")
Back to Journals » International Journal of General Medicine » Volume 15
A Patient with Moderate Intellectual Disability and 49, XXXYY Karyotype
Authors Verhoeven WMA , Egger JIM, Mergler S, Meijer TAA, Pfundt R, Willemsen MH
Received 10 November 2021
Accepted for publication 24 February 2022
Published 10 March 2022 Volume 2022:15 Pages 2799—2806
DOI https://doi.org/10.2147/IJGM.S348844
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Willem MA Verhoeven,1– 3 Jos IM Egger,3– 5 Sandra Mergler,6 Ton AA Meijer,7 Rolph Pfundt,4,8 Marjolein H Willemsen8
1Department of Psychiatry, Erasmus University Medical Center, Rotterdam, The Netherlands; 2Centre for Consultation and Expertise, Utrecht, The Netherlands; 3Vincent van Gogh Centre of Excellence for Neuropsychiatry, Venray, The Netherlands; 4Donders Institute for Brain, Cognition and Behaviour, Radboud University, Nijmegen, The Netherlands; 5Stevig, Specialized and Forensic Care for People with Intellectual Disabilities, Dichterbij, Oostrum, The Netherlands; 6ASVZ, Centre for People with Intellectual Disabilities, Sliedrecht, The Netherlands; 7Department of Internal Medicine, Albert Schweitzer Hospital, Dordrecht, The Netherlands; 8Department of Human Genetics, Radboud University Medical Centre, Nijmegen, The Netherlands
Correspondence: Willem MA Verhoeven, Centre of Excellence for Neuropsychiatry, Stationsweg 46, Venray, 5803 AC, The Netherlands, Tel +31651156556, Fax +31478584765, Email [email protected]
Abstract: Klinefelter syndrome is a chromosomal disorder in which one extra X chromosome is present (47,XXY). Several other numeric variants of this syndrome are described that comprise one or more additional sex chromosomes such as 48,XXXY, 48,XXYY and 49,XXXXY. These rare conditions are often associated with increased risk for congenital malformations, additional medical problems, and a more complex psychological phenotype. Since 1963, apart from two infants, only four adult patients with a XXXYY pentasomy have been published as case report. The present paper critically reviews the existing literature and provides detailed assessments of a 25-year-old male with intellectual disability and autism. For the first time, this very rare pentasomy is now recorded using all information about developmental history as well as findings from genetic, somatic, endocrinological and neuropsychological examination. It is concluded that children born with abnormalities of the external genitalia should always be evaluated for genetic abnormalities in order to avoid unwanted delay of appropriately designed multidisciplinary medical and psychological treatment.
Keywords: 49, XXXYY pentasomy, Klinefelter syndrome, testosterone, autism, treatment design
Introduction
Klinefelter syndrome (KS) was first described in 1942 by Harry F. Klinefelter and colleagues as an endocrine disorder in males with karyotype 47,XXY characterized by a constellation of features like gynecomastia, testicular dysgenesis, eunuchoidism, increased secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), androgen deficiency, and slight developmental delay.1 It is the most common sex chromosome disorder in man and a relatively common cause of male infertility and hypogonadism. KS may be associated with an array of somatic disorders like a tendency to develop visceral obesity, dyslipidemia, hypertension, diabetes mellitus due to insulin resistance resulting in metabolic syndrome, cardiovascular disease, thromboembolic disorders, immune disorders and malignancies.2,3 As reviewed by Giagulli et al, KS may also be associated with a variety of neuropsychiatric disorders such as autism, anxiety, attention deficit disorder, obsessive compulsive disorder and psychosis.4
Nowadays, the term KS describes a group of chromosomal disorders in which at least one extra X chromosome is present to a normal male karyotype, 46,XY. The majority of KS patients shows a 47,XXY karyotype. Less common are other numeric sex chromosome abnormalities such as 48,XXXY, 48,XXYY and 49,XXXXY.5,6 However, as summarized by Tartaglia and colleagues, these sex chromosome aneuploidy conditions have an increased risk for congenital malformations and other medical problems and a more complex psychological phenotype.7 It concerns among others the variable degree of facial dysmorphisms that are often more distinct than in the original 47,XXY Klinefelter syndrome as well as significant dental problems, congenital anomalies like congenital heart defects, hip dysplasia, and kidney dysgenesis. Concerning the neuropsychiatric conditions, males with such karyotypes are generally more affected with respect to symptoms from the autism spectrum as well as developmental and speech delays, particularly in expressive language.8 Also, other domains are reported to be more affected, especially social-emotional functioning.9
Apart from the above-mentioned numeric sex chromosome anomalies, from 1986 until present, six case reports on the 49,XXXYY chromosome anomaly have been published, ie, four male adults and two children. All were diagnosed with intellectual disability (Table 1). This chromosomal condition is extremely rare and its prevalence is estimated to be <1/1.000.000.10 Here, we describe in detail the developmental history and findings at genetic, somatic, endocrinological and neuropsychological examination of the fifth intellectually disabled adult male with this very rare pentasomy. The patient is 25 year-old. Written informed consent for the present case report was provided by both parents. Institutional approval was not required; the patient has never been institutionalized and still lives at his parents’ home.
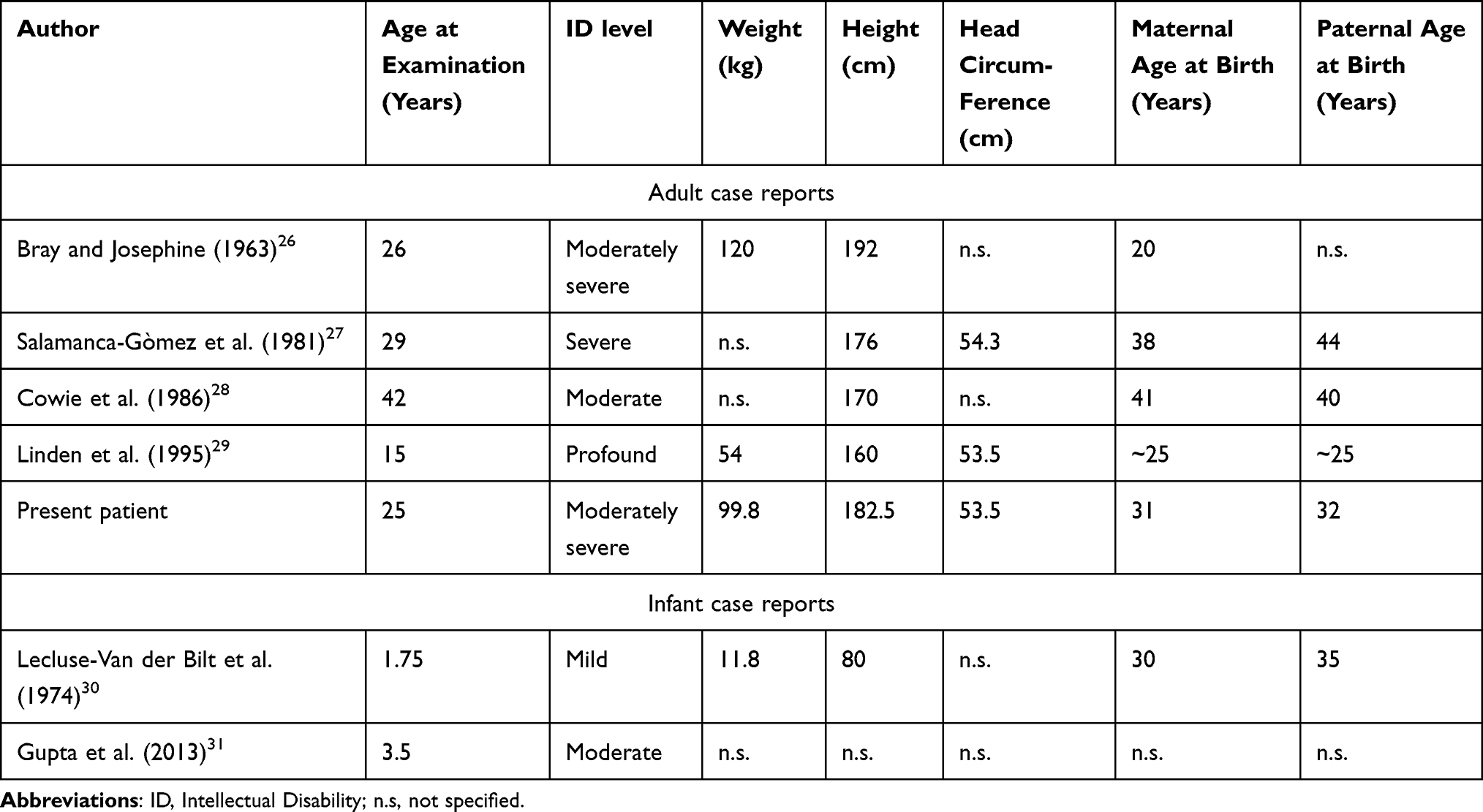 |
Table 1 Published Males with 49,XXXYY Sex Chromosome Anomaly |
Case Description and Assessments
Pregnancy and delivery were uncomplicated. His birth weight was about 3500 grams. He has one older brother and two younger sisters, all healthy and well educated. His father had completed professional higher education (teacher) and his mother intermediate vocational education (nurse). His birth weight was about 3500 grams. He has one older brother and two younger sisters, all healthy and well educated. With respect to family history, his mother uses levothyroxine for primary hypothyroidism. Neonatally, minor facial dysmorphisms, general hypotonia and excessive sleep were noticed. His psychomotor and speech/language development were delayed and his behaviour was characterized by some mild head banging and detailed interest for turning objects and toys. At the age of five years, he was referred to a child psychiatrist for evaluation of his significantly delayed development, chronic lack of energy and disinhibited behaviours. Apart from mild intellectual disability, a diagnosis of autism was made. Karyotyping showed a 49, XXXYY karyogram. He followed elementary school for a couple of days only and switched directly to a school for special education until the age of 19 years. Aged 10 years, he was referred again to a child psychiatrist because of panic attacks and aggressive outburst with self-biting, hitting and screaming at sudden contextual changes, especially during school time and when staying one day weekly in his guest house. At examination, the patient then displayed rigid thinking, nearly absence of eye contact and emotionally reciprocity, as well as high distractibility and weak impulse control especially when disturbed in his activities. A diagnosis of autism was reaffirmed and the parents were advised to maintain a structured and supportive approach and to continue logopedic training and physiotherapeutic support in order to stimulate language and motor development. In addition, a low dose of risperidone was started for behavioural control. Aged fifteen years, he attended again an outpatient department for child and adolescent psychiatry. At that time his developmental age was estimated to correspond with that of a five-year-old child. Once more a diagnosis of autism was made and his social-emotional developmental age was considered to be 18 months with a total IQ of maximally 45 with a high risk of overestimation. Because of substantial weight gain, one year later, symptomatic treatment with risperidone was replaced by 5mg aripiprazole. Aged 21 years, subclinical hypothyroidism was established and subsequently, treatment with levothyroxine was started in a daily dose of 25µgr. Despite symptomatic treatment with 5mg aripiprazole per day, his disinhibited behaviours with temper tantrums increasingly disturbed the family situation at home. Therefore, he was gradually admitted to a care farm where he was occupied during the day in a subgroup with simple work (ie, feeding animals, cleaning, sorting, carrying supplies), maintaining structured personal guidance aimed among others to increase his self-esteem. During his daytime activities, however, unexpected environmental changes frequently evoked temper tantrums with aggressive incidents and screaming which he regretted afterwards. Ultimately, however, these could no longer be corrected by the staff members and as a consequence, at the age of 23 years, he had to return to his parental home. Subsequently, he followed five days weekly elementary daytime activities at a wood and metal company (cleaning and sorting of second-hand materials) with individual guidance that, unfortunately, had to be temporarily stopped after eight months due to the COVID-19 restrictions. After restart, because of accumulation of challenging behaviours necessitating permanent individualized care, the patient was referred for expert consultation.
Results
At referral, the patient had recently restarted his daytime activities at the wood and metal company. In addition to symptomatic treatment with 5mg aripiprazole daily, 25µgr levothyroxine was still prescribed. Because of lack of symptomatic effect, aripiprazole was tapered off gradually. Relevant hematological and biochemical parameters as well as vitamin status were all normal. His hormonal panel revealed normal values for TSH, FT4, FT3, prolactin, and estradiol but increased values for FSH (16 U/l; normal range: 1–9 U/l) and LH (14 U/l; normal range: 1.7–9 U/l) and a significant lower value for testosterone (1.12 nmol/l; normal range: 5.70–26.10 nmol/l).
Etiological and Somatic Examination
Etiological analysis demonstrated again the pentasomy with karyotype 49, XXXYY (Figure 1). To exclude other genetic abnormalities, whole exome sequencing was performed which showed no pathogenic sequence variants or copy number variants associated with intellectual disability. At examination, height, weight and head circumference were 182,5cm, 99,8kg (BMI: 29,8) and 53,5cm (−2,5 SDS), respectively (Table 2). Somatic and neurological examination disclosed no abnormalities. Minor facial dysmorphisms were noticed ie, broad nasal tip, full lips, small simple shaped ears, brachycephaly, mild prognathism and slightly flat midface. In addition, he had small shoulders and marked truncal overweight. With respect to his extremities, he had relatively small hands and feet, four finger line, strikingly lateral position of the patella at his left knee and marked flexible flatfeet. His penis was short with minor scrotum and descended testes with a diameter of 3cm as well as nearly absent pubic hair-growth.
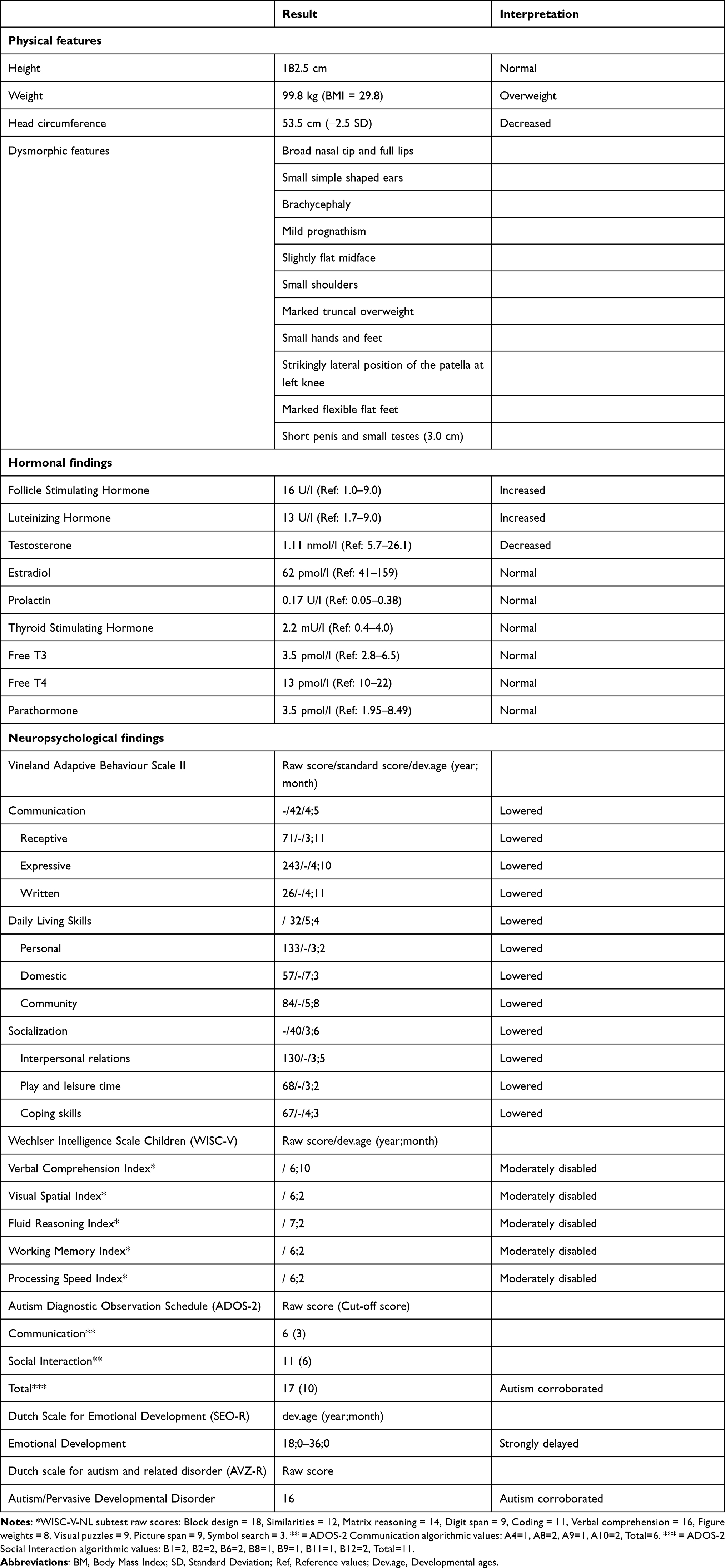 |
Table 2 Summary of Actual Findings in the Patient with 49, XXXYY Karyotype |
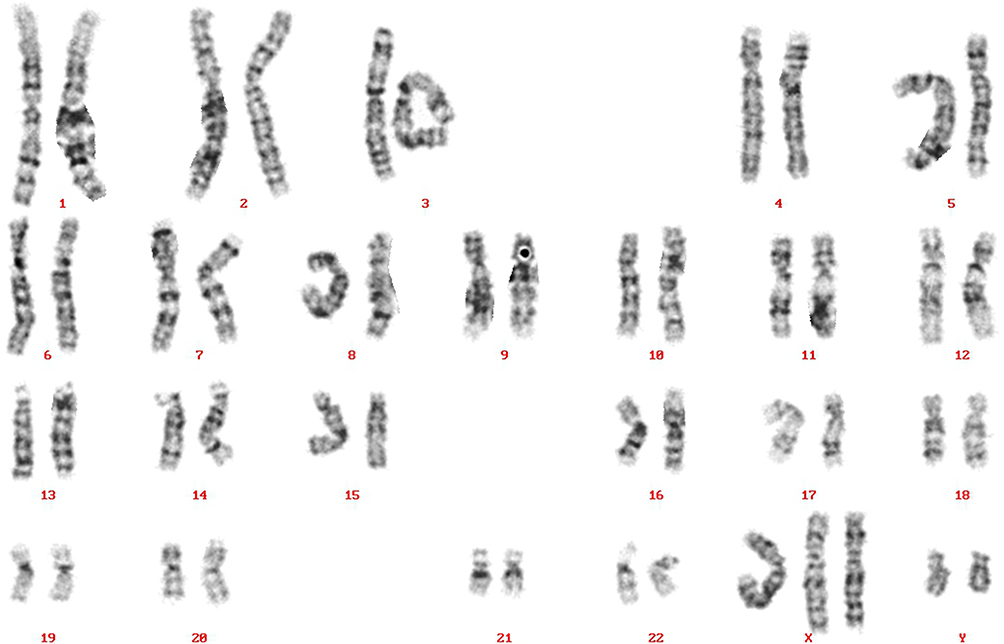 |
Figure 1 Karyogram of the patient with 49,XXXYY sex chromosome anomaly. |
Neuropsychological Examination
Behaviour was characterized by lack of energy and initiative as well as short periods with temper tantrums. There were clear autistic traits with echolalia, perseverations, repetitive and compulsive behaviours and weak impulse control. With the Wechsler Intelligence Scale for Children (WISC-V-NL11) for the indexes (a) Verbal Comprehension, (b) Visual Spatial, (c) Fluid Reasoning, and (d) Working Memory and Processing Speed, scores corresponding with an age of 6;10, 6;2, 7;2 and less than 6;2 (years;months) were found. As assessed with the Vineland Adaptive Behaviour Scale (VABS12), for the factors communication, daily performance and socialization, a mean developmental age of 4;5, 5;4 and 3;6 (years;months), respectively, was established. By using the Dutch Scale for Emotional Development in People with intellectual disability (SEO-R13), his emotional developmental age was established to be maximally 18–36 months. With the Dutch scale for autism and related disorder (AVZ-R14), covering the autism parameters social, communicative and stereotyped behaviour, a total score of 16 was established (range 0–19; scores 10–19: autism/pervasive developmental disorder). With the Autism Diagnostic Observation Schedule (ADOS-215), a total score of 17 (cut-off score = 10) corroborated a diagnosis of autism.
Based on the results of these combined assessments (see Table 2 for an overview), a diagnosis of autism was definitively confirmed as well as moderate intellectual disability. Two weeks after discontinuation of aripiprazole, the behavioural repertoire, lack of initiative and general well-being of the patient were significantly improved whereas temper tantrums occurred incidentally only. However, few weeks later, challenging behaviours intensified again upon which the physician for intellectual Disabilities decided to restart treatment with aripiprazole 5mg daily. Re-evaluation of his hormonal status revealed for LH, FSH and testosterone values of 13 U/l (1.7–9 U/L; range: 1.7–9 U/l)), 16 U/l (range: 1–9 U/L) and 1.11 nmol/l (range: 5.70–26.10 nmol/l) respectively, and normal values for prolactin as well as for TSH and FT4. Based on these hormonal results, the endocrinologist originally planned to start with transdermal testosterone gel once daily 12.5mg but instead decided to start with 5mg daily for two weeks and a more gradual dose increasing of 5mg each two weeks up to 20mg. This resulted in more stable behavioural and emotional functioning and a better sleep pattern. A further increase of testosterone dosage to the standard dose of 40–60mg daily will be contingent upon stability and course in relation to testosterone blood level.
Discussion
In the present paper we described in detail the developmental history and the findings obtained from genetic, endocrinological and neuropsychological examination in a 25-year-old intellectually disabled adult male with a 49,XXXYY chromosome anomaly. In addition to two young children, this is until now the fifth adult male individual worldwide in whom such a pentasomy is demonstrated (Table 1). As far as can be reconstructed from earlier documents, surprisingly, measurement of hormonal parameters, except TSH, FT4 and prolactin, has previously never been done.
As can be inferred from Table 2, somatic examination disclosed minor dysmorphic features, especially regarding head circumference. His hormonal panel was characterized by a significantly decreased level of testosterone and enhanced values for LH and FSH corresponding with a diagnosis of primary hypogonadism. Extensive neuropsychological evaluation of the patient with objective testing confirmed a diagnosis of autism. His behavioural profile differed not essentially from that described in patients with other numeric sex chromosomal disorders albeit that in the presented case, autism, level of intellectual disability, and contextually determined disinhibited behaviours alternated by apathy seemed to be more pronounced. As to treatment, since some atypical antipsychotics (risperidone and paliperidone, but as far as known not aripiprazole) are identified to have a decreasing effect on the testosterone level due to elevated prolactin levels,16,17 the endocrinologist decided to start suppletion with testosterone after aripiprazole was fully discontinued for at least two weeks in order to adjust the correct dosage.
The facial dysmorphisms and the severity of cognitive and psychiatric dysfunctions in the present patient are in accordance with results from other studies that found supernumerary sex chromosomes to be associated with further reduction of skull surface area,18–20 higher frequency of brain structure abnormalities,21 and increased vulnerability toward psychiatric and psychosocial disturbances.7,22 In general, individuals with sex chromosome aneuploidies have a higher risk to develop metabolic and cardiovascular disorders.23 Since the scientific information on syndromes with multiple additional sex chromosomes is rather limited, for those disorders it seems best to follow the recently published guidelines on Klinefelter syndrome of the European Academy of Andrology in collaboration with the European Society of Endocrinology.24,25
Conclusion
This report is an addition to the existing limited data on the very rare 49,XXXYY pentasomy. It demonstrates that children born with any abnormality of the external genitalia have to be always evaluated for chromosomal abnormalities, results of which can also guide the timely detection of intellectual and cognitive disabilities, somatic anomalies, and androgen deficiency. This approach will help avoiding underdiagnosis of delayed development of language and speech as well as of autistic disorder which are all crucial for the prompt deployment of specific pedagogical measures and medical interventions.
Acknowledgments
The authors are indebted to the parents of the patient for their kind cooperation and their written informed consent for publication of the developmental history of their son. We thank Mr. Leo van Mil, coordinator Centre for Consultation and Expertise region West, who referred the patient for expert consultation. Psychological tests were performed by Mrs. Aleika Jansen and Mrs. Tara Bonekamp from the Regional Team Diagnostics and Treatment of ASVZ in Sliedrecht.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Klinefelter HF, Reifenstein EC, Albright F. Syndrome characterized by gynecomastia, aspermatogenesis without A-Leydigism and increased excretion of follicle stimulating hormone. J Clin Endocrinol Metab. 1942;2:615–627.
2. Shiraishi K, Matsuyama H. Klinefelter syndrome: from pediatrics to geriatrics. Reprod Med Biol. 2019;18:140–150.
3. Spaziani M, Radicioni AF. Metabolic and cardiovascular risk factors in Klinefelter syndrome. Am J Med Genet C Semin Med Genet. 2020;184:334–343.
4. Giagulli VA, Campone B, Castellana M, et al. Neuropsychiatric aspects in men with Klinefelter syndrome. Endocr Metab Immune Disord Drug Targets. 2019;19:109–115.
5. Visootsak J, Graham JM. Klinefelter and other sex chromosomal aneuploidies. Orphanet J Rare Dis. 2006;1:42.
6. Frühmesser A, Kotzot D. Chromosomal variants in Klinefelter syndrome. Sex Dev. 2011;5:109–123.
7. Tartaglia N, Ayari N, Howell S, et al. 48,XXYY, 48XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome. Acta Paediatr. 2011;100:851–860.
8. Tartaglia NR, Wilson R, Miller JS, et al. Autism spectrum disorder in males with sex chromosome aneuploidy: XXY/Klinefelter syndrome, XYY and XXYY. J Dev Behav Pediatr. 2017;38:197–207.
9. Visootsak J, Graham JM. Social function in multiple X and Y chromosome disorders: XXY, XYY, XXYY, XXXY. Dev Disabil Res Rev. 2009;15:328–332.
10. Singh J. The portal for rare diseases and orphan drugs. J Pharmacol Pharmacother. 2013;4:168–169.
11. Hendriks MPH, Ruiter S, Schittekatte M, et al. WISC-V-NL, Wechsler Intelligence Scale for Children-V. Amsterdam: Pearson; 2018.
12. De Bildt A, Kraijer D. Dutch Adaptation of the Vineland Adaptive Behavior Scale of Sparrow, Balla, and Cicchetti. Leiden: PITS; 2003.
13. Claes L, Verduyn A. Scale for Emotional Development in People with Intellectual Disability [SEO-R: Schaal voor Emotionele Ontwikkeling bij Mensen Met Een Verstandelijke Beperking – Revised]. Antwerpen-Apeldoorn: Garant; 2012.
14. Kraijer D. AVZ-R Manual.
15. De Bildt A, De Jonge M, Greaves-Lord K. ADOS-2 Autism Diagnostic Observation Schedule (Dutch Version). Amsterdam: Hogrefe; 2013.
16. Konarzewska B, Galińska-Skok B, Waszkiewicz N, et al. Association between serum testosterone levels, body mass index (BMI) and insulin in male patients with schizophrenia treated with atypical antipsychotics – olanzapine or risperidone. Neuroendocrinol Lett. 2014;35:50–57.
17. Tasaki M, Ysui-Furukori N, Yokoyana S, et al. Hypoprolactinemia and hyperprolactinemia in male schizophrenia patients treated with aripiprazole and risperidone and their relationships with testosterone levels. Neuropsychopharmacol Rep. 2021;41:379–384.
18. Chang S, Skakkebaek A, Trolle C, et al. Anthropometry in Klinefelter syndrome – multifactorial influences due to CAG length, testosterone treatment and possibly intrauterine hypogonadism. J Clin Endocrinol Metab. 2015;10:E508–E517.
19. Raznahan A, Raitano Lee N, Greenstein D, et al. Globally different but locally convergent X- and Y-chromosome influences on cortical development. Cerebral Cortex. 2016;26:70–79.
20. Skakkebaek A, Gravholt CH, Chang S, et al. Psychological functioning, brain morphology, and functional neuroimaging in Klinefelter syndrome. Am J Med Genet. 2020;184C:506–517.
21. Reardon PK, Clasen L, Giedd JN, et al. An allometric analysis of sex and sex chromosome dosage effects on subcortical anatomy in humans. J Neurosci. 2016;36:2438–2448.
22. Blumling AA, Martyn K, Talboy A, et al. Rare sex chromosome variation 48,XXYY: an integrative review. Am J Med Genet. 2020;184C:386–403.
23. Spaziani M, Radicioni AF. Metabolic and cardiovascular risk factors in Klinefelter syndrome. Am J Med Genet. 2020;184C:334–343.
24. Gravholt CH, Tartaglia N, Diateche C. Sex chromosome aneuploidies in 2020 – the state of care and research in the world. Am J Med Genet. 2020;184C:197–201.
25. Zitzmann M, Askglaede L, Corona G, et al. European Academy of Andrology guidelines on Klinefelter syndrome. Andrology. 2021;9:145–167.
26. Bray P, Josephine A. An XXXYY sex-chromosome anomaly. JAMA. 1963;184:113–116.
27. Salamanca-Gòmez F, Cortès R, Sànchez K, et al. Brief clinical report: a 49,XXXYY male. Am J Med Genet. 1981;10:351–355.
28. Cowie VA, Rameshwari K, Wheater K. 49,XXXYY Chromosome anomaly in a mentally retarded man. Br J Psychiatry. 1986;148:210–212.
29. Linden MG, Bender B, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics. 1995;96:672–682.
30. Lecluse-van der Bilt FA, Hagemeijer A, Smit EME, et al. An infant with an XXXYY karyotype. Clin Genet. 1974;5:263–270.
31. Gupta A, Kumar P, Gupta S, et al. Multiple XY syndrome: a case study. Int J Res Appl Nat Social Sci. 2013;3:87–90.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.