Back to Journals » Drug Design, Development and Therapy » Volume 15
A Promising Single Oral Disintegrating Tablet for Co-Delivery of Pitavastatin Calcium and Lornoxicam Using Co-Processed Excipients: Formulation, Characterization and Pharmacokinetic Study
Authors Teaima MH ![]() , Abdel-Haleem KM
, Abdel-Haleem KM ![]() , Osama R, El-Nabarawi MA, Elnahas OS
, Osama R, El-Nabarawi MA, Elnahas OS ![]()
Received 4 August 2021
Accepted for publication 17 September 2021
Published 7 October 2021 Volume 2021:15 Pages 4229—4242
DOI https://doi.org/10.2147/DDDT.S332729
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Mahmoud H Teaima,1 Khaled M Abdel-Haleem,2 Rewan Osama,2 Mohamed A El-Nabarawi,1 Osama S Elnahas2
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Cairo, Egypt; 2Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, October 6 University, Giza, Egypt
Correspondence: Mahmoud H Teaima
Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Kasr El-Aini Street, Cairo, 11562, Egypt
Tel +201005840264
Email [email protected]
Significance: Statins are an important class of drugs that help to control hyperlipidemia, and one of these statins recently used is pitavastatin calcium (PITA). Nevertheless, the most reported adverse effect of statins is myopathy. Therefore, combining statins with non-steroidal anti-inflammatory drugs (NSAIDs) as Lornoxicam (LORNO) can help in the management of statin-induced myopathy.
Purpose: This study aimed to formulate and evaluate different oral disintegrating tablets (ODTs) containing PITA using different co-processed excipients. The best PITA-ODT was selected and reformulated with the addition of LORNO, forming a single ODT comprising both drugs. The pharmacokinetic parameters of PITA and LORNO in a single ODT were compared to those of the marketed products (Lipidalon® and Lornoxicam®).
Methods: Eight PITA-ODTs were prepared via direct compression. The prepared PITA-ODTs were evaluated for their weight variation, thickness, breaking force, friability, drug content, and wetting time (WT). In-vitro disintegration time (DT) and dissolution were also evaluated and taken as parameters for selection of the best formula based on the criteria of scoring the fastest DT and highest Q10 min. LORNO was added to the selected PITA-ODT, forming a single ODT (M1) comprising both drugs, which was subjected to an in-vivo pharmacokinetic study using rats as an animal model and liquid chromatography-mass spectrometry (LC-MS/MS) for analysis of both drugs in rat plasma.
Results: Results showed that all PITA-ODTs had acceptable physical properties in accordance with pharmacospecial standards. PITA-ODT prepared with Pharmaburst® (F2) had significantly (p< 0.05) the fastest DT (6.66± 1.52 s) and highest Q10 min (79.07± 2.02%) and was chosen as the best formula. The in-vivo pharmacokinetic study of M1 formula showed higher percent relative bioavailability (%RB) of 286.7% and 169.73% for PITA and LORNO, respectively, compared with the marketed products.
Conclusion: The single ODT comprising PITA and LORNO was promising for instant co-delivery of both drugs with higher %RB when compared with the marketed products.
Keywords: oral disintegrating tablets, co-processed excipients, pitavastatin calcium, Lornoxicam, in-vivo pharmacokinetic study, liquid chromatography-mass spectrometry
Introduction
Pitavastatin calcium (PITA) is a new addition to statins, which is chemically known as monocalcium (3R, 5S, 6E)-7-[2-cyclopropyl-4-(4-fluorophenyl)-3-quinolinyl]-3,5-dihydroxy-6-heptanoic acid.1,2 It has a unique pharmacological effect on the reduction of low-density lipoprotein-cholesterol (LDL-C) and the improvement of high-density lipoprotein-cholesterol (HDL-C) via competitive inhibition of the liver enzyme, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase.1,3 Nevertheless, the overuse of statin therapy may cause myopathy, which is the most reported side effect of statin.4 Therefore, combining statins with non-steroidal anti-inflammatory drugs (NSAIDs) can help in the management of statin-induced myopathy. NSAIDs are among the most commonly used medications to assist in the management of muscle pain.5 Lornoxicam (LORNO) is a well-known NSAID that has a strong anti-inflammatory effect with a better tolerability profile than other NSAIDs.6 Despite the huge developments in drug delivery, the oral route remains the most favored route of administration for numerous reasons, which are mainly summarized in ease of administration, accurate dosing, self-medication, and affordability, which have resulted in improving patient compliance.7 Recently, a new technology hit the tablet market, which is the oral disintegrating tablet (ODT).8 ODTs have been developed as an alternative to conventional oral tablets, to overcome the difficulty of swallowing in geriatrics via rapid disintegration (30s) upon administration in the mouth, allowing faster onset of action and hence improving patient compliance. Furthermore, the rapid disintegration of ODTs in the mouth allows pre-gastric drug absorption, which enhances the bioavailability of the drug as it passes the first-pass metabolism.9
Co-processing of excipients is a promising method used in the preparation of ODTs, where two or more excipients are combined to improve the physical properties of the tablet, such as flowability, disintegration time, and dissolution stability, rather than using a physical mixture of the same excipients. These types of excipients contain diluents, binders, glidants, disintegrants, and lubricants. Among the co-processed excipients are Pharmaburst®, Ludiflash®, F-melt®, Prosolv HD 90®, Prosolv ODT G2®, Pearlitol flash®, Starlac®, and Lactochem® microfine.10,11
This study aimed to formulate and evaluate PITA-ODTs using different co-processed excipients via direct compression. The best PITA-ODT was selected and reformulated with the addition of LORNO, forming a single ODT comprising both drugs, where this combination can provide great potential for limiting myopathy caused by statin overuse, and hence improving patient compliance. To our knowledge, the combination of both drugs into a single ODT using co-processed excipients has not been studied yet. An in-vivo study was performed on rats to assess the pharmacokinetic parameters and relative bioavailability (RB) of both drugs in a single ODT compared with the marketed products (Lipidalon® and Lornoxicam®).
Materials and Methods
Materials
Pitavastatin calcium (PITA) and Lipidalon® tablets (1 mg) were a kind gift sample from Mash Premiere for Pharmaceutical Industry (New Cairo, Egypt). Lornoxicam (form II) (LORNO) and Lornoxicam® tablets (4 mg) were obtained as a gift sample from Global Napi drug company Ltd (6th October City, Egypt). Pharmaburst® 500 and Lubripharm® Ssf (sodium stearyl fumarate) were a gift from SPI Pharma (Wilmington, DE, USA). F-melt® Type C was provided by Fuji Chemical Industry Ltd. (Toyama-Pref, Japan). Prosolv ODT G2® and Prosolv HD 90® were a gift from JRS Pharma GmbH and Co., KG (Rosenberg, Germany). Ludiflash® was provided by BASF (Ludwigshafen, Germany). Lactochem® Microfine was a gift sample from (Borculo Domo, Netherlands). Starlac® and Pearlitol flash® were obtained from Roquette (Lestrem, France). Mannitol, potassium dihydrogen phosphate, and disodium hydrogen phosphate were purchased from (El-Nasr Pharmaceutical Chemicals Co., Cairo, Egypt). Diethyl ether was from (El-Goumhouria Co., Cairo, Egypt). Torsemide (internal standard) was purchased from (Multi-Apex Pharma, Cairo, Egypt). Acetonitrile, ammonium acetate, and ethyl acetate (HPLC grade) were from Merck (Darmstadt, Germany).
Methods
Fourier Transform Infrared Spectroscopy (FT-IR)
The FT-IR study was done to allow the identification of functional groups of various chemicals as well as incompatibilities between the drug and excipients. Pure PITA and the physical mixture of PITA with co-processed excipients were subjected to FT-IR analysis using the potassium bromide (KBr) pellet method. A mortar and pestle were used to finely grind 3 mg of sample and 300 mg of KBr. A small amount of the mixture was placed under a hydraulic press and compressed at 10 kg/cm, forming a transparent pellet. The pellet was kept in the sample holder and scanned from 4000 to 500 cm−1 in FT-IR (IR Affinity-1, Shimadzu, Kyoto, Japan).12,13
Micromeritic Characterization of the Powder Blend Used in the Formulation of ODTs
Prior to compression, the flowability and compressibility parameters of powder blends were assessed via measuring the angle of repose for determination of the former and Carr’s index and Hausner ratio for the latter12,14 according to the following equations:11,12

where h is the height and r is the radius.
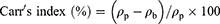

Where ρp is the tapped density and ρb is the bulk density.
Formulation of PITA-ODTs
The composition of all PITA-ODTs is shown in Table 1. The ODTs were prepared via direct compression using a single punch tablet machine (Talleres Sanchez SRL, Model SC1, Buenos Aires, Argentina), with a flat-faced 7 mm punch and die set. All powder blends corresponding to 40 tablets of each formula were mixed using a bench-scale V-shape mixer, where the powder was filled in glass vials (powder loading not exceeding 50% of the vial volume) which were fitted inside the V-shape mixer using adhesive tape, then mixed continuously for 10 min. The powder blend was fed into the die and compressed with a constant compression force.15,16 Finally, each ODT was compressed to a final weight of 100 mg containing 1 mg of PITA.
 |
Table 1 The Composition of PITA-ODTs and the Single ODT Containing PITA and LORNO |
Characterization of the Prepared PITA-ODTs
Weight Variation
The test was performed according to the British Pharmacopeia, where twenty ODTs were selected haphazardly from each formulation and weighed individually, then compared with the mean weight, and the SD was calculated.12,17
Thickness
The thickness of the ODT was measured by placing it between the two arms of the vernier calipers. The findings were presented as a mean value ± SD based on five measurements.10,18
Breaking Force
Three ODTs from each formula were tested for breaking force using a Monsanto hardness tester (Model HT-50P, Thermonik, Campbell Electronics, Veer Savarkar Marg, Mumbai). Each ODT was positioned horizontally in the space provided on the device and the breaking force value was recorded upon ODT breakage.12,19
Friability
About 6.5 g of the total tablet weight from each formula was weighed (initial weight) and put into the friability tester at 25 rpm rotation speed for 4 min. The tablets were removed and then re-weighed (final weight) after the rotation cycle was done and the percent friability was then calculated. The difference between the previous weights represents the friability value, which can be expressed as a percentage of the initial weight of the tablet, as shown in the following equation:11
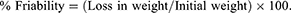
Tablet friability value was considered good enough when the percent friability ranged from 0.1% to 0.9%.12,19
Drug Content
The drug content was evaluated by dissolving each ODT in 100 mL simulated saliva fluid (SSF, pH 6.8). The solution was filtered, and then the absorbance was measured spectrophotometrically at the maximum wavelength (λmax=245nm), and the concentration was obtained from a pre-constructed calibration curve in SSF (pH 6.8). The experiment was done in triplicates to determine the mean drug content of each formula.19,20
Wetting Time (WT)
A piece of tissue paper was doubled twice and placed in a small petri dish containing 6 mL of methylene blue aqueous solution (dye). Each ODT was carefully positioned on the paper surface, and the time required for the dye to reach the upper surface of the tablet was taken as the WT. A time limit of 180s was set, and a higher value was deemed inconvenient for an ODT.19
In-vitro Disintegration Time (DT)
Each ODT was placed into a beaker containing 5 mL of SSF (pH 6.8) maintained at 37±0.5°C. The time in seconds consumed for full disintegration of the tablet was observed and recorded.11,19
In-vitro Dissolution Study
The dissolution study of the PITA-ODTs was performed using a USP dissolution system, Distek (Model 2500i Type II, TCS-0500 Scheduler, New Jersey, USA) at 37±0.5°C and 50 rpm using 100 mL of SSF (pH 6.8) as a dissolution medium. At predetermined time intervals (2, 4, 6, 8, 10, 20 min), 5 mL aliquots were withdrawn and immediately replaced with an equal volume of fresh SSF (pH 6.8) solution kept at the same temperature. The samples were filtered via 0.45-µm membrane filters. The absorbencies of the filtered samples were spectrophotometrically measured at λmax 245nm.20
Statistical Analysis
The experimental results were analyzed using statistical software of statistical package for social sciences (SPSS®) version 26. One-way analysis of variance (ANOVA) followed by post hoc multiple comparisons using the least square difference (LSD) was carried out to cut off for the significance between the prepared ODTs at P<0.05 in terms of in-vitro DT and dissolution.
Selection of the Best Formula
Based on the results of the in-vitro DT and dissolution tests. The ODT that scored the fastest DT and highest Q10 min was chosen as the best formula, which was then subjected to further investigations.
Scanning Electron Microscopy (SEM) of the Selected PITA-ODT
The surface topography and morphology of the sectioned surface of the selected ODT were determined using SEM. The internal matrix of the selected ODT was examined using SEM at a voltage of 20 kV after the tablet was cut transversely. To prepare the cross-section sample, a thin slice of the tablet was cut using a scalpel.12,21
Spectrophotometric Analysis of PITA and LORNO Mixture
Overlying for both PITA and LORNO scanning was done by using the data stored on the computer for the two drugs. Derivative spectroscopy for simultaneous determination of PITA and LORNO in SSF (pH 6.8), calibration curve construction for PITA in SSF (pH 6.8) by second derivative spectroscopy, and recovery study of PITA and LORNO mixtures using SSF (pH 6.8) as a solvent were carried out.22
FTIR
The FT-IR study was performed for pure LORNO, the physical mixture of PITA and LORNO, and the physical mixture of the selected PITA-ODT with LORNO using the same technique that was previously mentioned.
Formulation of a Single ODT Containing PITA and LORNO
The selected PITA-ODT was reformulated via a direct compression technique as mentioned before with the addition of LORNO, forming a single ODT (M1) comprising 1 mg of PITA and 4 mg of LORNO per tablet as shown in Table 1.
Characterization of the Single ODT Containing PITA and LORNO
The M1 ODT was subjected to the characterization tests that were previously carried out for PITA-ODTs using the same techniques. The in-vitro dissolution study of M1 ODT was compared with that of the marketed products (Lipidalon® and Lornoxicam®).
In-vivo Pharmacokinetic Study of the Single ODT Containing PITA and LORNO
The study protocol was approved by the Institutional Animal Ethics Committee (PI 2646), Faculty of Pharmacy, Cairo University, Egypt. Twelve male Wistar rats (weighing between 300 and 350 g) were used for this study. The rats were randomly divided into two groups, each with six animals: group I received the M1 formula orally at doses of (1 mg/kg)23,24 for PITA, and (1.6 mg/kg)25 for LORNO, and group II received the marketed products, Lipidalon® (1mg) and Lornoxicam® (4 mg), with equivalent doses of 1 mg/kg for the former and 1.6 mg/kg for the latter, according to rat body weight. The marketed tablets were dispersed in distilled water,26 where 0.9 mL contained the proper dose of PITA (1 mg/kg) and 1.2 mL contained the needed dose of LORNO (1.6 mg/kg). The M1 ODT containing the adjusted dose of PITA (0.3 mg) and LORNO (0.48 mg) according to rat body weight was prepared using the previously mentioned technique. Following parallel design, each group was housed in its own cage in a temperature-controlled room with a 12 h light/dark cycle, free access to water, and a typical laboratory diet.27 The rats were fasted overnight for 12 h prior to the study, and their oral cavity was cleansed to remove any debris before placing the ODT upon the tongue by means of tweezers and 50–200 μL of water was added to aid tablet disintegration.28 The rats were fed 4 h after receiving the medication and had free access to water for the duration of the study. Blood samples (0.5 mL) were withdrawn from the retro-orbital venous plexus using a heparinized tube under light anesthesia with diethyl ether29 at the predetermined time intervals of 0, 0.25, 0.5, 1, 2, 4, 8, 24, and 48 h post-dose. The collected blood samples were instantly centrifuged (PLC-012, Gemmy Industrial Corp, Taiwan) at 4000 rpm for 5 min. The supernatant plasma was separated and analyzed with Liquid Chromatography-Mass Spectrometry (LC-MS/MS) (Shimadzu®, Japan) using a previously validated LC-MS/MS method of analysis.12,30–32
Sample Preparation
About 100 µL of torsemide (from a stock solution with a concentration of 200 ng/mL) was added to each sample (0.5 mL of rat plasma) as an internal standard. The plasma sample and torsemide were extracted using ethyl acetate (4 mL), vortexed for 2 min, and then centrifuged for 5 min at 5000 rpm (cooling centrifuge, TGL-20 MB). The supernatant was transferred to other vials filtered through a 0.22-µm membrane filter, then evaporated to dryness using a vacuum concentrator (Eppendorf Vacufuge plus, Germany). The dry residue was reconstituted in 0.5 mL of mobile phase and an aliquot of 20 µL of this solution was loaded into LC-MS/MS.30
LC-MS/MS Assay of PITA and LORNO
A validated LC-MS/MS method for analyzing plasma concentrations was employed using an LC-MS/MS system (Shimadzu®, Japan) coupled with a triple quadrupole detector (API-3200, AB Sciex, Foster, CA, USA). The mobile phase was composed of 80% acetonitrile, 20% 0.01% ammonium acetate. The chromatographic separation was performed on the C18 column (4.6×50 mm, 5 µm diameter; Agilent Zorbax, CA, USA). The injection volume was 20 µL, with an isocratic flow rate of 1.0 mL/min.31 The analytical data was processed by Analyst Software version 1.6 (Applied Biosystems Inc., Foster City, CA, USA).
Pharmacokinetic Parameters Analysis
A non-compartmental pharmacokinetic model was used to analyze the pharmacokinetic parameters of PITA and LORNO in the M1 ODT and the marketed products using Kinetica® software (version 5, Thermo Fisher Scientific Inc., Waltham, MA).12,30 The values of the maximum PITA and LORNO plasma concentration (Cmax) (ng/mL), the time to reach Cmax (Tmax) (h), mean residence time (MRT) (h), half-life (T1/2) (h), the area under the curve (AUC0-48), and (AUC0-∞) (ng. h/mL) were calculated. Moreover, the relative bioavailability of the M1 ODT compared with the marketed products was calculated as a percentage value:

Statistical Analysis of the Pharmacokinetic Results
A parametric one-way ANOVA test was used to show the significant or non-significant difference of Cmax, AUC0-48, and AUC0-∞ between the M1 ODT and the marketed products. A non-parametric Kruskal–Wallis test was performed to compare the Tmax data obtained from the M1 ODT and the marketed products using statistical software of SPSS® version 26. Differences are significant at p<0.05.
Results and Discussion
FT-IR
The FT-IR spectra of pure PITA and the physical mixture of PITA with co-processed excipients are displayed in Figure 1. Pure PITA showed a characteristic peak at 1411.06 cm−1 due to C=C bending, a peak at 1554.46 cm−1 indicated C=O stretching, a peak at 2937.06 cm−1 indicated the presence of C-H stretching, a peak at 3350.30 cm−1 indicated O-H stretching, and a peak at 3864.50 cm−1 represented N-H stretching.1 The characteristic peaks of PITA in the physical mixture of PITA with co-processed excipients existed in their original positions without shifting, so no incompatibility arises between the drug and co-processed excipients.
 |
Figure 1 FT-IR spectra of (A) pure PITA, (B) PITA with co-processed excipients, (C) pure LORNO, (D) physical mixture of (PITA/LORNO), and (E) M1 formula physical mixture. |
Micromeritic Characterization of the Powder Blend Used in the Formulation of ODTs
As shown in Table 2, the results of the micromeritic examination of the powder blend revealed good flowability, with an angle of repose value ranging from 31.11±0.42 to 37.17±0.87°. The lower compressibility index and Hausner ratio, which ranged from 3.56±1.52 to 25.23±2.08% for the former and 1.03±0.01 to 1.33±0.03 for the latter, further confirmed this.
 |
Table 2 Micromeritic Properties of the Powder Blend Used in the Formulation of ODTs |
Characterization of the Prepared PITA-ODTs
Table 3 shows the results of the characterization tests performed on the prepared PITA-ODTs, all of which meet the pharmacopeial requirements. The mean weight ranged from 93.66±0.30 to 98.66±0.35 mg. The thickness values of the ODTs were confined between 1.39±0.02 to 2.34±0.01 mm and the breaking force ranged from 3.13±0.15 to 3.96±0.11 kg. The friability findings showed less than 1% friability, indicating that all prepared ODTs had high mechanical resistance.33,34 The drug content of all ODTs ranged between 91.97±0.57 to 103.37±1.57%, which was within the pharmacopeial limits.
 |
Table 3 Characterization of the Prepared PITA-ODTs and the Single ODT Containing PITA and LORNO |
Wetting Time
Wetting time is an important parameter for the disintegration properties of the tablets. Lower WT usually infers a faster disintegration.35 The results of WT shown in Table 3 revealed that all prepared PITA-ODTs had acceptable WT ranged from 3.33±1.52 to 41.00±1.00s. Pharmaburst® (F2) recorded the shortest WT (3.33±1.52s), while Ludiflash® (F5) recorded the longest WT (41.00±1.00s), despite the fact that both are mannitol-based excipients together with crospovidone as a superdisintegrant. The difference in the obtained results might be due to the presence of a mixture of sorbitol and mannitol in Pharmaburst®, which promotes hydration compared to mannitol alone in Ludiflash®. The favorable hydration capacity of sorbitol occurred due to the presence of equatorial OH on the C-2 atom, which has the preference to have two hydrogen-bonded contacts, resulting in a high wetting capacity and hence lowering the WT compared to mannitol, which contains an axial OH on the C-2 atom that tends to have only one hydrogen bond.35 Our results are in harmony with the work reported by Tayel et al,35 on Sumatriptan succinate orodispersible tablets showed that ODT prepared with pharmaburst® had the fastest WT, but ODT prepared with Ludiflash® had the longest WT.
In-vitro Disintegration Time
Table 3 displays the in-vitro DT results of all ODTs. According to the European Pharmacopeia, the limit for the DT of ODTs is 3 min.20 As a result, all ODTs had acceptable DT values ranging from 6.66±1.52 to 54.66±1.52s. The results revealed that Pharmaburst® (F2) had significantly (p<0.05) the fastest DT, while Ludiflash® (F5) had the longest DT when compared with other formulae, which was in good correlation with WT results. Similar findings were reported by EL-Nabarawi et al; Tayel et al.11,35
Although Pharmaburst®, Ludiflash®, Prosolv ODT G2®, and F-melt® consist of crospovidone as a superdisintegrant, they had different DT values. This could be attributed to the presence of a mixture of sorbitol and mannitol in Pharmaburst® (F2) in contrast to mannitol alone in Ludiflash® (F5) as previously mentioned.35 The same rationale was given for Prosolv ODT G2® (F3), as it also contains mannitol alone. Moreover, it has a complex matrix (crospovidone, microcrystalline cellulose (MCC), mannitol, and fructose), which might be the reason for its disintegration delay. In addition, the presence of MCC lowers the water uptake into the tablet.10,19 When it comes to F-melt® (F1), its disintegration delay could be attributed to the fact that it had less surface area than Pharmaburst®.19 Our result complies with those of Moqbel et al, who reported that ODTs prepared using Prosolv ODT G2® and F-melt® showed delayed DT in contrast to Pharmaburst® in preparation of chlorzoxazone ODTs.19
Concerning Pearlitol flash® (F4) and Starlac® (F6), the results showed that they had longer DT when compared with Pharmaburst® (F2). This might be due to the difference in superdisintegrant type. Pharmaburst® contains crospovidone, whereas Pearlitol Flash® and Starlac® contain maize starch as disintegrant, which has a lower water uptake capacity than crospovidone. In addition to the previous findings, Pearlitol flash® had a non-wetting rigid central core that delayed its DT.19,35 Similar results were also reported by Tayel et al, who found that Pearlitol Flash had slower disintegration than Pharmaburst due to the presence of a non-wetting, hard compact central core of Pearlitol-based formulae.35
On the other hand, the results revealed that Pearlitol flash® (F4) scored a faster DT than Starlac® (F6). However, both have the same disintegrant (maize starch). This might be due to the difference in the diluent type where mannitol was available in the former and lactose in the latter, which is notorious for delaying the wetting of the tablet and hence increasing the DT as reported by Moqbel et al.19 The same justification was applied for Lactochem® Microfine (F8) as it contains highly processed lactose,11,19 which was likely to be the reason of the DT delay when compared with Pearlitol flash®.
In case of Prosolv HD90® (F7), it had a faster DT compared with Prosolv ODT G2® (F3). The difference could be attributed to the presence of silicified microcrystalline cellulose, which provides a five-fold greater surface area than MCC (present in Prosolv ODT G2®) and allows for faster disintegration. Similar outcomes were stated by El-Nabarawi et al.10
In-vitro Dissolution Study
Figure 2 displays the in-vitro dissolution profile of PITA from the different ODTs. The amount of PITA dissolved after 10 min (Q10 min) was taken as a parameter for comparison between the different ODTs.19 The results showed that pharmaburst® (F2) had significantly (p<0.05) the highest Q10 min value (79.07±2.02%), whereas Ludiflash® (F5) had the lowest Q10 min value (28.80±0.31%). The results were in accordance with those obtained from WT and DT. The highest % PITA dissolved recorded by pharmaburst® might be due to the higher capacity of crospovidone as a superdisintegrant, as it had high capillary activity and marked hydration with little propensity for gel-formation.19 Furthermore, crospovidone particles are granular and highly porous, allowing water to wick into the tablet, resulting in rapid disintegration, and thus enhancing the drug dissolution.20 The lowest % of PITA dissolved recorded by Ludiflash® could be attributed to the existence of the extended-release excipient kollicoat® SR 30D in its composition.11 These findings fall in agreement with those obtained by Tayel et al.35
 |
Figure 2 In-vitro dissolution profile of PITA from different ODTs in SSF (pH 6.8). |
Selection of the Best Formula
The best formula was PITA-ODT that contained Pharmaburst® (F2), which had significantly (p<0.05) the fastest DT (6.66±1.52s) and the highest Q10 min (79.07±2.02%).
SEM of the Selected PITA-ODT
The SEM of the cross-section view of the selected ODT (F2) is shown in Figure 3. In the cross-section view of the tablet, the micrograph revealed numerous large voids, which appear to be the inner structure. This porous nature of the F2 formula aided in water penetration through the tablet matrix, resulting in rapid disintegration in the oral cavity, and this was confirmed by the previous findings of WT and DT.20,36
 |
Figure 3 Scanning electron micrograph of the selected F2 ODT (Magnification X 500). |
Spectrophotometric Analysis of PITA and LORNO Mixture
Figure 4 displays that at λmax 376 nm of LORNO, there was no reading for PITA giving zero absorption, so there was no interference for measurement of LORNO in the presence of PITA. On the other hand, at λmax 245 nm of PITA, there was a reading for LORNO, indicating overlapping between the investigated drugs, so second derivative spectroscopy was adopted to measure PITA in the presence of LORNO. The absorbance and second derivative spectra for both PITA and LORNO solutions in SSF (pH 6.8) are shown in Figure 5. The zero-crossing method revealed that the zero-crossing of LORNO was at λ 246.2 nm, so this could be suitable for the measurement of PITA with no interference from LORNO. The degree of interference was calculated from the recovery percent of the mixtures. No significant interference was detected in different mixtures.
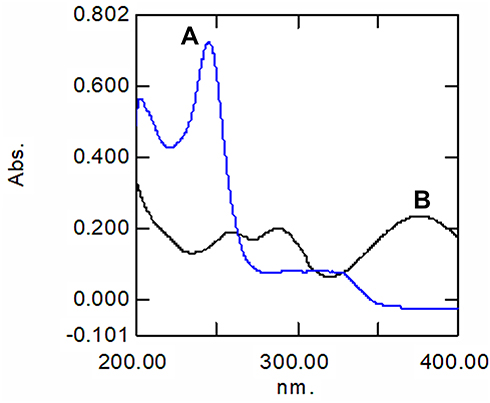 |
Figure 4 UV spectra overly of (A) PITA and (B) LORNO in SSF (pH 6.8). |
 |
Figure 5 Second derivative spectra of (A) PITA and (B) LORNO in SSF (pH 6.8). |
FT-IR
The FT-IR spectra of pure LORNO, the physical mixture of PITA and LORNO, and the physical mixture of the selected formula with LORNO (M1 formula) are displayed in Figure 1. LORNO showed a characteristic peak at 3053.20 cm−1 corresponding to–NH stretching vibration. An intense absorption peak was found at 1630.63 cm−1 due to the stretching vibration of the C═O group in the primary amide. Other peaks were observed at 1589.81 cm−1 and 1529.59 cm−1 showed the bending vibrations of the N–H group in secondary amide. Peaks obtained at 1137.27 cm−1, 1376.08 cm−1, and 1316.67 cm−1 were due to stretching vibrations of the O═S═O group. Other prominent peaks appeared at 828.40 cm−1 corresponding to–CH aromatic ring bending and at 779.73 cm-1 due to C–Cl bending vibration.30 The characteristic peaks of PITA and LORNO in the physical mixture of both drugs and the physical mixture of the M1 formula existed in their original positions without shifting, indicating the absence of incompatibility.12
Characterization of the Single ODT Containing PITA and LORNO
Table 3 reveals the outcomes of the characterization tests performed on the M1 ODT, where the obtained results met the pharmacopeial standards. Figure 6 displays the in-vitro dissolution profile of PITA and LORNO from M1 ODT and the marketed products, where Q10 min for M1 ODT was 79.20±0.88% and 73.88±1.23% for PITA and LORNO, respectively, and this was higher than the marketed products that showed Q10 min 53.58±0.92% for Lipidalon® and 45.72±1.17% for Lornoxicam®.
 |
Figure 6 In-vitro dissolution profile of PITA and LORNO from M1 ODT and the marketed products (Lipidalon® and Lornoxicam®) in SSF (pH 6.8). |
In-vivo Pharmacokinetic Study of the Single ODT Containing PITA and LORNO
It is obvious from the results shown in Table 4 and Figure 7 that the mean values of Cmax of the M1 formula and Cmax of the marketed products were significantly different (p<0.05), where the recorded values showed 1.49 and 1.44 folds increase for PITA and LORNO, respectively, compared with the marketed products. The statistical analysis of Tmax showed that the mean value of Tmax of M1 formula was shorter and significantly different (p<0.05) from the marketed products and the mean value of AUC (0- ∞) of the M1 formula was 2.86 times for PITA and 1.69 times for LORNO, which were significantly (p<0.05) greater than that of the marketed products. Moreover, the % RB of M1 was 286.7% for PITA and 169.73% for LORNO with respect to the marketed products. Based on the AUC (0-∞), Tmax, and % RB values, the M1 formula outperforms the marketed products. The faster absorption rate and the enhanced bioavailability of the two drugs compared to the marketed products might be attributed to the partial drug absorption through the membrane of the oral cavity and esophagus, as evident by the higher Cmax and the shorter Tmax.19 Furthermore, the rapid absorption of the two drugs from M1 ODT correlates well with the results of in-vitro dissolution, which showed that PITA and LORNO were rapidly dissolved from ODT (M1).
 |
Table 4 Mean Pharmacokinetic Parameters and Relative Bioavailability of the Single ODT Containing PITA and LORNO in Comparison with Lipidalon® and Lornoxicam® |
 |
Figure 7 Mean plasma concentration profiles of (A) M1 and Lipidalon®, (B) M1 and Lornoxicam® in rats following oral administration. |
Conclusion
PITA-ODTs were successfully prepared using different co-processed excipients via a direct compression technique. The addition of co-processed excipients facilitates tablet compression and improves the disintegration and dissolution properties of the ODT. PITA-ODT (F2) that contained Pharmaburst® showed the fastest DT and highest Q10 min and was selected as the best formula. Combining LORNO with PITA into a single ODT was carried out to help in the management of myopathy caused by statin overuse. This combination could be promising for reducing the need for multiple medications and hence improving patient compliance. The in-vivo pharmacokinetic study in rats showed that the single ODT (M1) was found to be appropriate for co-delivery of both drugs with higher bioavailability and faster drug absorption rate when compared to the marketed products. However, further pharmacokinetic and pharmacodynamic clinical studies should be conducted to support the obtained results and to investigate the potential of co-delivery of PITA and LORNO to relieve myalgia caused by statin overuse and to evaluate the risk of hepatotoxicity.
Statement of Animal Rights
All institutional and national guidelines for the care and use of laboratory animals were followed and approved by the research ethics committee at the Faculty of Pharmacy, Cairo University (PI 2646).
Disclosure
No conflict of interest was declared by the authors.
References
1. Dasari N, Maruvajala V. Preparation and evaluation of fast dissolving tablets of pitavastatin by 32 full factorial design. Int J App Pharm. 2020;12(1):108–114.
2. Niranjani S, Venkatachalam K. Method development and validation of pitavastatin calcium and its degradation behavior under varied stress conditions by UV spectrophotometric methods. Dhaka Univ J Pharm Sci. 2019;18(2):159–169. doi:10.3329/dujps.v18i2.43258
3. Kumar P, Mangla B, Singh S. Pitavastatin: a potent drug pitavastatin: a potent drug. Int J Pharma Res Health Sci. 2018;6(1):2070–2074.
4. Ward NC, Watts GF, Eckel RH. Statin toxicity: mechanistic insights and clinical implications. Circ Res. 2019;124(2):328–350. doi:10.1161/CIRCRESAHA.118.312782
5. Hatt KM, Vijapura A, Maitin IB, Cruz E. Safety Considerations in Prescription of NSAIDs for Musculoskeletal Pain: a Narrative Review. PM R. 2018;10(12):1404–1411. doi:10.1016/j.pmrj.2018.06.011
6. Tayal S. The role of lornoxicam in pain and inflammation: a review. Curr Res Pharm Sci. 2012;01:1–4.
7. Rai PK, Modi K. Orally disintegrating tablets: a novel approach for medication. Int j Med Health Res. 2019;5(1):84–91.
8. Pinho LA, Temer AC, Ribeiro C, Sá-Barreto LL, Cunha-Filho MSS. the popularization of orodispersible tablets in the pharmaceutical market. Infarma Ciências Farm. 2018;30(2):77–84. doi:10.14450/2318-9312.v30.e2.a2018.pp77-84
9. Tawfeek HM, Faisal W, Soliman GM. Enalapril maleate orally disintegrating tablets: tableting and in vivo evaluation in hypertensive rats. Pharm Dev Technol. 2018;23(5):496–503. doi:10.1080/10837450.2017.1329318
10. El-Nabarawi MA, Teaima MH, Hamid MMA, Shoman NA, Mohamed AI, El-Sahar A. Formulation, evaluation and antioxidant activity of caffeine fast melt tablets. Res J Pharm Technol. 2018;11(7):3131–3138.
11. El-Nabarawi MA, El-Monem RAA; Inas Essam Ibrahim Al-Samadi. Effect of co-process excipients in formulation of ODTs using a model drug. Int J Pharm Sci Res. 2019;10(5):2172–2181.
12. Elmeshad AN, Abdel-Haleem KM, Gawad NAA, El-nabarawi MA, Sheta NM. Core in Cup Ethylmorphine Hydrochloride Tablet for Dual Fast and Sustained Pain Relief: formulation, Characterization, and Pharmacokinetic Study. AAPS PharmSciTech. 2020;21(7):1–11. doi:10.1208/s12249-020-01759-0
13. Karsono Tanuwijaya J, Fatma D. Formulation of ibuprofen orally disintegrating tablets (ODTs) by Lyophilization method using gelatin and mannitol. Int J PharmTech Res. 2014;6(3):996–1002.
14. Phulzalke SB, Kate BA, Bagade MY, Shete RV. Formulation development and evaluation of orodispersible tablets of quetiapine fumarate by sublimation method. Asian J Biomed Pharm Sci. 2016;6(57):22–31.
15. Patra CN, Kumar AB, Pandit HK, Singh SP, Devi MV. Design and evaluation of sustained release bilayer tablets of propranolol hydrochloride. Acta Pharm. 2007;57:479–489. doi:10.2478/v10007-007-0038-0
16. Panda N, Reddy AV, Reddy GVS, Sultana A. Formulation design and in vitro evaluation of bilayer sustained release matrix tablets of doxofylline. Int J Pharm Sci. 2015;7(10):74–83.
17. Aher SS, Saudagar RB, Shinde MS. Review: fast dissolving tablet. Int J Curr Pharm Res. 2018;10(2):5–12. doi:10.22159/ijcpr.2018v10i2.25876
18. Panda S, Hemalatha N, Shankar PU, Baratam SR. Formulation and evaluation of orodispersible tablets (ODTS) of diclofenac sodium by using superdisintegrant from natural origin. Int J App Pharm. 2019;11(6):190–197. doi:10.22159/ijap.2019v11i6.33480
19. Moqbel HA, ElMeshad AN, El-Nabarawi MA. A pharmaceutical study on chlorzoxazone orodispersible tablets: formulation, in-vitro and in-vivo evaluation. Drug Deliv. 2016;23(8):2998–3007. doi:10.3109/10717544.2016.1138340
20. Moutasim MY, ElMeshad AN, El-Nabarawi MA. A pharmaceutical study on lornoxicam fast disintegrating tablets: formulation and in vitro and in vivo evaluation. Drug Deliv Transl Res. 2017;7(3):450–459. doi:10.1007/s13346-017-0367-6
21. Shukla V, Manvi FV. Effect of Kollidon Cl on release behavior of isoniazid and rifampicin combination dispersible tablets for oral treatment of tuberculosis. Int J Drug Dev Res. 2010;2(4):892–906.
22. El-Nabarawi MA, Makky AM, El-Setouhy DA, Abd-Elmoniem R, Amin MG, Jasti BA. Fabrication, evaluation and preliminary clinical study of bi-layer orobuccal devices containing ketorolac tromethamine and chlorhexidine HCL for treatment of oral inflammation. Int J Pharm Sci. 2014;6(2):851–857.
23. Mukhtar RYA, Reid J, Reckless JPD. Statin-induced myositis: a commonly encountered or rare side effect? Drug Focus. 2005;59(2):239–252.
24. Ansari JA, Bhandari U, Haque SE, Pillai KK. Enhancement of antioxidant defense mechanism by pitavastatin and rosuvastatin on obesity-induced oxidative stress in Wistar rats. Toxicol Mech Methods. 2012;22(1):67–73. doi:10.3109/15376516.2011.603391
25. Li F, Song S, Guo Y, et al. Preparation and pharmacokinetics evaluation of oral self-emulsifying system for poorly water-soluble drug Lornoxicam. Drug Deliv. 2015;22(4):487–498. doi:10.3109/10717544.2014.885615
26. Habib BA, Abd El-Samiae AS, El-Houssieny BM, Tag R. Formulation, characterization, optimization, and in-vivo performance of febuxostat self-nano-emulsifying system loaded sublingual films. Drug Deliv. 2021;28(1):1321–1333. doi:10.1080/10717544.2021.1927247
27. Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16(2):109–110. doi:10.1016/0304-3959(83)90201-4
28. Bae J, Johnston TA, Chaiittianan R, Sutthanut K, Jay M, Marson L. Characterization and in vivo efficacy of a heptapeptide ODT formulation for the treatment of neurogenic bladder dysfunction. Int J Pharm. 2018;536(1):397–404. doi:10.1016/j.ijpharm.2017.11.036
29. Cocchetto DM, Bjornsson TD. Methods for vascular access and collection of body fluids from the laboratory rat. J Pharm Sci. 1983;72(5):465–492.
30. Abdelmonem RA, Abd El Galil RM, El-Setouhy DA, El-Miligi MF, El-Nabarawi MA. Dissolution enhancement and formulation of film coated tablets of lornoxicam by phase transition method: in vitro and in vivo evaluation. Int J App Pharm. 2020;12(3):74–85. doi:10.22159/ijap.2020v12i3.36867
31. Zaid AN, Mousa A, Jaradat N, Bustami R. Lornoxicam Immediate-Release Tablets: formulation and Bioequivalence Study in Healthy Mediterranean Volunteers Using a Validated LC-MS/MS Method. Clin Pharmacol Drug Dev. 2017;6(6):564–569. doi:10.1002/cpdd.333
32. Yin T, Liu Q, Zhao H, et al. LC-MS/MS assay for pitavastatin in human plasma and subsequent application to a clinical study in healthy Chinese volunteers. Asian J Pharm Sci. 2014;9(6):348–355. doi:10.1016/j.ajps.2014.07.002
33. Gupta A, Jain AK, Patel NN, Parmar J, Chaudhary R. Formulation & evaluation of orodispersible tablets of diazepam. Pharm Res. 2002;7(2):23–32.
34. Dave V, Yadav RB, Ahuja R, Yadav S. Formulation design and optimization of novel fast dissolving tablet of chlorpheniramine maleate by using lyophilization techniques. Bull Fac Pharmacy Cairo Univ. 2017;5(1):31–39. doi:10.1016/j.bfopcu.2016.12.001
35. Tayel SA, El Nabarawi MA, Amin MM, AbouGhaly MHH. Comparative Study Between Different Ready-Made Orally Disintegrating Platforms for the Formulation of Sumatriptan Succinate Sublingual Tablets. AAPS PharmSciTech. 2017;18(2):410–423. doi:10.1208/s12249-016-0517-z
36. Ahmed IS, Shamma RN, Shoukri RA. Development and optimization of lyophilized orally disintegrating tablets using factorial design. Pharm Dev Technol. 2013;18(4):935–943. doi:10.3109/10837450.2011.619543
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.