, Janssen, Lundbeck, Otsuka, and Servier. Alexander McIntyre has received research grants from AstraZeneca. He has received honoraria from or participated in advisory boards for AstraZeneca, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen-Ortho and Lundbeck, Otsuka, and Pfizer. He has performed research for or received grants from Eli Lilly, Janssen-Ortho, Shire, Otsuka, Takeda, Pfizer, and Pharmaboost. Shane Raines is an employee of AstraZeneca. Willie Earley and Hans Eriksson are former employees of AstraZeneca.
References
Masand PS. Tolerability and adherence issues in antidepressant therapy. Clin Ther. 2003;25(8):2289–2304. | |
Howland RH. Sequenced Treatment Alternatives to Relieve Depression (STAR*D). Part 2: Study outcomes. J Psychosoc Nurs Ment Health Serv. 2008;46(10):21–24. | |
AstraZeneca Pharmaceuticals (Ireland). Seroquel XR: summary of product characteristics for Ireland. 2013. Available from: http://www.medicines.ie/medicine/13032/SPC. Accessed August 1, 2013. | |
AstraZeneca Pharmaceuticals. Once-daily Seroquel XR (quetiapine fumarate) extended-release tablets. 2012. Available from: http://www1.astrazeneca-us.com/pi/seroquelxr.pdf. Accessed July 9, 2012. | |
AstraZeneca Pharmaceuticals. Seroquel XR product monograph (Canada). 2011. Available from: http://www.astrazeneca.ca/documents/ProductPortfolio/SEROQUEL%20XR_PM_en.pdf. Accessed July 21, 2011. | |
AstraZeneca Pharmaceuticals. Seroquel XR Australian prescribing information. 2013. Available from: http://www1.astrazeneca-australia.com/pi/seroquelxr.pdf. Accessed. | |
Bortnick B, El-Khalili N, Banov M, et al. Efficacy and tolerability of extended release quetiapine fumarate (quetiapine XR) monotherapy in major depressive disorder: a placebo-controlled, randomized study. J Affect Disord. 2011;128(1–2):83–94. | |
Cutler AJ, Montgomery SA, Feifel D, Lazarus A, Åström M, Brecher M. Extended release quetiapine fumarate monotherapy in major depressive disorder: a placebo- and duloxetine-controlled study. J Clin Psychiatry. 2009;70(4):526–539. | |
Weisler R, Joyce M, McGill L, Lazarus A, Szamosi J, Eriksson H. Extended release quetiapine fumarate monotherapy for major depressive disorder: results of a double-blind, randomized, placebo-controlled study. CNS Spectr. 2009;14(6):299–313. | |
Bauer M, Pretorius HW, Constant EL, Earley WR, Szamosi J, Brecher M. Extended-release quetiapine as adjunct to an antidepressant in patients with major depressive disorder: results of a randomized, placebo-controlled, double-blind study. J Clin Psychiatry. 2009;70(4):540–549. | |
El-Khalili N, Joyce M, Atkinson S, et al. Extended-release quetiapine fumarate (quetiapine XR) as adjunctive therapy in major depressive disorder (MDD) in patients with an inadequate response to ongoing antidepressant treatment: a multicentre, randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2010;13(7):917–932. | |
Liebowitz M, Lam RW, Lepola U, Datto C, Sweitzer D, Eriksson H. Efficacy and tolerability of extended release quetiapine fumarate monotherapy as maintenance treatment of major depressive disorder: a randomized, placebo-controlled trial. Depress Anxiety. 2010;27(10):964–976. | |
Katila H, Mezhebovsky I, Mulroy A, et al. Randomized, double-blind study of the efficacy and tolerability of extended release quetiapine fumarate (quetiapine XR) monotherapy in elderly patients with major depressive disorder. Am J Geriatr Psychiatry. 2013;21(8):1769–1784. | |
Glick ID, Suppes T, DeBattista C, Hu RJ, Marder S. Psychopharmacologic treatment strategies for depression, bipolar disorder, and schizophrenia. Ann Intern Med. 2001;134(1):47–60. | |
AstraZeneca. Efficacy of Seroquel SR in Combination With an Antidepressant in Treatment of Major Depressive Disorder (AMBER). Available from: http://clinicaltrials.gov/ct2/show/NCT00351169. NLM identifier:NCT00351169. Accessed November 18, 2013. | |
Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (MINI): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59 Suppl 20:22–33. | |
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. | |
Montgomery SA, Åsberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. | |
European Agency for the Evaluation of Medicinal Products. Committee for Proprietary Medicinal Products (CPMP): Note for guidance on clinical investigation of medicinal products in the treatment of depression. 2002. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003526.pdf. Accessed November 6, 2013. | |
Forest Pharmaceuticals. Escitalopram prescribing information. 2009. Available from: http://www.frx.com/pi/lexapro_pi.pdf. Accessed November 6, 2013. | |
National Institute of Mental Health. CGI: clinical global impressions. In: Guy W, Bonato RR, editors. Manual for the ECDEU Assessment Battery. Rockville (MD): NIMH; 1970:1–6. | |
Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959;32(1):50–55. | |
Endicott J, Nee J, Harrison W, Blumenthal R. Quality of Life Enjoyment and Satisfaction Questionnaire: a new measure. Psychopharmacol Bull. 1993;29(2):321–326. | |
Buysse DJ, Reynolds CF 3rd, Monk TH, Berman SR, Kupfer DJ. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res. 1989;28(2):193–213. | |
Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl. 1970;212:11–19. | |
Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154:672–676. | |
Clayton AH, McGarvey EL, Clavet GJ. The Changes in Sexual Functioning Questionnaire (CSFQ): development, reliability, and validity. Psychopharmacol Bull. 1997;33(4):731–745. | |
Michelson D, Fava M, Amsterdam J, et al. Interruption of selective serotonin reuptake inhibitor treatment. Double-blind, placebo-controlled trial. Br J Psychiatry. 2000;176:363–368. | |
Rosenbaum JF, Fava M, Hoog SL, Ascroft RC, Krebs WB. Selective serotonin reuptake inhibitor discontinuation syndrome: a randomized clinical trial. Biol Psychiatry. 1998;44(2):77–87. | |
Khan A, Schwartz K. Study designs and outcomes in antidepressant clinical trials. Essent Psychopharmacol. 2005;6(4):221–226. | |
Yang H, Cusin C, Fava M. Is there a placebo problem in antidepressant trials? Curr Top Med Chem. 2005;5(11):1077–1086. | |
Fava M, Evins AE, Dorer DJ, Schoenfeld DA. The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach. Psychother Psychosom. 2003;72(3):115–127. | |
Kobak KA, Kane JM, Thase ME, Nierenberg AA. Why do clinical trials fail? The problem of measurement error in clinical trials: time to test new paradigms? J Clin Psychopharmacol. 2007;27(1):1–5. | |
Vieta E, Pappadopulos E, Mandel FS, Lombardo I. Impact of geographical and cultural factors on clinical trials in acute mania: lessons from a ziprasidone and haloperidol placebo-controlled study. Int J Neuropsychopharmacol. 2011;14(8):1017–1027. | |
Dunlop BW, Thase ME, Wun CC, et al. A meta-analysis of factors impacting detection of antidepressant efficacy in clinical trials: the importance of academic sites. Neuropsychopharmacology. 2012;37(13):2830–2836. | |
Undurraga J, Baldessarini RJ. Randomized, placebo-controlled trials of antidepressants for acute major depression: thirty-year meta-analytic review. Neuropsychopharmacology. 2012;37(4):851–864. | |
Yildiz A, Vieta E, Tohen M, Baldessarini RJ. Factors modifying drug and placebo responses in randomized trials for bipolar mania. Int J Neuropsychopharmacol. 2011;14(7):863–875. | |
Papakostas GI, Fava M. Does the probability of receiving placebo influence clinical trial outcome? A meta-regression of double-blind, randomized clinical trials in MDD. Eur Neuropsychopharmacol. 2009;19(1):34–40. | |
Posternak MA, Zimmerman M, Keitner GI, Miller IW. A reevaluation of the exclusion criteria used in antidepressant efficacy trials. Am J Psychiatry. 2002;159(2):191–200. | |
Stein DJ, Baldwin DS, Dolberg OT, Despiegel N, Bandelow B. Which factors predict placebo response in anxiety disorders and major depression? An analysis of placebo-controlled studies of escitalopram. J Clin Psychiatry. 2006;67(11):1741–1746. | |
US Food and Drug Administration. Antidepressant use in children, adolescents and adults. 2010. Available from: http://www.fda.gov/cder/drug/antidepressants/default.htm. Accessed May 1, 2011. | |
American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596–601. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.