Back to Journals » Drug Design, Development and Therapy » Volume 14
A Pharmacokinetic Drug Interaction Between Fimasartan and Linagliptin in Healthy Volunteers
Authors Kang WY ![]() , Lee HW
, Lee HW ![]() , Gwon MR, Cho S
, Gwon MR, Cho S ![]() , Shim WS
, Shim WS ![]() , Lee KT
, Lee KT ![]() , Yang DH, Seong SJ, Yoon YR
, Yang DH, Seong SJ, Yoon YR ![]()
Received 3 February 2020
Accepted for publication 27 April 2020
Published 26 May 2020 Volume 2020:14 Pages 2101—2111
DOI https://doi.org/10.2147/DDDT.S248205
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yan Zhu
Woo Youl Kang,1,2,* Hae Won Lee,1,2,* Mi-Ri Gwon,1,2 Seungil Cho,1,2 Wang-Seob Shim,3 Kyung-Tae Lee,3 Dong Heon Yang,4 Sook Jin Seong,1,2 Young-Ran Yoon1,2
1Department of Molecular Medicine, School of Medicine, Kyungpook National University, Daegu, Republic of Korea; 2Department of Clinical Pharmacology, Kyungpook National University Hospital, Daegu, Republic of Korea; 3Kyung Hee Drug Analysis Center, Kyung Hee University, Seoul, Republic of Korea; 4Division of Cardiology, Department of Internal Medicine, School of Medicine, Kyungpook National University, Daegu 41944, Republic of Korea
*These authors contributed equally to this work
Correspondence: Young-Ran Yoon
Department of Molecular Medicine, School of Medicine, Kyungpook National University, Daegu, Republic of Korea
Tel +82-53-420-4950
Fax +82-53-420-5218
Email [email protected]
Sook Jin Seong
Department of Clinical Pharmacology, Kyungpook National University Hospital, Daegu, Republic of Korea
Tel +82-53-200-6351
Fax +82-53-420-5218
Email [email protected]
Objective: Fimasartan, an angiotensin II type 1 receptor blocker, and linagliptin, a dipeptidyl-peptidase-4 inhibitor, are frequently coadministered to treat patients with hypertension and diabetes, respectively. This study sought to evaluate the pharmacokinetic interactions between fimasartan and linagliptin after co-administration in healthy Korean subjects.
Methods: The overall study was divided into two separate parts, with each part designed as an open-label, multiple-dose, two-period, and single-sequence study. In Part A, to investigate the effect of linagliptin on fimasartan, 25 subjects received 120 mg fimasartan alone once daily for seven days during Period I, and 120 mg fimasartan with 20 mg linagliptin for seven days during Period II. In Part B, to examine the effect of fimasartan on linagliptin, 12 subjects received only linagliptin once daily for seven days during Period I, followed by concomitant administration of fimasartan for seven days during Period II, at the same doses used in Part A. Serial blood samples were collected at scheduled intervals for up to 24 h after the last dose to determine the steady-state pharmacokinetics of both drugs.
Results: Thirty-six subjects completed the study. The geometric mean ratio and 90% confidence intervals for maximum plasma concentration at steady state (Cmax,ss) and area under the concentration–time curve at steady state (AUCτ,ss) of fimasartan with or without linagliptin were 1.2633 (0.9175– 1.7396) and 1.1740 (1.0499– 1.3126), respectively. The corresponding values for Cmax,ss and AUCτ,ss of linagliptin with or without fimasartan were 0.9804 (0.8480– 1.1336) and 0.9950 (0.9322– 1.0619), respectively. A total of eight adverse events (AEs) were reported and the incidence of AEs did not increase significantly with co-administration of the drugs.
Conclusion: Our results suggest that there are no clinically significant pharmacokinetic interactions between fimasartan and linagliptin when co-administered. Treatments were well tolerated during the study, with no serious adverse effects.
Clinical Trial Registry: http://clinicaltrials.gov, NCT03250052.
Keywords: fimasartan, pharmacokinetics, drug–drug interaction, linagliptin, safety
Introduction
Fimasartan (Kanarb®, Boryung Pharmaceutical Co. Ltd., Seoul, Republic of Korea), an angiotensin II receptor blocker (ARB), lowers blood pressure (BP) through the inhibition of the vasoconstricting and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the type 1 (AT1) receptor.1 Fimasartan, approved by the Korean Ministry of Food and Drug Safety for the treatment of mild to moderate hypertension in 2010, is administered once daily at doses of 30–60 mg.2 Previous randomized Phase III clinical trials showed that the antihypertensive effect of fimasartan (60 mg/120 mg) was comparable to that of losartan (50 mg/100 mg) or candesartan (8 mg) in lowering BP with a good safety profile in patients with mild to moderate hypertension, after 12 weeks of treatment.3,4 After oral administration, fimasartan is rapidly absorbed, with the mean time to peak concentration (tmax) of 0.5–3.0 h, and terminal half-life (t1/2) of 5–16 h.2–4 The majority of administered fimasartan is excreted in its parent form, primarily via bile, with a glucuronide conjugate (20%) transformed by UDP-glucuronosyltransferases.5,6 Although hepatic metabolism is a minor route of clearance, and CYP-mediated metabolism of fimasartan is minimal, the major metabolite desulfo-fimasartan is formed mainly by CYP3A4.2,7
Linagliptin, a selective and potent dipeptidyl-peptidase (DPP)-4 inhibitor, shows glucose-lowering efficacy through an effect on two key incretin hormones, active glucagon-like peptide-1 and gastric inhibitory peptide.8,9 Linagliptin is administered once daily (5 mg) for the treatment of type 2 diabetes mellitus.8 Multi-national phase III clinical trials conducted with linagliptin as a monotherapy, and in combination with metformin, glimepiride, pioglitazone, and/or insulin, demonstrated the safety and efficacy of linagliptin for the treatment of type 2 diabetes mellitus.8 Peak plasma concentrations of linagliptin are reached in approximately 1.5–2 hours, with a long t1/2 (>100 h), after oral administration of a single 5 mg dose in healthy subjects.8,10 Following oral administration, the majority (90%) of linagliptin is excreted unchanged, via the enterohepatic system or in urine. As such, the parent compound is pharmacologically active, and hepatic metabolism represents a minor elimination pathway, with a small fraction of absorbed parent drug metabolized to a pharmacologically inactive metabolite.8,10
Hypertension and diabetes are modifiable risk factors of cardiovascular disease and frequently coexist in the same patients.11,12 Consequently, concomitant treatment with ARBs and DPP-4 inhibitors is increasingly common.13 The renoprotective effect of combined DPP-4 inhibitor and ARB treatment is superior to ARB treatment alone in patients with type 2 diabetic nephropathy, as evidenced by the higher proteinuria reduction and lower eGFR decline.14 According to the report by Yang et al, insulin secretion is increased by fimasartan treatment in patients with type 2 diabetes and hypertension, compared with amlodipine.15 These findings suggest that fimasartan combined with linagliptin would be more beneficial in such patients. However, to our knowledge, there are no reports of studies on the interaction between fimasartan and linagliptin. Therefore, we investigated whether there is a potential pharmacokinetic interaction between fimasartan and linagliptin.
Methods
Study Design and Subjects
This study was conducted at the Kyungpook National University Hospital Clinical Trial Center (CTC), in accordance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice. The study protocol was approved by the Institutional Review Board of Kyungpook National University Hospital (KNUH, Daegu, Republic of Korea), and all subjects provided written informed consent to participate in this study before entering the study.
This study consisted of two parts: in Part A, an investigation of the effect of linagliptin on fimasartan was performed and in Part B, an exploration of the effect of fimasartan on linagliptin. Each part was designed as a randomized, open-label, two-period, two treatment study (administration of one drug in the absence or presence of the other drug). After screening, the subjects enrolled in the trial were randomized into Part A or Part B in the order of enrollment. In Part A of the study, twenty-five subjects were enrolled, who were given 120 mg of oral fimasartan (Kanarb®; lot no. F017; expiration date, November 2019; Boryung Pharmaceutical Co. Ltd., Seoul, Republic of Korea) alone once daily for seven days during the first period, followed by the concomitant administration of fimasartan (120 mg) and 5 mg of linagliptin (Tradjenta®; lot no. 661246A; expiration date, August 2019; Boehringer Ingelheim Korea Ltd., Seoul, Republic of Korea) once daily for seven days during the second period. In Part B of the study, twelve subjects received 5 mg of oral linagliptin alone once daily for seven days during Period I, and linagliptin with 120 mg of fimasartan once daily for seven days during Period II. A schematic diagram of the study design is shown in Figure 1.
 |
Figure 1 (A and B) Study design. |
In each part of the study, subjects visited the KNUH CTC each morning on the first 6 days of the first period (days 1–6) to receive the study drug administered orally with 150 mL of water under supervision of the investigator. On day 6, participants were admitted to the study center at 6 pm and were confined until 24 h after the last dose. After an overnight fast of 10 h, subjects received the last maintenance dose with 150 mL of water in the morning of day 7. After pharmacokinetic (PK) sampling for 24 h after the last dose, subjects received the first dose of the second period on day 8 and were discharged from the KNUH CTC. Thereafter, the study subjects visited the study center for 5 days (days 9–13) for drug administration. On day 13, subjects were admitted to the KNUH CTC at 6 pm, disallowed food for at least 10 h, and held until the completion of PK sampling. Participants received the last maintenance dose of drug with 150 mL of water on the morning of day 14, and serial blood samples collected for 24 h until discharge. Subjects were required to abstain from medication, either prescribed or over-the-counter, vitamins or health supplements, any foods that may have affected the metabolism of the study drugs, strenuous physical exercise, smoking, and beverages containing alcohol or caffeine during the study.
Eligible subjects were males between 19 and 50 years old with body mass index of 18.5–27.0 kg/m2 and were considered healthy as determined by medical history, physical examination, vital signs, 12-lead electrocardiogram (ECG), and clinical laboratory tests (hematology, blood chemistry, urinalysis, and serology) within 21 days prior to study drug administration. Exclusion criteria included the following: a medical history of clinically significant hypersensitivity to study drug; any history or evidence of clinically significant disease (hepatobiliary, renal, respiratory, gastrointestinal, hemato-oncology, endocrine, cardiovascular, central nervous system, psychiatric, and/or musculoskeletal system); a systolic blood pressure ≥140 or <115 mmHg, or a diastolic blood pressure ≥90 or <70 mmHg; active liver disease or levels of alanine aminotransferase, aspartate aminotransferase, or total bilirubin >1.5 × the upper limit of normal; and creatinine clearance <80 mL/min (calculated by the Cockcroft–Gault formula using serum creatinine). Other circumstances for exclusion were participation in any other study within three months of the first administration of drug in this study (the completion date of the previous study was the day of the last dose of any prior study drug); donation of whole blood within two months or donation of any blood component within one month of the first dose of drug in this study; and/or abnormal diet that may affect the absorption, distribution, metabolism and excretion of drugs (eg, grapefruit juice ≥1 L/day within seven days prior to administration of study drug).
Determination of Fimasartan and Linagliptin Plasma Concentrations
Serial blood samples for the determination of plasma concentrations were collected at the scheduled time points for fimasartan: 0 (pre-dose), 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, and 24 h after dosing on days 7 and 14. Blood samples were obtained for linagliptin pharmacokinetics at 0 (pre-dose), 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 10, 12, and 24 h after dosing on days 7 and 14. At each sampling time point, five mL of blood was collected into a tube containing sodium heparin and centrifuged at 4°C (3500 rpm, 10 min) to obtain plasma. The plasma samples (1.0 mL/tube) were transferred to two separate microcentrifuge tubes, and stored frozen at −70°C until analysis at the Kyung Hee Drug Analysis Center of Kyung Hee University (Seoul, Republic of Korea).
The plasma concentrations of fimasartan and linagliptin were determined by a validated method using an Agilent 1200 series high-performance liquid chromatography (HPLC) system (Agilent Technologies, Santa Clara, CA, USA) coupled to an MDS SCIEX API-4000 triple quadrupole mass spectrometer (Applied Biosystems, Framingham, MA, USA).
For fimasartan, chromatographic separations were performed on a Luna C18 column (3.0 μm particle size, 2.0 mm i.d. x 50 mm; Phenomenex, Torrance, CA, USA) at a flow rate of 250 μL/min. The mobile phase consisted of a 20:80 (v/v) mixture of 0.05% formic acid in distilled water and 0.05% formic acid in acetonitrile. Multiple reaction monitoring (MRM) transitions were performed at mass-to-charge ratios (m/z) of 502.252 → 207.100 and 526.354 → 207.100 for fimasartan and the internal standard (IS) BR-A-563, respectively. Frozen plasma samples were thawed at room temperature, and vortexed for at least 10 sec or more. The IS (20 μL of BR-A-563 [1000 ng/mL]) was added to a disposable borosilicate glass culture tube containing a 50 μL aliquot of plasma, before the addition of 50 μL of 1% formic acid and 1 mL of organic solvent (ethyl acetate: hexane at a ratio of 8:2), and the mixture vortexed for 10 min. After centrifugation of the mixture at 3500 rpm for 10 min at 4°C, 800 μL of the upper layer was transferred to a disposable borosilicate glass culture tube, and dried with a stream of nitrogen gas at 50°C. The residue was reconstituted with 1 mL of 90% acetonitrile solution (distilled water: acetonitrile in the ratio 1:9 [v/v]). After vortexing for 10 min, the tube was centrifuged at 3500 rpm for 15 minutes at 4°C. A 3 μL aliquot of the upper layer was injected into the HPLC-coupled to tandem mass spectrometry (MS/MS) system for analysis.
For linagliptin, chromatographic separations were performed on a Halo C18 column (2.7 μm particle size, 2.1 mm i.d. x 100 mm; Advanced Materials Technology, Inc., Wilmington, DE, USA) at a flow rate of 250 μL/min. The mobile phase consisted of a 40:60 (v/v) mixture of 10 mM ammonium formate and 100% acetonitrile. The MRM transitions were performed at m/z of 473.137 → 420.300 and 477.231 → 420.300 for linagliptin and the IS linagliptin-13C-d3, respectively. Frozen plasma samples were thawed at room temperature, and then vortexed for >10 sec. The IS (20 μL of linagliptin-13C-d3 [30 ng/mL]) was then added to the disposable borosilicate glass culture tube containing a 200 μL aliquot of plasma, before adding 20 μL of 10% ammonium hydroxide and 3 mL of organic solvent (ethyl acetate: hexane in an 8:2 ratio), and vortexing for 10 min. After centrifugation of the mixture at 3500 rpm for 10 min at 4°C, 2400 μL of the upper layer was transferred to a disposable borosilicate glass culture tube and dried with a stream of nitrogen gas at 50°C. The residue was reconstituted with 200 μL of 50% acetonitrile solution, vortexed for 10 min, centrifuged at 3500 rpm for 1 min at 4°C, and transferred into an Axygen 2.0 mL microtube. After centrifugation of the mixture at 14,000 rpm for 10 min at 4°C, 120 μL of the upper layer was transferred to a clean vial. A 10 μL aliquot of the upper layer was injected into the HPLC-MS/MS system for analysis.
The calibration curves were linear (r > 0.998) over the concentration ranges from 2 to 2000 ng/mL for fimasartan and from 0.05 to 20 ng/mL for linagliptin. The overall intra-day accuracy ranged from 86.8% to 109.9% for fimasartan, and from 98.9% to 111.6% for linagliptin. The overall inter-day accuracy ranged from 98.5% to 101.3% and 100.3% to 107.9% for fimasartan and linagliptin, respectively. The intra-day precision (% coefficient of variation, CV) ranged from 0.6% to 11.9% for fimasartan and from 1.0% to 7.2% for linagliptin. The inter-day precision (% CV) ranged from 1.4% to 12.9% and from 2.5% to 6.7% for fimasartan and linagliptin, respectively. The lower limit of quantification was 2 ng/mL for fimasartan and 0.05g/mL for linagliptin.
Pharmacokinetic Evaluations
Fimasartan and linagliptin PK analyses were performed using non-compartmental methods with WinNonlin Pro 5.3 (Pharsight Corporation, Mountain View, CA, USA). The area under the plasma concentration–time curve over the dosing interval (τ) after repeated dose administration at steady state (AUCτ,ss) was calculated using the linear trapezoidal method. The maximum plasma concentration of drug at steady state (Cmax,ss) and the time to reach Cmax,ss (Tmax,ss) were taken directly from the observed plasma concentrations over time. The apparent clearance was calculated by dividing the dose by the AUCτ,ss.
Safety of Subjects
In all subjects who received at least one or more doses of fimasartan and/or linagliptin, safety was assessed throughout the study period by evaluating any undesirable signs, symptoms, or medical conditions occurring on or after administration of the first dose (treatment emergent adverse event, TEAE). Subjects voluntarily reported any subjective symptoms, and the investigators observed and recorded any objective signs, including vital signs, physical examinations, and results from clinical laboratory tests and from 12-lead electrocardiogram testing. All laboratory tests were performed at an accredited laboratory (Department of Laboratory Medicine, KNUH, Daegu, Republic of Korea).
Statistical Analyses
The sample sizes necessary for Part A and Part B of the trial were calculated based on the intra-subject coefficient of variation of the PK exposure of fimasartan (24.2%) and linagliptin (11.7%), respectively, from earlier PK studies.2,16 In a two-way crossover design, 20 subjects for Part A and 7 subjects for Part B were required to detect a statistical difference of ≥20% in the log-transformed PK parameters between the two different treatments, with a geometric mean ratio (GMR) of 1.0, a significance level of 0.05, and a statistical power of 80%. Therefore, a total of 25 subjects for Part A and 12 subjects for Part B were needed for enrollment, respectively, based on the recorded attrition rate.
Demographic data, PK parameters, and safety data were summarized using descriptive statistics. Results are presented as mean ± standard deviation (SD), except for Tmax,ss values, which are expressed as the median, minimum and maximum values. Pharmacokinetic parameters were compared between the two treatment groups (concomitant administration and individual administration) using paired t-tests, or the Wilcoxon signed-rank test. To determine the associated effect of concomitant administration of fimasartan and linagliptin on the steady-state pharmacokinetics of each agent alone, the GMRs and 90% confidence intervals (CIs) of log-transformed AUCτ,ss and Cmax,ss of fimasartan and linagliptin for the two treatment groups (concomitant administration/individual administration) in Part A or B were assessed using a linear mixed effect model. All statistical analyses were performed using SPSS for Windows software (ver. 18.0; SPSS Korea, Seoul, Korea). A p-value below 0.05 was deemed to indicate statistical significance.
Results
Subjects
In total, 37 healthy Korean male volunteers were enrolled in this study after screening and randomly assigned to one of two different groups (Part A, n = 25; Part B, n = 12). The demographic characteristics of the enrolled subjects are summarized in Table 1. In Part A, one subject withdrew consent during Period I; therefore, 24 subjects completed the study. In Part B, two subjects who withdrew consent before the initiation of Period I were replaced by other subjects from the waiting list, leaving a total of 12 subjects in Part B. Accordingly, a total of 36 subjects who completed the study were included in the PK analyses. All subjects receiving fimasartan and/or linagliptin at least once (Part A, n = 25; Part B, n = 12) were included in the safety assessment. The demographic characteristics of the study subjects who completed the study are summarized in Table 1.
 |
Table 1 Demographic Characteristics of the Subjects Who Completed the Study |
Pharmacokinetic Properties
Effect of Linagliptin on the Pharmacokinetics of Fimasartan
After 7-day repeated once-daily administration of fimasartan alone, or the co-administration of fimasartan and linagliptin, plasma fimasartan levels reached a peak at about 0.5 h (Figure 2A). The PK parameters of fimasartan with and without linagliptin are presented in Table 2. The GMR for Cmax,ss was 1.2633 (90% CI, 0.9175–1.7396), indicating that co-administration with linagliptin increased the Cmax,ss of fimasartan by 1.26-fold (Table 3). The GMR for AUCτ,ss was 1.1740 (90% CI, 1.0499–1.3126), indicating a similar increase in the fimasartan AUCτ,ss by 1.17-fold when it was administered with linagliptin (Table 3).
 |
Table 2 Steady-State Pharmacokinetic Parameters Following Administration of Fimasartan (120 mg) and Linagliptin (5 mg) as Concomitant Administration versus Individual Administration Under Fasted Conditions in Healthy Male Subjects |
 |
Table 3 Geometric Mean Ratio (90% CIs) for the Log-Transformed AUC τ,ss and Cmax, ss Following Administration of Fimasartan (120 mg) and Linagliptin (5 mg) as Concomitant Administration versus Individual Administration in Healthy Male Subjects |
 |
Figure 2 Mean plasma concentration–time profiles in healthy subjects (A) for fimasartan after 7-day multiple oral administrations of fimasartan (120 mg) alone or coadministration of fimasartan (120 mg) and linagliptin (5 mg), (B) for linagliptin after 7-day multiple oral administrations of linagliptin (5 mg) alone of coadministration of linagliptin (5 mg) and fimasartan (120 mg). |
Effect of Fimasartan on the Pharmacokinetics of Linagliptin
After 7-day repeated once-daily administration of linagliptin alone, or the co-administration of linagliptin and fimasartan, the linagliptin PK profiles did not significantly differ when linagliptin was administered alone or when co-administered with fimasartan, with the median Tmax,ss of 0.5 h (Figure 2B). The PK parameters of linagliptin with and without fimasartan are presented in Table 2. The GMRs for Cmax,ss and AUCτ,ss were 0.9804 (90% CI, 0.8480–1.1336) and 0.9950 (90% CI, 0.9322–1.0619), respectively, indicating no significant differences in GMR (linagliptin with fimasartan to linagliptin alone), as presented in Table 3.
Safety
All 37 subjects (Part A, n = 25; Part B, n = 12) who received at least one dose of the study drugs were included in the safety assessment. As presented in Table 4, a total of eight TEAEs (five in Part A and three in Part B) were reported in seven of the 37 subjects (Part A, n = 4 participants; Part B, n = 3 participants) during our study. All AEs were thought to be related to the medication. Most AEs were transient and mild in intensity, with none considered serious. All AEs spontaneously resolved without specific treatment.
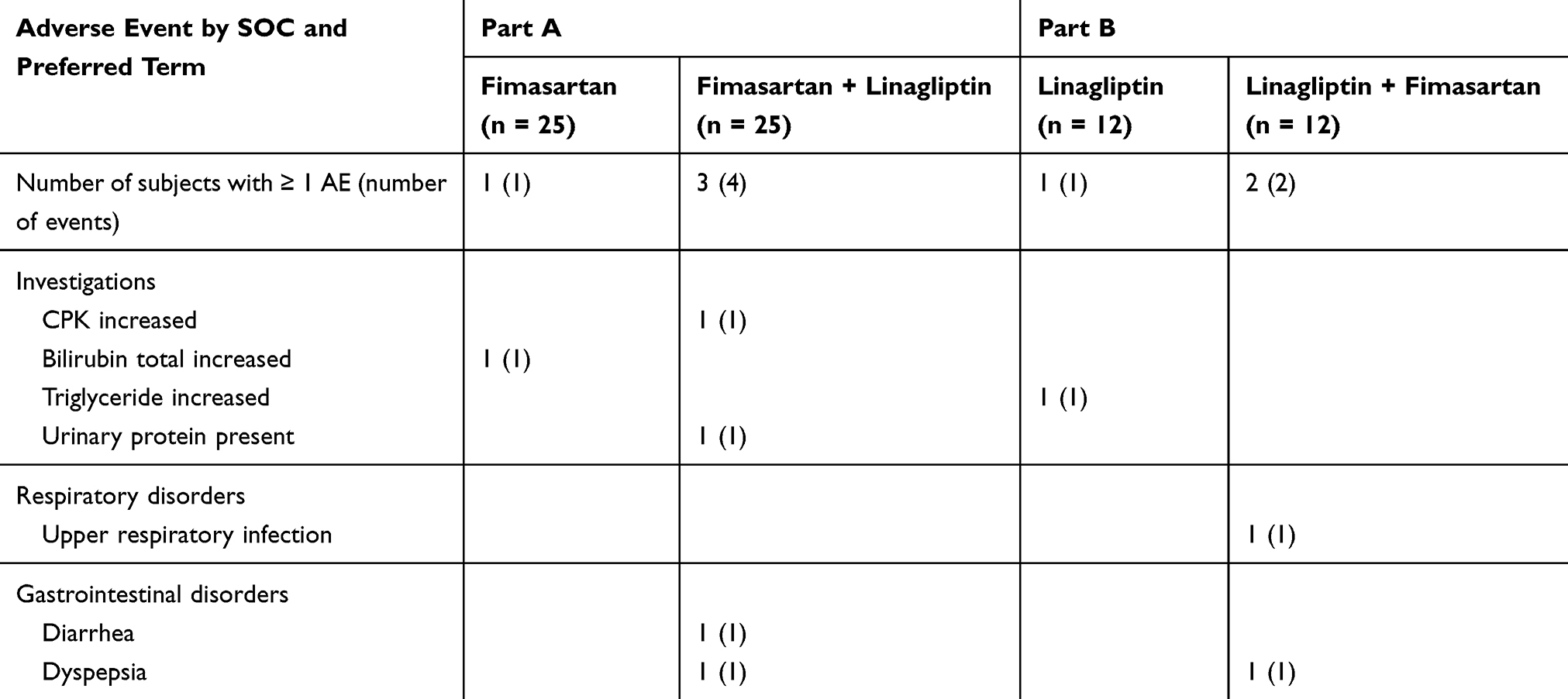 |
Table 4 Adverse Events That Were Reported at Least Twice Following Multiple Oral Administration of 120 mg of Fimasartan and/or 5 mg of Linagliptin in Healthy Subjects |
Discussion
In the present study, we investigated the pharmacokinetic interaction between fimasartan and linagliptin in healthy male subjects. Peak plasma concentration and systemic exposure of fimasartan and linagliptin at steady state were compared between administration of the individual drug and concomitant administration of both drugs after 7 days of repeated dosing. As recommended by the guidelines for clinical drug interaction studies, this study was designed as parallel, two-arm studies instead of two-way crossover studies, because linagliptin has a long (>100 hours) t1/2.17 The doses used in this study were the maximum limit of the dosing regimens of the full prescribing information for fimasartan (120 mg) and the daily recommended dose for linagliptin (5 mg).2,8 Therefore, the study design was more likely to identify any changes in PK parameters resulting from drug–drug interactions.
Although the t1/2 of linagliptin is long and related to the saturable binding of linagliptin to DPP-4, steady-state plasma concentrations of linagliptin 5 mg are reached by the third dose after once-daily dosing.8 Accordingly, the dosing period of 7 days for linagliptin in this study was appropriate for providing the steady-state PK. There was no statistically significant difference in Ctrough values between administration of the individual drug and concomitant administration of both drugs, supporting that steady-state plasma concentrations of linagliptin are reached in this study (Table 2).
For fimasartan, the mean AUCτ,ss and Cmax,ss values from our study (723.9 ng∙h/mL and 243.2 ng/mL, respectively) were comparable to those of previous reports (746.5–813.5 ng∙h/mL, and 241.1–258.0 ng/mL, respectively) following multiple oral doses of 120 mg fimasartan for seven days.3,18 The intra-subject variability (% CV) values of fimasartan obtained in this study (22.8% for AUCτ,ss and 56.7% for Cmax,ss) were analogous to earlier findings from the literature.3,18 The respective GMRs (90% CIs) for AUCτ,ss and Cmax,ss of fimasartan with or without linagliptin in this study indicated that AUCτ,ss and Cmax,ss of fimasartan increased by 17% and 26%, respectively, in the presence of linagliptin. The systemic clearance (CLss/F) of fimasartan was significantly decreased (p = 0.0205) after co-administration of fimasartan and linagliptin.
For linagliptin, the mean AUCτ,ss and Cmax,ss values from our study (90.6 ng∙h/mL and 7.5 ng/mL, respectively) following multiple oral doses of 5 mg linagliptin for seven days were comparable to those of previous reports (73.2–96.0 ng∙h/mL, and 5.4–6.7, respectively).16,19 The 90% CIs of the GMRs for AUCτ,ss and Cmax,ss from our study (0.9322–1.0619 and 0.8480–1.1336, respectively) fell within the range of 0.8–1.25, indicating that fimasartan administered concomitantly did not influence the rate and extent of linagliptin absorption at steady state. The CLss/F of linagliptin was not changed (p = 0.8104).
According to several in-vitro and in-vivo studies, only minimal amounts of fimasartan are metabolized by CYP3A4, and fimasartan is transported by organic anion transporter 1 (OAT1) in the kidneys, and by organic anion-transporting polypeptide (OATP) 1B1, OATP2B1, and OATP1B3 in the hepatobiliary system.7,20,21 Jeong et al (2015) reported that fimasartan glucuronide is a substrate for P-glycoprotein (Pgp) and breast cancer resistance protein (BCRP).6 In vivo studies indicated that linagliptin showed the potential to cause drug–drug interactions with substrates of CYP3A4, CYP2C9, CYP2C8, Pgp, and organic cationic transporter (OCT).8 The plausible explanation for the increased AUCτ,ss and Cmax,ss of fimasartan in the presence of linagliptin in our study is that when fimasartan and linagliptin are administered concomitantly, linagliptin might competitively inhibit CYP3A4-mediated metabolism of fimasartan, and Pgp-mediated efflux of the glucuronide conjugate of fimasartan, resulting in significantly decreased CLss/F of fimasartan. According to the review by the Mudra et al, bioavailability and clearance of the substrate drugs of both CYPs and Pgp can be affected by CYP3A4/Pgp interplay in a non-linear manner.22 Accordingly, the extent to which clearance or bioavailability can be affected by CYP/Pgp interplay would be more difficult to be assessed, compared to Pgp substrate drugs without CYP metabolism.23
For the unchanged CLss/F of linagliptin, the renal excretion of linagliptin mediated by Pgp may increase to compensate for the competitive inhibition of the CYP3A4-mediated metabolism by fimasartan. Further studies are needed to investigate the exact mechanisms of the pharmacokinetic interaction between these two drugs.
According to the investigator’s brochure, in a study to evaluate the antihypertensive efficacy and safety of fimasartan at 20, 60, 120, and 240 mg doses following oral administration for 8 weeks in patients with mild to moderate essential hypertension, the BP-lowering effect by 240 mg fimasartan was similar to those seen in 60 and 120 mg doses, respectively.2 Furthermore, there were no statistically significant differences in the proportions of subjects with AE among the dose groups. Accordingly, in spite of the increased GMRs in AUCτ,ss and Cmax,ss of fimasartan in the presence of linagliptin, it is unlikely that there are clinically significant pharmacokinetic interactions between fimasartan and linagliptin when co-administered.
Multiple doses of fimasartan and linagliptin, whether administered individually or concomitantly, were well tolerated by all subjects enrolled in this study, and coadministration of fimasartan and linagliptin did not affect the safety results in both parts (Part A, p=0.6092; Part B, p=0.6092).
This study was conducted in a relatively small size of healthy male Korean subjects, which limits its application. To generalize the result clinically, studies should be evaluated in hypertensive patients with type 2 diabetes mellitus. Considering the short dosing period of 7 days in healthy subjects, the interaction effects noted in our study should be interpreted with caution.
Conclusions
To our knowledge, this study is the first to investigate the pharmacokinetic interaction between fimasartan and linagliptin. There are no clinically significant pharmacokinetic interactions between fimasartan and linagliptin when co-administered. Multiple doses of 120 mg fimasartan or 5 mg linagliptin, administered alone and in combination, were well tolerated by healthy male Korean subjects.
Abbreviations
BP, blood pressure; tmax, time to peak concentration; t1/2, terminal half-life; DPP-4, dipeptidyl-peptidase-4; CTC, clinical trial center; KNUH, Kyungpook National University Hospital; PK, pharmacokinetic; ECG, electrocardiogram; HPLC, high-performance liquid chromatography; MS/MS, tandem mass spectrometry; MRM, multiple reaction monitoring; IS, internal standard; CV, coefficient of variation; AUCτ,ss, area under the concentration–time curve over a dosing interval τ at steady state; Cmax, ss, maximum plasma concentration at steady-state; Ctrough, ss, trough plasma concentration at steady state; Tmax, ss, time from last dosing to maximum plasma concentration at steady-state; CLss/F, apparent clearance at steady state; TEAE, treatment emergent adverse event; GMR, geometric mean ratio; SD, standard deviation; CI, confidence interval; OAT1, organic anion transporter 1; OATP, organic anion-transporting polypeptide; Pgp, P-glycoprotein; OCT1, organic cation transporter 1.
Data Sharing Statement
We, the authors, intend to share individual de-identified participant data. However, there must be a limit on our data sharing, because this study was sponsored by a pharmaceutical company. Young-Ran Yoon should be contacted for the sharing of the data.
Acknowledgments
The authors are grateful to all study participants and volunteer subjects. This study was sponsored by Boryung Pharmaceutical Co. Ltd., Seoul, Korea, and was supported by the grants from Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI14C2750, HI15C0001) as well as the Industrial Core Technology Development Program (10051129, Development of the system for ADME assessment using radiolabeled compounds) funded by the Ministry of Trade, Industry and Energy (MOTIE, Korea).
Disclosure
The authors report no conflicts of interest regarding the content of this article.
References
1. Kim JH, Lee JH, Paik SH, Kim JH, Chi YH. Fimasartan, a novel angiotensin II receptor antagonist. Arch Pharm Res. 2012;35(7):1123–1126. doi:10.1007/s12272-012-0700-z
2. Kanarb® (fimasartan) tablets [Investigator’s Brochure]. Seoul, Republic of Korea: Boryung Pharmaceutical Co. Ltd.; 2018.
3. Ghim JL, Paik SH, Hasanuzzaman M, et al. Absolute bioavailability and pharmacokinetics of the angiotensin II receptor antagonist fimasartan in healthy subjects. J Clin Pharmacol. 2016;56(5):576–580. doi:10.1002/jcph.618
4. Kang WY, Kim EH, Seong SJ, et al. Pharmacokinetic drug interaction study using fimasartan and rosuvastatin in healthy volunteers. Int J Clin Pharmacol Ther. 2016;54(12):992–1003. doi:10.5414/CP202615
5. Kim TH, Shin S, Bashir M, et al. Pharmacokinetics and metabolite profiling of fimasartan, a novel antihypertensive agent, in rats. Xenobiotica. 2014;44:913–925. doi:10.3109/00498254.2014.915359
6. Jeong ES, Kim YW, Kim HJ, et al. Glucuronidation of fimasartan, a new angiotensin receptor antagonist, is mainly mediated by UGT1A3. Xenobiotica. 2015;45:10–18. doi:10.3109/00498254.2014.942810
7. Kim JW, Yi S, Kim TE, et al. Increased systemic exposure of fimasartan, an angiotensin II receptor antagonist, by ketoconazole and rifampicin. J Clin Pharmacol. 2013;53:75–81. doi:10.1177/0091270011433328
8. Tradjenta® (linagliptin) tablets [prescribing information]. Binger Strasse, Germany: Boehringer Ingelheim Pharmaceuticals, Inc. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/201280s005lbl.pdf.
9. Langley AK, Suffoletta TJ, Jennings HR. Dipeptidyl peptidase IV inhibitors and the incretin system in type 2 diabetes mellitus. Pharmacotherapy. 2007;27(8):1163–1180. doi:10.1592/phco.27.8.1163
10. Ceriello A, Inagaki N. Pharmacokinetic and pharmacodynamics evaluation of linagliptin for the treatment of type 2 diabetes mellitus, with consideration of Asian patient populations. J Diabetes Investig. 2017;8(1):19–28.
11. Hong KN, Fuster V, Rosenson RS, Rosendorff C, Bhatt DL. How low to go with glucose, cholesterol, and blood pressure in primary prevention of CVD. J Am Coll Cardiol. 2017;70(17):2171–2185. doi:10.1016/j.jacc.2017.09.001
12. Ali MK, Bullard KM, Saaddine JB, et al. Achievement of goals in U.S. diabetes care, 1999–2010. N Engl J Med. 2013;368:1613–1624. doi:10.1056/NEJMsa1213829
13. Cooper ME, Perkovic V, Groop P-H, et al. Hemodynamic effects of the dipeptidyl peptidase-4 inhibitor linagliptin with renin–angiotensin system inhibitors in type 2 diabetic patients with albuminuria. J Hypertens. 2019;37(6):1294–1300. doi:10.1097/HJH.0000000000002032
14. Qiu DD, Liu J, Shi JS, et al. Renoprotection provided by dipeptidyl peptidase-4 inhibitors in combination with angiotensin receptor blockers in patients with type 2 diabetic nephropathy. Chin Med J (Engl). 2018;131(22):2658–2665. doi:10.4103/0366-6999.245277
15. Yang YS, Lim MH, Lee SO, et al. Fimasartan increases glucose-stimulated insulin secretion in patients with type 2 diabetes and hypertension compared with amlodipine. Diabetes Obes Metab. 2018;20(7):1670–1677. doi:10.1111/dom.13282
16. Friedrich C, Metzmann K, Rose P, Mattheus M, Pinnetti S, Woerle HJ. A randomized, open-label, crossover study to evaluate the pharmacokinetics of empagliflozin and linagliptin after coadministration in healthy male volunteers. Clin Ther. 2013;35(1):A33–A42. doi:10.1016/j.clinthera.2012.12.002
17. US Food and Drug Administration. [webpage on the Internet] Clinical drug interaction studies – study design, data analysis, and clinical implications guidance for industry; 2017. Available from: https://www.fda.gov/media/82734/download.
18. Yi S, Kim TE, Yoon SH, et al. Pharmacokinetic interaction of fimasartan, a new angiotensin II receptor antagonist, with amlodipine in healthy volunteers. J Cardiovasc Pharmacol. 2011;57(6):682–689. doi:10.1097/FJC.0b013e31821795d0
19. Cao CQ, Xiang YF, Zhou ZG. The clinical application of linagliptin in Asians. Ther Clin Risk Manag. 2015;11:1409–1419. doi:10.2147/TCRM.S64402
20. Shin KH, Kim TE, Kim SE, et al. The effect of the newly developed angiotensin receptor II antagonist fimasartan on the pharmacokinetics of atorvastatin in relation to OATP1B1 in healthy male volunteers. J Cardiovasc Pharmacol. 2011;58:492–499. doi:10.1097/FJC.0b013e31822b9092
21. Gu N, Kim BH, Lim KS, et al. The effect of fimasartan, an angiotensin receptor type 1 blocker, on the pharmacokinetics and pharmacodynamics of warfarin in healthy Korean male volunteers: a one-sequence, two-period crossover clinical trial. Clin Ther. 2012;34:1592–1600. doi:10.1016/j.clinthera.2012.06.004
22. Mudra DR, Desino KE, Desai PV. In silico, in vitro and in situ models to assess interplay between CYP3A and P-gp. Curr Drug Metab. 2011;12(8):750–773. doi:10.2174/138920011798356999
23. Akamine Y, Yasui-Furukori N, Uno T. Drug-drug interactions of P-gp substrates unrelated to CYP metabolism. Curr Drug Metab. 2019;20(2):124–129. doi:10.2174/1389200219666181003142036
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.