Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 9 » Issue 1
A randomized controlled trial of inhaled corticosteroids (ICS) on markers of epithelial–mesenchymal transition (EMT) in large airway samples in COPD: an exploratory proof of concept study
Authors Singh Sohal S, Soltani A, Reid D, Ward C, Wills K, Muller HK, Walters EH
Received 11 March 2014
Accepted for publication 9 April 2014
Published 27 May 2014 Volume 2014:9(1) Pages 533—542
DOI https://doi.org/10.2147/COPD.S63911
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sukhwinder Singh Sohal,1,* Amir Soltani,1,* David Reid,1,2 Chris Ward,1,3 Karen E Wills,1,4 H Konrad Muller,1 Eugene Haydn Walters1
1National Health and Medical Research Council Centre of Research Excellence for Chronic Respiratory Disease, School of Medicine, University of Tasmania, Hobart, Tasmania, Australia; 2Iron Metabolism Laboratory, Queensland Institute of Medical Research, Brisbane, Queensland, Australia; 3Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, Tyne and Wear, UK; 4Department of Biostatistics, Menzies Research Institute Tasmania, University of Tasmania, Hobart, Tasmania, Australia
*These authors contributed equally to this work
Background: We recently reported that epithelial–mesenchymal transition (EMT) is active in the airways in chronic obstructive pulmonary disease (COPD), suggesting presence of an active profibrotic and promalignant stroma. With no data available on potential treatment effects, we undertook a blinded analysis of inhaled corticosteroids (ICS) effects versus placebo on EMT markers in previously obtained endobronchial biopsies in COPD patients, as a “proof of concept” study.
Methods: Assessment of the effects of inhaled fluticasone propionate (FP; 500 µg twice daily for 6 months) versus placebo in 34 COPD patients (23 on fluticasone propionate and eleven on placebo). The end points were epidermal growth factor receptor (EGFR; marker of epithelial activation) and the biomarkers of EMT: reticular basement membrane (Rbm) fragmentation (“hallmark” structural marker), matrix metalloproteinase-9 (MMP-9) cell expression, and S100A4 expression in basal epithelial and Rbm cells (mesenchymal transition markers).
Results: Epithelial activation, “clefts/fragmentation” in the Rbm, and changes in the other biomarkers all regressed on ICS, at or close to conventional levels of statistical significance. From these data, we have been able to nominate primary and secondary end points and develop power calculations that would be applicable to a definitive prospective study.
Conclusion: Although only a pilot “proof of concept” study, this trial provided strong suggestive support for an anti-EMT effect of ICS in COPD airways. A larger and fully powered prospective study is now indicated as this issue is likely to be extremely important. Such studies may clarify the links between ICS use and better clinical outcomes and protection against lung cancer in COPD.
Keywords: pilot trial, reticular basement membrane, S100A4, EGFR, MMP-9, lung cancer
Background
Smoking-related chronic obstructive pulmonary disease (COPD) is the fourth most common cause of chronic disability and death in developed countries.1 Our limited insights into the details of airway remodeling in the airway wall in COPD mainly date from the 1960s, and our knowledge about the effects of inhaled corticosteroids (ICS) on such airway remodeling is scanty. This is especially true of data from endobronchial biopsies done in nonoperative volunteer patients, which contrasts with copious such studies on asthma.2
We have recently reported novel characteristics of airway remodeling in active smokers. These changes are especially marked in those with established COPD. The subepithelial reticular basement membrane (Rbm) was variably thickened but also markedly fragmented with clefts or elongated spaces within it,3–8 which is quite different from that in asthma (Figure 1), where the Rbm is homogeneously thickened and hyaline in appearance.9 These elongated clefts in the Rbm in COPD are usually not empty, but frequently contain cells.3 These cells positively express the mesenchymal markers S100A4 and vimentin, as well as matrix metalloproteinase-9 (MMP-9),3 and do not penetrate inflammatory or other immune cells.4 These changes are “classic” hallmarks of the process termed epithelial–mesenchymal transition (EMT),10,11 defined as the transition of epithelial cells to a mesenchymal phenotype with the potential to digest and migrate through the Rbm to the subepithelial lamina propria.10,11 The epithelium in COPD is also highly activated, with upregulation of epidermal growth factor receptor (EGFR).3 In general, these changes were most marked in currently smoking COPD subjects compared with ex-smoking COPD and physiologically normal smokers. We have also demonstrated hypervascularity in and around the Rbm,9,12 which would characterize the EMT in COPD as being Type III.5–8,11,13
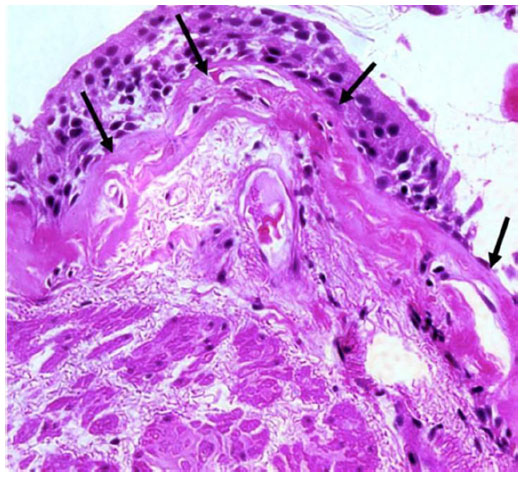 | Figure 1 Rbm fragmentation in COPD. |
Vessel changes seemed much the same in both smoker groups (with COPD or not), and much less marked in ex-smoker COPD subjects, which suggests that vessel changes were mainly effects of smoking. Type III EMT is recognized as producing a highly dangerous pro-cancerous stroma immediately under the epithelium. Of course, we recognize that smoking, and especially COPD is closely related to lung cancer development,14 and such promalignant stroma has been described as being important in the development of other common epithelial malignancies.15 ICS have become standard treatment in severe COPD, on the basis of positive empirical results from large studies, demonstrating short-term improvement in lung function and longer term benefits in terms of quality of life, exacerbation rates, long-term physiological decline, and possibly mortality.16 Importantly, there are also reports about the effects of ICS on inflammatory stromal airway changes in COPD;17 there is also epidemiological and circumstantial evidence of a protective effect of ICS use against lung cancer development in the highly vulnerable COPD population.18,19
To our knowledge, there have been no longitudinal trials published about the effects of ICS on EMT or other airway structural changes in COPD. In such a situation, the performance of a pilot trial has been advocated.20,21 Given published evidence linking EMT and epithelial cancers in general, and the potential effects of ICS on lung cancer risk, we believed that a pilot randomized trial evaluating the effects of ICS on EMT markers in COPD was warranted and so this “proof of concept” study was undertaken. This was a prospective analysis under blinded conditions that utilized tissue available from a previous study mainly directed at documenting changes in airway inflammatory cells,17 although it was always a stated intention to address airway remodeling at an appropriate time.
Methods
Using retrospectively obtained tissue, this study still constituted a prospective, double-blinded, randomized, and placebo-controlled study (Figure 2), but was intended as a pilot investigation given the paucity of data on the likely effects of ICS on the parameters of interest. A pilot study does not require a formal sample size calculation but is a precursor exercise to test whether the components of subsequent larger studies can work well together. Pilot studies provide valuable information such as that on patient recruitment and retention, the practicability of performing investigations, and an estimate of effect sizes, where no other information exists. The pilot study data may be analyzed as an “external pilot”, and it is understood that results from hypothesis testing should be treated as preliminary, with robust but pragmatic statistical management, and therefore, cautious interpretation.21 The details of the study design (full protocol available from the authors) and data on inflammatory cell profiles have been published previously.17
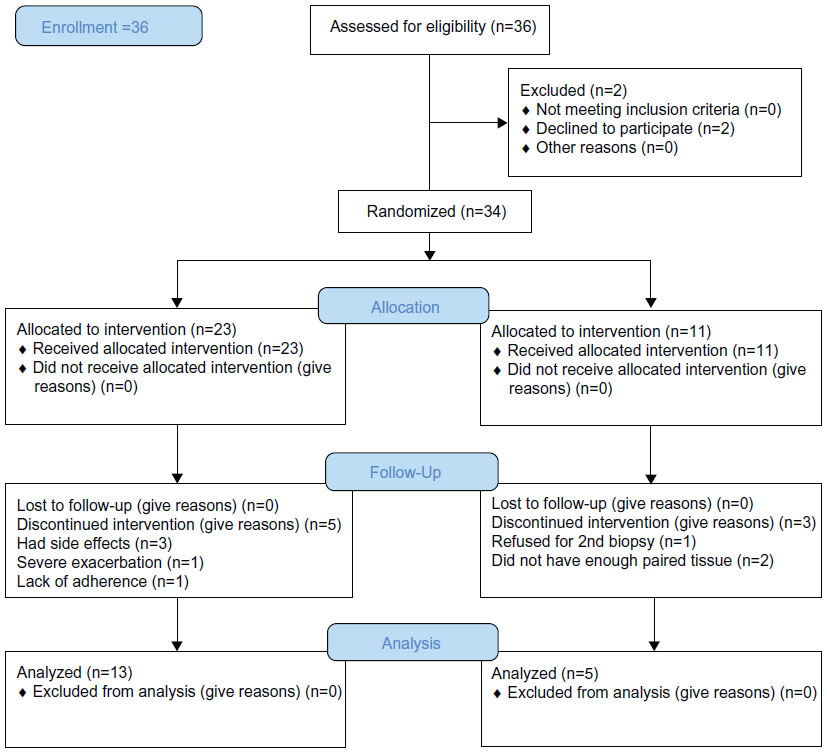 | Figure 2 Study design. |
Subjects
The demographics of the study groups are presented in Table 1. The study was approved by “The Human Research Ethics Committee (Tasmania) Network” and “The Alfred Health Human Ethics Committee” Melbourne (The Alfred Hospital), and is registered with the national trials registry (ANZCTR, Trial ID: ACTRN12612001111864). All subjects gave written, informed consent prior to participation. Details of subjects were published previously.17,22 The clinical study was performed over 2000–2002, with an initial analysis of inflammatory cells,17 although it was always intended to analyze “remodeling” changes. Time has been taken up as we cross-sectionally analyzed the details and relevance of the structural changes we subsequently observed,3,4 and with the previous use of tissue, it meant that more limited matched pairs of biopsy sections were available for this set of analyses. Further, randomization was originally done on a 2:1 basis of active to placebo treatment because at the time it was considered that the most useful data would be obtained in the active (ICS) limb, and this too limited our statistical power in the between-group comparisons. The control tissue was available from clinically and physiologically normal, age-matched non-smoker individuals who had volunteered for research bronchoscopy over a number of years, and their endobronchial biopsies had been stored and then processed in the same way as for the longitudinal intervention subjects.3 Exclusion criteria include: 1) subjects with a history suggestive of asthma, which includes symptoms in childhood, related atopic disorders, eczema or hay fever, substantial day-to-day variability or prominent nocturnal symptoms, or a history of wheeze rather than progressive breathlessness, and any who had previously used ICS (oral or inhaled); 2) no major comorbidities such as uncontrolled diabetes, angina, or cardiac failure, nor other coexisting respiratory disorders including pulmonary fibrosis, lung cancer, or bronchiectasis, and subjects were not on medication; and 3) subjects who were unable to give written informed consent. Inclusion criteria include: 1) current-smokers with COPD aged 40–70 years with a smoking history equal to or more than 15 pack-years; subsequently obtained bronchoalveolar lavage (BAL) fluid had to be free of culturable bacteria; forced expiratory volume in 1 second (FEV1) 40%–80% predicted, with forced expiratory ratio (FER; ratio of FEV1 to forced vital capacity [FVC]) ≤70% post-bronchodilator with definite scalloping out of the descending limb of the flow–volume loop on spirometry. COPD in ex-smokers with 6 months of smoking cessation were included; and 2) normal healthy never smoking controls also underwent bronchoscopic examination and physiological evaluation. They were at least 18 years old with a FEV1/FVC ratio of 70% or higher and FEV1% predicted of 80% or higher; they had no history of respiratory illness.
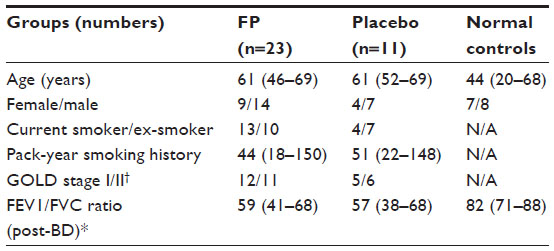 | Table 1 Demographics |
Bronchoscopy
Bronchoscopy and endobronchial biopsies were performed in the standard way, as previously described.2
Immunostaining
Tissue was processed as previously described.3,4 Following removal of paraffin, followed by hydration, sections were stained with these antibodies: monoclonal-anti-MMP-9 (R&D Systems, Inc., Minneapolis, MN, USA; MAB911; 1:50 for 2 hours), monoclonal-anti-EGFR (Chemicon S/A, São Paulo, Brazil; CBL417; 1:40 for 60 minutes), and anti-S100A4 polyclonal antibody (Dako, Glostrup, Denmark; A5114; 1:2,500 for 90 minutes). In each case, a non-immune immunoglobulin (Ig)G1 negative control (Dako; X0931 clone DAK-GO1) was performed to eliminate false positive staining and a positive tissue control (surgically resected lung tissue) was also used. Bound antibodies were elaborated using peroxidase-labeled EnVision™ + (Dako; K4001) and liquid diaminobenzidine (DAB) + (Dako; K3468).
Biopsy analysis
Computer-assisted image analysis was performed with a Leica DM 2500 microscope (Leica Microsystems, Wetzlar, Germany), Spot insight 12 digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI, USA), and Image Pro (v5.1; Media Cybernetics, Inc., Rockville, MD, USA) software. All slides were coded and randomized by an independent person and then counted by a single experienced observer (SSS) blinded to the subject and diagnosis, with quality assurance on randomly selected slides provided by a clinical and academic pathologist (Professor Hans Konrad Muller; International Academy of Pathology and Royal College of Pathologists of Australasia, Sydney, Australia).3,4
Quantification of indices was performed before and after treatments to compare the effects of interventions. Cellular staining changes in the basal epithelium and Rbm were documented. Cell numbers positive for S100A4 and MMP-9 and the number of vessels within the Rbm were normalized for purposes of comparisons relative to the total length of basement membrane assessed. EGFR was measured as a percentage of epithelium stained for EGFR over the total basement membrane length. Although the full picture of fragmentation of the Rbm includes linear “clefts” or elongated spaces/cracks in the Rbm, as fragments of Rbm commonly “hang off” and with similar pieces completely separated from the remainder (Figure 1), we have used Rbm linear splitting (ie, cleft formation) as a quantitative measure for the observation. Thus, the total length of splits was summed and normalized as a percentage of the length of Rbm. Normal control data were provided from physiologically normal, non-smoking, age-matched individuals, obtained previously in a published cross-sectional study.3
Statistical analyses
At the time of developing this pilot investigation, there were no data on which to base power calculations. In keeping with the principles of such a pilot study, our analyses were predominantly exploratory by definition, with no formal sample size calculable. Recommendations for good practice are that where possible, approximately 20–30 patients at baseline should be evaluated, as in the present study, to investigate the distribution of outcomes of key study parameters.21,23 A limited set of hypotheses were tested, complementing descriptive data. We had one end point on epithelial activation, one on a structural change classically associated with EMT, and two putative mesenchymal biomarkers of EMT (S100A4 and MMP-9 expression, with the former analyzed separately in basal epithelium and Rbm cells).
The data subsequently reported were generally skewed, so results are presented as medians and ranges and non-parametric analyses of variance were performed. The effects of the interventions were assessed using Wilcoxon related-samples tests to compare the indices for each of the biomarkers before and after treatment within treatment groups and also as changes within the groups (active versus [vs] placebo). The differences between the indices for the placebo and ICS treatment groups after intervention and differences from normal controls were compared using the Mann–Whitney test. These statistical analyses were performed using SPSS (v15.0 for Windows; IBM Corporation, Armonk, NY, USA) with a two-tailed P≤0.05 considered statistically significant. In this exploratory analysis, no primary or secondary end points have been discriminated, and as regarded reasonable under the conditions of a pilot study,20,21 no statistical allowance was made for the potential problem of multiple and collinear comparisons.
Sample size and power calculations, with putative primary and secondary outcomes
From the data obtained, we estimated the sample size required for a definitive study that would include “full” statistical analysis of drug affect, including allowance for multiple comparisons. We prospectively specified a three-stage, fixed-sequence statistical testing procedure and adjusted “alpha” (power) for multiplicity accordingly.24 Firstly, we would specify EGFR expression in the epithelium as the primary “driving” outcome measure and test the null hypothesis of no difference using alpha =0.05. If the null hypothesis cannot be rejected there would be a rationale for no further testing. However, given the mechanistic uncertainty between EGFR upregulation and EMT, we have nominated a coprimary hypothesis related to the main descriptive structural hallmark of EMT, namely the aggregate lengths of splitting in the Rbm that could therefore also be tested using alpha =0.05. If appropriate, ie, if the null hypothesis is rejected, in the third stage of testing we would treat the remaining outcomes, (basal cell S100A4, Rbm cell S100A4, and MMP-9 expression) as a cluster of related end points with an overall and shared alpha, so that individual hypotheses would be tested using an alpha of 0.0167; subsequently, larger and/or more consistent changes would be required.
Results
Studying markers of EMT in airway biopsies proved practicable in a randomized trial setting. Recruitment, randomization and allocation concealment, 6 months of treatment, and assessments including two bronchoscopies, were all practicable and acceptable to patients.22 The groups were reasonably well-matched for demographics and lung function (Table 1). All five pathological features of interest at baseline were significantly abnormal in COPD patients compared to normal controls.3
EGFR expression (as marker of activation) in the airway epithelium
The percentage area of EGFR staining in the epithelium was statistically significantly lower after treatment for the ICS group (median [range]: 34% [14.6–59.5] before vs 5.8% [2.6–43.8] after; P<0.03) but not for the placebo group (14.4% [3.6–38.2] before vs 10.3% [1.3–39.1] after treatment; P=0.3; Figure 3). The active arm was essentially normalized after the ICS treatment (P=0.9 vs normal controls). There was modest evidence for aggregate change over time on ICS for EGFR relative to changes on placebo (P=0.06), but with the removal of a single extreme outlier, this was statistically significant (P<0.03).
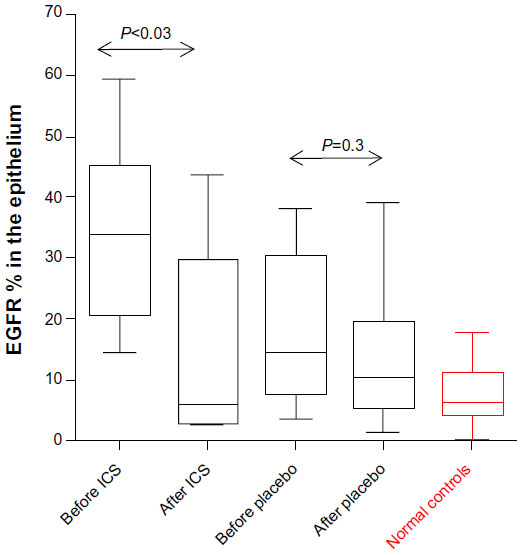 | Figure 3 EGFR expression. |
Fragmentation within the Rbm
The lengths of splitting of the Rbm (length of splitting/total Rbm length ×100) was statistically significantly lower after treatment for the ICS group (median [range]: 19.2% [0.2–42.8] before vs 1.9% [0–29.2] after treatment; P<0.03) but not for the placebo group (24.0% [6.6–100] before vs 26.9% [2.5–48.5] after treatment; P=0.4; Figure 4). Posttreatment, the active treatment arm had statistically significantly less splitting than the placebo group (P<0.02; Figure 4), and Rbm splitting for the ICS group was not statistically significantly different from normal controls (P=0.4). There was modest evidence for an effect of ICS on aggregate change in fragmentation relative to placebo (P=0.06), but again, after the removal of a single outlier, this was again statistically significant (P<0.03).
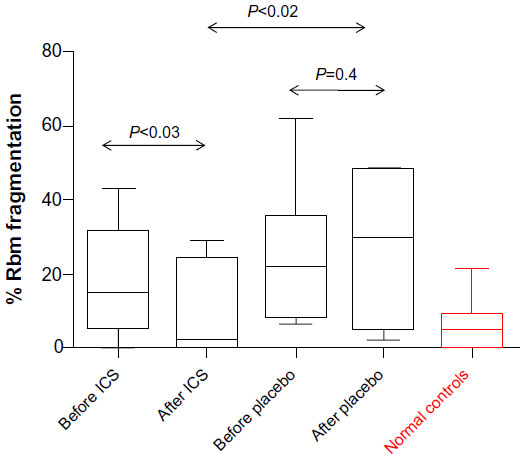 | Figure 4 Percentage Rbm fragmentation. |
S100A4 positive cells in the basal epithelium
Cell staining (Figure 5) for S100A4 within the basal layers of the airway epithelium was statistically significantly lower after treatment for the ICS group (median [range]: 25.8 per mm [2.4–55.3] before vs 12.3 per mm [0.6–24.9] after; P<0.004) but not for the placebo group (19.8 per mm [2.9–31.6] before vs 17.4 per mm [10.3–35.5] after treatment; P=0.4). However, at the end of the treatment phase, the active arm was still showing statistically significantly higher numbers for S100A4 than for normal controls (P<0.03), ie, they were not fully normalized. Changes over time on ICS were also statistically significant compared to those on placebo (P<0.009; Figure 5).
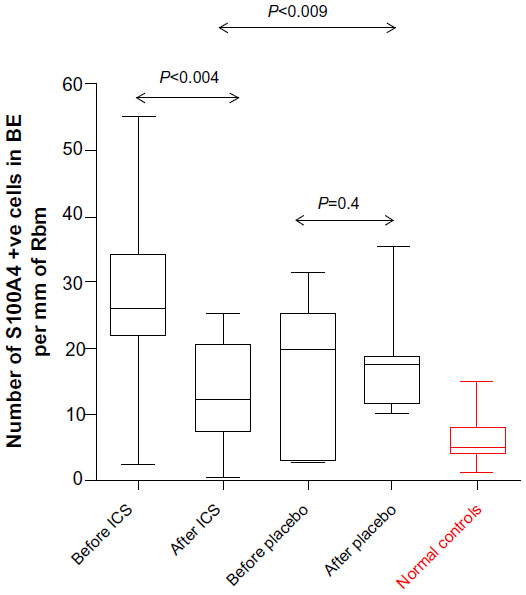 | Figure 5 S100A4 in basal epithelium. |
S100A4 positive cells in the Rbm
Cell numbers in the Rbm positive for S100A4 were statistically significantly fewer after treatment for the ICS group (median [range]: 44.4 per mm [15.3–92.6] before vs 20.8 per mm [2.6–60.7] after; P<0.02) but not on placebo (23.1 per mm [14–82.9] before vs 29.3 per mm [3.6–48.1] after treatment; P=0.6; Figure 6). However, even after ICS, the cell numbers were still statistically significantly higher compared to normal controls (P<0.004), ie, they were not fully normalized. Aggregate individual changes on ICS were also statistically significantly greater than on placebo (P<0.002; Figure 6).
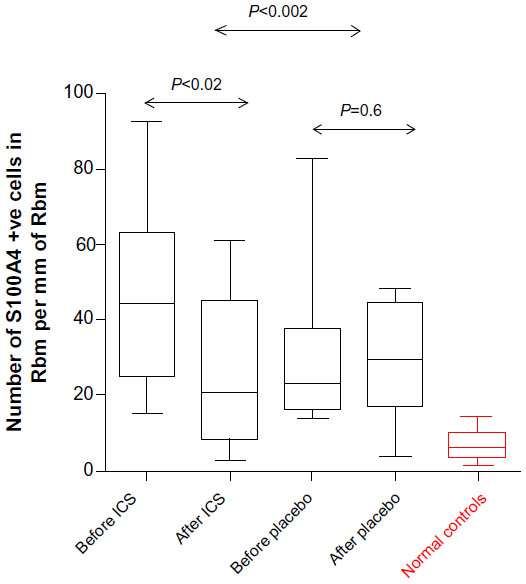 | Figure 6 S100A4 expression in Rbm. |
MMP-9 positive cells in the Rbm
MMP-9 positive cells decreased statistically significantly in the Rbm in the treatment arm (median [range]: 0.5 per mm [0–4.3] before vs 0 per mm [0–0.6] after treatment; P<0.02) but did not change within the placebo group (1.1 per mm [0–4.1] before vs 1.3 per mm [0–2.7] after treatment; P=0.8; Figure 7). After treatment, the active ICS treatment arm had statistically significantly fewer cells positive for MMP-9 in Rbm than did the placebo group (P<0.05; Figure 7). Further, MMP-9 positive cell numbers in the active arm became quite similar to normal control values after treatment (P=0.3). However, individual changes in MMP-9 expression over 6 months on ICS were not statistically significantly different to changes on placebo (P=0.3).
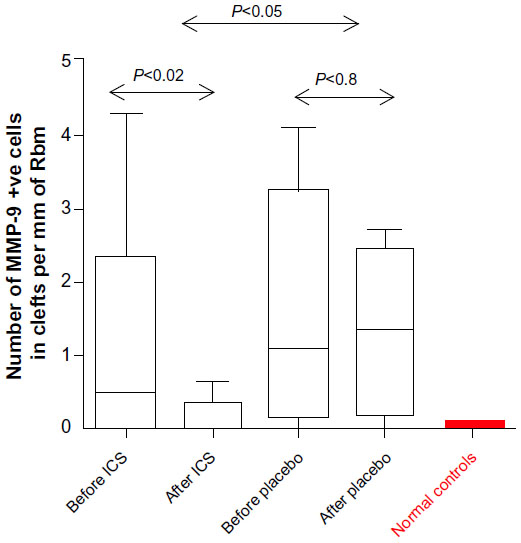 | Figure 7 MMP-9 expression in Rbm. |
Post hoc power calculations
Based on our pilot data for 22 individuals, for the nominated primary outcome of change in EGFR, a total sample size of 36 (18 per balanced treatment group) would provide 80% power to detect the observed difference in mean change. This sample size would provide 65% power (α=0.05) to detect the observed difference in Rbm splitting (for 80% power, 52 would be required), 97% power (α=0.0167) for basal S100A4 (for 80% power, only 26 would be required), 48% power for Rbm S100A4, and 31% power for MMP-9. The sample sizes required to achieve 80% power for each of these secondary outcomes individually would be 26, 70, and 104 respectively, given the stringent conditions imposed by allowing for multiple comparisons (Table 2).
 | Table 2 From the data obtained in the pilot study: observed mean and SD for changes in epithelial activation and EMT biomarkers for the placebo and ICS groups over 6 months of intervention and sample sizes required for 80% power for change for each end point in future potential studies (the secondary end points stringently share an α of 0.05) |
Discussion
We have previously shown that EMT is likely to be an active process in smokers’ airways, but is especially marked in current smokers with COPD.3,4 These findings were based on immunostaining for acknowledged “classic” empiric EMT markers10,11,25,26 and based especially on criteria for in vivo research on EMT as suggested by Zeisberg and Neilson.10 Recent papers by Milara et al27 and Wang et al28 have confirmed the potential for EMT as a major mechanism for small airway fibrosis (EMT Type II) in COPD. We have hypothesized that large airway EMT, which we have found is also associated with Rbm hypervascularity, may be important in both fibrosis and cancer pathogenesis and progression, as this is typical of what has been described as procancerous for epithelial tumors elsewhere.5–8,15 In this pilot randomized controlled trial, we have provided consistent but still provisional evidence that ICS over 6 months suppresses such EMT-related changes. We feel that the study was successful, at the very least, as a “proof of concept” study.
Our pilot trial indicated that intervention studies using bronchoscopic airway material for studying EMT in COPD are practicable, and we were able to provide reliable data regarding baseline levels and reproducibility of markers of EMT, as well as changes associated with ICS treatment, albeit in a pilot setting. For developing power calculations and sample size, we have now defined our primary end points as Rbm fragmentation (a classic structural hallmark of EMT) and epithelial activation, and the other downstream EMT biomarkers of MMP-9 cell expression, in addition to S100A4 cell expression in the basal epithelium and Rbm, as secondary end points. The results of this analysis would suggest that if powering a study for these primary end points, then rather modest numbers are required. If S100A4 expression in the basal epithelium was treated as an independent variable, it would provide the most sensitive outcome, and perhaps a primary end point of combining S100A4 staining in both basal epithelial cells and Rbm together would be worth considering in numerically limited intervention studies.
The present study supports the concept that the epithelium in smokers/COPD is activated with high levels of EGFR expression. We nominated this as a primary outcome in the power calculations because it may well be that activation of the epithelium is a primary mechanistic event in inducing EMT and could also be important in cancer induction.29 This pilot trial showed a reduction in EGFR expression in the airway epithelium in the active ICS arm compared to placebo, and again, it was essentially normalized. EGFR is overexpressed in many types of cancers, including non-small-cell lung cancer,29 and we have shown that this is highly expressed in the epithelium as part of the “COPD–EMT phenotype signal”.3 This is the first report of the effects of ICS on epithelial activation in COPD.
Taken together, these data provide strong suggestive evidence that fluticasone propionate could have significant anti-epithelial activation and anti-EMT effects in COPD, but is seemingly not able to completely eliminate all manifestations of EMT activity in COPD airways in all subjects over this 6-month timescale. We did not have sufficient numbers of samples to adequately address the question of whether current smoking status influences the effects of ICS on EMT markers; however, there was no obvious difference between these two groups in any of the biomarker changes on active treatment.
Given that this study was treated as a pilot for “proof of concept” and investigative power-determination purposes, with assumed insufficient numbers of participants to be a definitive study, we have avoided some statistical finesse, such as allowing for multiple comparisons, or treating primary versus secondary end points differently. Under such pilot circumstances, this is regarded as quite acceptable,20,21 but we have now provided evidence for how a larger definitive study could be designed.
The presence of COPD per se increases the risk of developing lung cancer by four- to fivefold when the smoking history is controlled for.18 Furthermore, up to 70% of lung cancer occurs in the context of mild-to-moderate COPD rather than end-stage.30 This implies that mechanisms specific to the relatively early pathogenesis of COPD may be involved in the development of lung cancer. Potential shared biological mechanisms in COPD and lung cancer include: chronic inflammation, matrix degradation, cell proliferation and anti-apoptosis, abnormal wound repair, and angiogenesis.31 All of these are associated with EMT, especially EMT Type III, which is recognized as a promalignant condition in other situations of potential epithelial malignancy.11 Indeed, the relationship between COPD pathology and carcinogenesis may reflect a more general paradigm of epithelial instability and cancer etiology, bearing in mind that epithelial cancers make up 90% of all malignancies.15
A number of observational studies in the literature have demonstrated that ICS reduce local and systemic inflammation among patients with COPD.17,32 ICS also improves lung function and the frequency of exacerbation in long-term users.33 Animal models of smoking-induced COPD have demonstrated that glucocorticoids strikingly inhibit the development of smoking-related lung cancer.34–36 In human epidemiological research, a US veterans cohort study of 10,474 patients in primary care clinics found that use of ICS, albeit only at high doses (as used in our study), was associated with appreciable (50%) decreased risk of lung cancer.18 Similar findings were reported in ex-smokers with COPD.19 However, the Towards a Revolution in COPD Health (TORCH) study37 of ICS in severe COPD failed to show this effect, but it was not powered to pick this up, and was a study on severe rather than mild-to-moderate COPD, where lung cancer tends to occurs in particular.30 TORCH would have excluded obvious lung cancer at entry and the study participants could be regarded as a survivor population. There is an essential need for both in vivo and in vitro human studies to understand the mechanistic link between COPD and airway cancer and how ICS and other drugs may affect it.38,39
Conclusion
This relatively small pilot study using retrospectively obtained tissue has actually provided quite strong suggestive evidence for an effect of ICS in downregulating epithelial activation and selected EMT biomarkers in COPD airways. We hope to have provided a strong stimulus for a larger confirmatory study, and have provided estimates of sample sizes needed for adequate power for appropriate statistical analyses. The numbers of COPD subjects needed to be recruited for such a study are quite feasible.
Disclosure
The authors report no conflicts of interest in this work.
References
Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet. 2004;364(9434):613–620. | |
Feltis BN, Wignarajah D, Zheng L, et al. Increased vascular endothelial growth factor and receptors: relationship to angiogenesis in asthma. Am J Respir Crit Care Med. 2006;173(11):1201–1207. | |
Sohal SS, Reid D, Soltani A, et al. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15(6):930–938. | |
Sohal SS, Reid D, Soltani A, et al. Evaluation of epithelial mesenchymal transition in patients with chronic obstructive pulmonary disease. Respir Res. 2011;12:130. | |
Sohal SS, Walters EH. Epithelial mesenchymal transition (EMT) in small airways of COPD patients. Thorax. 2013;68(8):783–784. | |
Sohal SS, Walters EH. Role of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD). Respir Res. 2013;14:120. | |
Sohal SS, Ward C, Danial W, Wood-Baker R, Walters EH. Recent advances in understanding inflammation and remodeling in the airways in chronic obstructive pulmonary disease. Expert Rev Respir Med. 2013;7(3):275–288. | |
Sohal SS, Soltani A, Weston S, Wood-Baker R, Walters EH. Intermediate filament vimentin and potential role in epithelial mesenchymal transition (EMT). In: De Mello RA, editor. Vimentin Concepts and Molecular Mechanisms. New York: Nova Publishers; 2013:37–61. | |
Soltani A, Muller HK, Sohal SS, et al. Distinctive characteristics of bronchial reticular basement membrane and vessel remodelling in chronic obstructive pulmonary disease (COPD) and in asthma: they are not the same disease. Histopathology. 2012;60(6):964–970. | |
Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119(6):1429–1437. | |
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. | |
Soltani A, Reid DW, Sohal SS, et al. Basement membrane and vascular remodelling in smokers and chronic obstructive pulmonary disease: a cross-sectional study. Respir Res. 2010;11:105. | |
Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776–1784. | |
Young R, Hopkins R, Etzel C, El-Zein R. Genetics of lung cancer susceptibility and COPD. Lancet Oncol. 2011;12(7):622–623. | |
Garber K. Epithelial-to-mesenchymal transition is important to metastasis, but questions remain. J Natl Cancer Inst. 2008;100(4):232–233, 239. | |
Calverley PM, Boonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. Eur Respir J. 2003;22(6):912–919. | |
Reid DW, Wen Y, Johns DP, Williams TJ, Ward C, Walters EH. Bronchodilator reversibility, airway eosinophilia and anti-inflammatory effects of inhaled fluticasone in COPD are not related. Respirology. 2008;13(6):799–809. | |
Parimon T, Chien JW, Bryson CL, McDonell MB, Udris EM, Au DH. Inhaled corticosteroids and risk of lung cancer among patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(7):712–719. | |
Kiri VA, Fabbri LM, Davis KJ, Soriano JB. Inhaled corticosteroids and risk of lung cancer among COPD patients who quit smoking. Respir Med. 2009;103(1):85–90. | |
Arain M, Campbell MJ, Cooper CL, Lancaster GA. What is a pilot or feasibility study? A review of current practice and editorial policy. BMC Med Res Methodol. 2010;10:67. | |
Lancaster GA, Dodd S, Williamson PR. Design and analysis of pilot studies: recommendations for good practice. J Eval Clin Pract. 2004;10(2):307–312. | |
Sohal SS, Reid D, Soltani A, et al. Changes in airway histone deacetylase2 in smokers and COPD with inhaled corticosteroids: a randomized controlled trial. PLoS One. 2013;8(5):e64833. | |
Richmond I, Booth H, Ward C, Walters EH. Intrasubject variability in airway inflammation in biopsies in mild to moderate stable asthma. Am J Respir Crit Care Med. 1996;153(3):899–903. | |
Luepker RV, Raczynski JM, Osganian S, et al. Effect of a community intervention on patient delay and emergency medical service use in acute coronary heart disease: The Rapid Early Action for Coronary Treatment (REACT) Trial. JAMA. 2000;284(1):60–67. | |
Ward C, Forrest IA, Murphy DM, et al. Phenotype of airway epithelial cells suggests epithelial to mesenchymal cell transition in clinically stable lung transplant recipients. Thorax. 2005;60(10):865–871. | |
Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119(6):1417–1419. | |
Milara J, Peiro T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68(5):410–420. | |
Wang Q, Wang Y, Zhang Y, Zhang Y, Xiao W. The role of uPAR in epithelial-mesenchymal transition in small airway epithelium of patients with chronic obstructive pulmonary disease. Respir Res. 2013;14:67. | |
Dasari V, Gallup M, Lemjabbar H, Maltseva I, McNamara N. Epithelial-mesenchymal transition in lung cancer: is tobacco the “smoking gun”? Am J Respir Cell Mol Biol. 2006;35(1):3–9. | |
Barnes PJ, Adcock IM. Chronic obstructive pulmonary disease and lung cancer: a lethal association. Am J Respir Crit Care Med. 2011;184(8):866–867. | |
Yang IA, Relan V, Wright CM, et al. Common pathogenic mechanisms and pathways in the development of COPD and lung cancer. Expert Opin Ther Targets. 2011;15(4):439–456. | |
Hattotuwa KL, Gizycki MJ, Ansari TW, Jeffery PK, Barnes NC. The effects of inhaled fluticasone on airway inflammation in chronic obstructive pulmonary disease: a double-blind, placebo-controlled biopsy study. Am J Respir Crit Care Med. 2002;165(12):1592–1596. | |
Chanez P, Bourdin A, Vachier I, Godard P, Bousquet J, Vignola AM. Effects of inhaled corticosteroids on pathology in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1(3):184–190. | |
Greenberg AK, Hu J, Basu S, et al. Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am J Respir Cell Mol Biol. 2002;27(3):320–328. | |
Yao R, Wang Y, Lemon WJ, Lubet RA, You M. Budesonide exerts its chemopreventive efficacy during mouse lung tumorigenesis by modulating gene expressions. Oncogene. 2004;23(46):7746–7752. | |
Wattenberg LW, Wiedmann TS, Estensen RD, Zimmerman CL, Steele VE, Kelloff GJ. Chemoprevention of pulmonary carcinogenesis by aerosolized budesonide in female A/J mice. Cancer Res. 1997;57(24):5489–5492. | |
Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356(8):775–789. | |
Miller YE, Keith RL. Inhaled corticosteroids and lung cancer chemoprevention. Am J Respir Crit Care Med. 2007;175(7):636–637. | |
van Gestel YR, Hoeks SE, Sin DD, et al. COPD and cancer mortality: the influence of statins. Thorax. 2009;64(11):963–967. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.