Back to Journals » International Journal of Nanomedicine » Volume 11
A novel type of self-assembled nanoparticles as targeted gene carriers: an application for plasmid DNA and antimicroRNA oligonucleotide delivery
Authors Zhu Y, Liang G ![]() , Sun B, Tian T, Hu F, Xiao Z
, Sun B, Tian T, Hu F, Xiao Z
Received 18 March 2015
Accepted for publication 11 August 2015
Published 27 January 2016 Volume 2016:11 Pages 399—411
DOI https://doi.org/10.2147/IJN.S84927
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Yanliang Zhu,1 Gaofeng Liang,2 Bo Sun,1 Tian Tian,3 Feihu Hu,1 Zhongdang Xiao1
1State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, 2School of Medical Technology and Engineering, Henan University of Science and Technology, Luoyang, 3Department of Neurobiology, Nanjing Medical University, Nanjing, People’s Republic of China
Abstract: In this study, a new type of amphiphilic cetylated polyethyleneimine (PEI) was synthesized, and then polylactic-co-glycolic acid (PLGA)/cetylated PEI/hyaluronic acid nanoparticles (PCPH NPs) were developed by self-assembly as a novel type of gene-delivering vehicle. The PCPH NPs showed good DNA-condensation ability by forming polyplexes with small particle size and positive zeta potential. The transfection efficiency and cytotoxicity of PCPH NPs were evaluated as plasmid DNA vectors to transfect HepG2 in vitro. PCPH NPs exhibited much lower cytotoxicity and higher gene-transfection efficiency than PEI (25,000) and commercial transfection reagents. Furthermore, PCPH NPs were used as an anti-miR-221 vector for transfecting HepG2 cells, and anti-miR-221 was effectively transfected into cells and produced a greater inhibitory effect on cancer-cell growth by PCPH NPs. These results demonstrate that PCPH NPs can be a promising nonviral vector for gene-delivery systems.
Keywords: gene delivery, hyaluronic acid, polyethyleneimine, anti-miR-221, PLGA
Introduction
Nucleic acid-based therapeutics could play a significant role in the treatment of a variety of diseases in the foreseeable future.1 This is because gene therapies are aimed at treating or eliminating the cause of disease, whereas most current drugs treat only the symptoms.2 However, a major challenge of gene therapy is developing a delivery system that can efficiently deliver DNA/RNA molecules to the target cells, with minimal toxicity and high bioavailability during the delivery process.3 Much evidence has suggested that small RNA molecules can fulfill relatively generic functions in cells,4 especially with research being conducted on noncoding small RNA, and are involved in many pathological and physiological processes.5,6 Therefore, small interfering (siRNA) and microRNA (miRNA) are being used more and more as a new category of agents.7 The biggest disadvantage, however, is that these small noncoding RNAs, which act as genetic medicine in vivo, are all unstable, so the search for an ideal gene-delivery system is becoming more important.
At present, commonly used gene vectors fall into two broad categories: viral vectors and nonviral vectors. Viral vectors are utilized for gene delivery with high transfection efficiency, but the potential infectivity and immunogenicity of those viral vectors has been a major shortcoming for their effective utilization.8 In contrast, nonviral delivery vectors have emerged as an alternative strategy for gene delivery in the context of avoiding the defects of the viral vectors.9 Through these vectors, genes can either be conjugated to carrier molecules, such as lipids, peptides, and polymers, or encapsulated into a cationic delivery system. The most conspicuous feature of nonviral gene-delivery systems is multivariate formulations, such as hydrogels, micelles, and nanoparticles (NPs).10 Among various formulations, NP gene-delivery systems have gradually gained attention and been extensively researched because of some excellent properties. In particular, for a NP gene-delivery system with a hydrophobic core–hydrophilic shell structure, the hydrophilic shells can actually prevent the NPs from aggregating and being trapped in the reticuloendothelial system. This would thereby ensure a longer circulation time in the blood.11
Polylactic-co-glycolic acid (PLGA) NPs were initially used to encapsulate genetic medicine after it was demonstrated they had the endolysosomal escape property, while being biodegradable and cell-compatible.12,13 However, the structure of nucleic acid molecules is likely to be damaged during the preparation of spheres. In addition, the negative zeta potential of PLGA NPs hampers the cellular uptake of NPs, making the transfection efficacy very low. To overcome this shortcoming, one effective approach is to use some cationic coating materials to modify the PLGA NP surface. Studies have shown that this method can be used effectively to enhance the loading efficiency and cellular uptake.14,15 In this study, in line with the objective of promoting PLGA NPs as gene carriers, we synthesized cetylated polyethyleneimine (PEI-cet; a novel amphiphilic polycation) and then introduced PEI-cetyl on the surface of PLGA NPs through hydrophobic and electrostatic interactions. This was done taking into consideration the nucleic acid-binding capacity, cellular uptake efficiency, and endosomal escape ability of the NPs.
Hyaluronic acid (HA) is a negatively charged biopolymer composed of alternating disaccharide units of d-glucuronic acid and N-acetyl-d-glucosamine with β(1–4)-glycosidic linkage. Research on the biological functions of HA has demonstrated that it takes part in a broad range of biological activities.14,15 Moreover, HA-binding CD44 receptors are also known to be involved in tumor metastasis, and are overexpressed on the surface of a variety of tumor cells. Therefore, HA has been studied as a targeting moiety for drug- and gene-delivery systems.16,17 In our study, in order to reduce the cytotoxicity of NPs and enhance the effectiveness of tumor-cell targeting, HA was introduced to the surface of PLGA/PEI-cet/gene nanocomposites through electrostatic self-assembly (Figure 1). The placement of HA on the outer layer of NPs has three main benefits: firstly, it can facilitate the intracellular uptake by cancer cells through HA receptor-mediated endocytosis; secondly, the hydrophilic HA might prevent the nanocomplex from interacting with blood serum components, thereby contributing to serum stability; thirdly, HA constitutes the outermost layer of the sphere, preventing PEI from interacting with the cell membrane, so HA can decrease the cytotoxicity of NPs.18
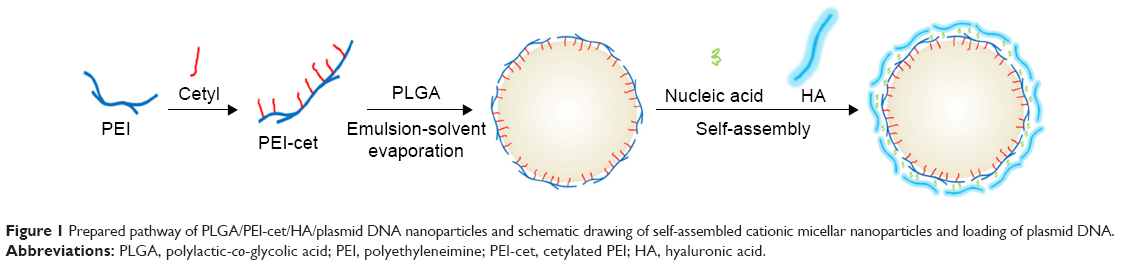 | Figure 1 Prepared pathway of PLGA/PEI-cet/HA/plasmid DNA nanoparticles and schematic drawing of self-assembled cationic micellar nanoparticles and loading of plasmid DNA. |
miRNAs are short (19–25 nucleotides) noncoding RNA molecules, which negatively modulate protein expression at the posttranscriptional level.19 miRNAs play a pivotal role in almost all aspects of biology, such as disease and biological development,20,21 and tumorigenesis is related to abnormal expression of some specific miRNAs.22 The aim of this work was to design a delivery carrier that could be applied to a broad range of gene-delivery applications, such as plasmid DNA (pDNA) or oligonucleotides. To assess this ability, we not only treated the NPs as pDNA carriers but also analyzed them as anti-miRNA oligonucleotide (AMO) carriers. As expected, the results showed that the prepared nano-gene carrier can be efficiently applied to various gene-delivery applications.
Materials and methods
Materials
D,L-Lactide/glycolide copolymer (PLGA, lactic:glycolic molar ratio 53:47, molecular weight [MW] 25 kDa) was purchased from Daigang Chemical Reagent Co Ltd (Jinan, People’s Republic of China [PRC]), PEI (branched, 25 kDa), and polyvinyl alcohol were purchased from Sigma-Aldrich Co (St Louis, MO, USA). HA (MW 10 kDa) was purchased from Lifecore Biomedical LLC (Chaska, MN, USA). Dulbecco’s Modified Eagle’s Medium, fetal bovine serum (FBS), penicillin–streptomycin, trypsin, and Dulbecco’s phosphate-buffered saline (PBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Plasmids (pEGFP-C1, 4.7 kb) containing a cytomegalovirus promoter and an EGFP reporter were obtained from BD Biosciences (San Jose, CA, USA). The human hepatoma cell line HepG2 was purchased from American Type Culture Collection (Manassas, VA, USA). The study was approved by the institutional review board (CWO) of College of Basic Medicine Science, southeast University, PR China. No ethics committee permission was required for the use of human hepatoma cell cancer cell line HepG2 because only certificated cell lines provided by ATCC were used. Hoechst 33342 was purchased from Thermo Fisher Scientific. The sequences of anti-miR-221 and fluorescein amidite (FAM)-labeled anti-miRNA were chemically synthesized by GenePharma Co Ltd (Shanghai, PRC) as follows: 5′-GAAACCCAGCAGACAAUGUAGCU-3′. The nontargeting sequence was 5′-CAGCUGAAGUAAAUACCGACCAG-3′. The siRNA used for silencing the CD44 gene (NCBI Ref Seq NM_000610.3) was purchased from GenePharma. The sequences of sense and antisense for CD44 siRNA were sense 5′-UAUUCCACGUGGAGAAAAAtt-3′ and antisense 5′-UUUUUCUCCACGUGGAAUAca-3′. Other reagents were of analytical grade were obtained from suppliers and used without purification.
Synthesis and characterization of PEI-cet
PEI-cet was synthesized via cetyl bromide alkylation of PEI (25 kDa), as described previously.21 Cetyl bromide (2.1 g, 7.0 mmol) was added to chloroform solution containing PEI (3 g, 70 mmol ethylenimine units), and the solution was refluxed for 16 hours at 65°C in the presence of triethylamine (0.729 g, 7.2 mmol). After the reaction, chloroform was evaporated under vacuum, and then the mixture was dissolved in ethanol, dialyzed against water using 6,000 MW-cutoff membrane to remove low-MW impurities, and freeze-dried. The obtained PEI-cet copolymer was characterized by 1H-nuclear magnetic resonance (NMR).
Surface modification of PLGA nanoparticles with PEI-cet
The preparation of PLGA/PEI-cet (PCP) NPs was carried out according to the double-emulsion solvent-evaporation technique evolved by Kumar et al.23 PLGA was dissolved in methylene chloride by overnight stirring at a concentration of 10% (w/v); it was then filtered by a 220 nm filter. After that, 1 mL of methylene chloride containing 12 mg PEI-cet was added to 1 mL of PLGA solution, and the mixed organic phase was poured into an aqueous phase of 20 mL of 0.5% (w/v) polyvinyl alcohol and stirred at 2,000 rpm to give an oil-in-water emulsion. This resulted in the formation of a water/oil/water emulsion that was stirred for at least 12 hours at room temperature, allowing the methylene chloride to evaporate. The resulting microspheres were washed twice in deionized water by centrifugation at 16,000 g and freeze-dried.
Preparation of PCP/DNA/HA complexes
The PCP/pDNA/HA charge ratio (nitrogen:phosphate [N:P]) was expressed as the mole ratio of the amine groups of PEI-cetyl to the phosphate of pDNA. The complexes were induced to self-assemble in 150 mM PBS buffer (pH 7.4) by mixing the DNA plasmid (0.1 mg/mL) with the NP solution (0.1 mg/mL) at certain charge ratios, keeping the amount of pDNA constant. The complexes were incubated for 10 minutes at room temperature. Then HA, a third the weight of PEI-cetyl, was added to the solution for attachment to the NP surface. The final suspension was incubated while being shaken for 30 minutes at room temperature.
Nanoparticle characterization
Hydrodynamic diameters and size distribution
Mean hydrodynamic diameters of PLGA NPs, PCP NPs, and PCP/pDNA/HA (PCPH) NPs were measured using an NP analyzer (Beckman Coulter Inc, Pasadena, CA, USA). The mean hydrodynamic diameter was determined via cumulative analysis.
Zeta potential
The zeta potential (surface charge) of each NP sample formed at various N:P ratios was determined at 25°C with a scattering angle of 90° using a potential measurement analyzer (90PLus; Brookhaven Instruments Corporation, Holtsville, NY, USA). Samples were prepared in PBS and diluted with deionized water to ensure that the measurements were performed under conditions of low ionic strength, where the surface charge of the particles can be measured accurately.
Surface morphology
The particle size and morphology of each sample were characterized via transmission electron microscopy (JEM-2100; JEOL, Tokyo, Japan).
Measurement of interactions between nucleic acid and nanoparticles
NPs were mixed with pDNA at various ratios. Varying amounts of NPs were added to 1 μg pDNA, then the resulting mixtures of NPs/pDNA with different N:P weight ratios were loaded onto a 1% (w/v) agarose gel containing 0.2 mg/mL ethidium bromide and electrophoresed at 90 V in TAE for 50 minutes. Images were acquired using a PeiQing gel imaging system (PeiQing, Shanghai, PRC). GelRed (Biotium, Hayward, CA, USA), an ultrasensitive nucleic acid dye, was used to examine the interactions of DNA with the nanocomplex to determine the optimal N:P ratio of the nanocomplex.
Cell culture
The HepG2 cells were grown in Dulbecco’s Modified Eagle’s Medium containing 10% heat-inactivated FBS (Biological Industries, Kibbutz Beit Haemek, Israel) at 37°C in a humidified atmosphere of 5% CO2.
In vitro cell viability
Cell viability was evaluated by MTT assay. HepG2 cells were seeded into a 96-well plate with 104 cells/well and incubated for 24 hours to allow cell attachment. Then, the cells were incubated with PCPH/pDNA, PCP/pDNA or PEI/DNA at different concentrations of nanocomplexes for 24 hours at 37°C and 5% CO2. Cells without incubation with test nanocomplexes were used as negative controls. Cells were then washed with PBS and reinsulated in 200 mL of medium containing FBS for 2 days. At the end of the transfection step, 20 μL of 2 mg/mL MTT solution in PBS was added to the plate and incubated at 37°C for an additional 4 hours. Then, the medium containing MTT was removed, and 300 μL of dimethyl sulfoxide was added to dissolve the formazan crystal formed by live cells. The optical density was measured at 540 nm with an ultraviolet spectrophotometer. Cell viability (%) was calculated using the following equation:
Cell viability (%) = (OD540 [sample]/OD540 [control]) ×100 |
where OD540 (sample) represents the optical density from the wells treated with PEI/pDNA, PCP/pDNA, or PCPH/pDNA complex, and OD540 (control) represents that from the wells treated with PBS.
Small interfering RNA for inhibition of CD44 expression
CD44 siRNA and Silencer Negative Control were transfected into HepG2 cells using Effectene transfection reagent (Qiagen NV, Venlo, the Netherlands) following the manufacturer’s protocol. The siRNA concentration for silencing CD44 expression was 40 nM for each cell line. Total cellular proteins were extracted from various cell groups at 48 hours posttransfection.
In vitro cellular uptake test
The in vitro transfection efficiency of PCPH NPs with pEGFP was compared with PEI, PCP, and the commercial reagents. The study was carried out under optimized conditions of N:P ratios in all forms of gene carriers. HepG2 cells were plated in 96-well microplates at 104 cells and allowed to adhere for 12 hours, then were washed with PBS and incubated in 200 mL of medium without FBS. Different pDNA solutions or DNA-loaded NP suspensions corresponding to 2 μg pDNA were incubated with cells for 12 hours. Cells were then washed and reincubated in 200 mL of medium containing FBS for 2 days. Expression of EGFP within cells was assessed after 48 hours by fluorescence microscopy. Lipofectamine 2000 was used as positive control and applied according to the manufacturer’s procedures. The uptake test of the CD44-downregulation group was processed as a normal cell group after 24 hours’ transfection of CD44 siRNA.
In vitro silencing of miR-221 by PCPH/anti-miR-221 nanocomplexes
Confocal imaging
HepG2 cells were seeded onto coverslips in a 24-well tissue-culture plate and then transfected with various NPs loaded with 20 nmol/L of FAM-anti-miRNA. Cells were washed three times in PBS, counterstained with Hoechst 33342, and imaged using an inverted fluorescence microscope (Nikon Corporation, Tokyo, Japan).
Quantification of miR-221 levels
HepG2 cells were transfected with anti-miR-221 or mismatched AMOs at final concentrations of 5 nM, 20 nM, 50 nM, and 100 nM using PCPH NPs while using free anti-miR-221 as control. We harvested RNA from each sample after 48 hours’ transfection, and then performed quantitative real-time polymerase chain reaction (qRT-PCR) analysis of miR-221 in each sample. Total RNA was obtained using RNAiso Plus (TaKaRa Bio, Tokyo, Japan) according to the manufacturer’s protocol. The approach of the experiment followed standard protocols on a 7500 Sequence Detection System (Thermo Fisher Scientific). Briefly, 1.25 μL of complementary DNA was added to 10 μL of the 2× SYBR green PCR master mix (TaKaRa), 200 nM of each primer, and water to 20 μL. The reactions were amplified for 15 seconds at 95°C and 1 minute at 60°C for 40 cycles. The thermal denaturation protocol was run to determine the number of the products that were present in the reaction at the end of the PCR. Reactions were typically run in triplicate. The cycle number at which the reaction crossed an arbitrarily placed threshold (Ct) was determined for each gene and the relative amount of each miRNA. Target-gene expression was normalized to the expression of the housekeeping gene U6 for each sample. Data were analyzed using the 2−ΔΔCT method.24
Western blot analysis
Cells were lysed in buffer containing 50 mM Tris HCl, pH 7.6, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% Nonidet P-40, and 0.5% sodium deoxycholate. Protein-concentration ratios were determined using a BCA Protein Assay Reagent Kit (Beijing CoWin Bioscience Co Ltd, Beijing, PRC), and aliquots of 15 μg protein were subjected to gel electrophoresis on 7.5% or 10% SDS polyacrylamide gel-electrophoresis gels and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 5% nonfat milk in PBS for 1 hour and then incubated overnight with primary antibody at 4°C. After the membrane had been washed with 0.1% Tween 20 in PBS five times, it was further incubated with horseradish peroxidase-conjugated secondary antibody for 1 hour. The membrane was washed and developed with enhanced chemiluminescence using ECL Plus (Beijing CoWin Bioscience) followed by autoradiography.
Apoptosis and cell-cycle study
Cell-cycle profiles of HepG2 cells transfected with different formations were performed using a FACSCalibur along with CellQuest software (BD Biosciences). HepG2 cells (5×104 per well) were seeded into 24-well plates. Cells were treated with different formulations at a concentration of 1 μg miRNA in serum containing medium at 37°C for 72 hours. Cells were washed once with PBS and then fixed in 75% ethanol, and after fixation were stained with propidium iodide according to the manufacturer’s instructions.
Results and discussion
Synthesis and characterization of PEI-cet copolymer
We succeeded in synthesizing amphiphilic polymer PEI-cet by alkylation of PEI. The structure of the synthesized compound was confirmed using 1H-NMR (Figure 2). The signals from PEI ethylene protons (–CH2CH2NH–) appeared at 2.4–3.0 ppm. The peak at 0.881 ppm corresponds to –CH3 from the cetyl substituents, and the multiplet between 1.15–1.56 ppm corresponds to the remaining protons from the cetyl substituents, except for the protons at the carbon atom linked to the amine group.
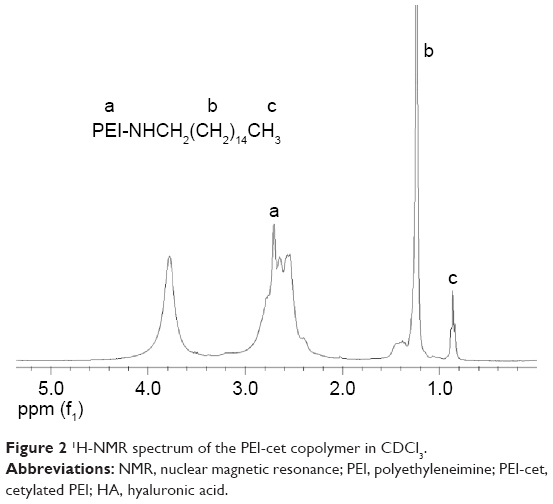 | Figure 2 1H-NMR spectrum of the PEI-cet copolymer in CDCl3. |
Characterization of NPs
Two of the most important parameters of gene-carrier NPs are particle size and zeta potential. They have been demonstrated to play important roles in determining cellular and tissue-uptake efficiency and toxic effect on cells.
The zeta potential and size of NPs not only determine their colloidal stability but also influence the effectiveness of their interaction with negatively charged cell membranes, which is the pivotal step for successful cellular uptake.25 Therefore, the particle size, polydispersity index, and zeta potential of NPs prepared at varying N:P molar ratios were investigated by dynamic light scattering (Table 1). It can be seen that the unmodified PLGA NPs had a slightly negative surface charge (−3.16 mv), due to the existence of carboxyl. Interestingly, the zeta potential of PCP NPs increased to 35.4 mv, indicating that PEI-cet, which has amino and imino groups, was located on the surface of NPs by electrostatic force and hydrophobic force. In addition, with increasing pDNA amounts, the positively charged zeta potential of PCPH/pDNA nanocomplexes declined, due to the presence of negatively charged pDNA located on the NP surface. However, with effective control of the proportion of various components, PCPH/pDNA nanocomplexes still had positive zeta potential (21.3 mv), which is necessary to ensure the uptake of complexes by cells due to electrostatic interactions between negatively charged cellular membranes and positively charged complexes.
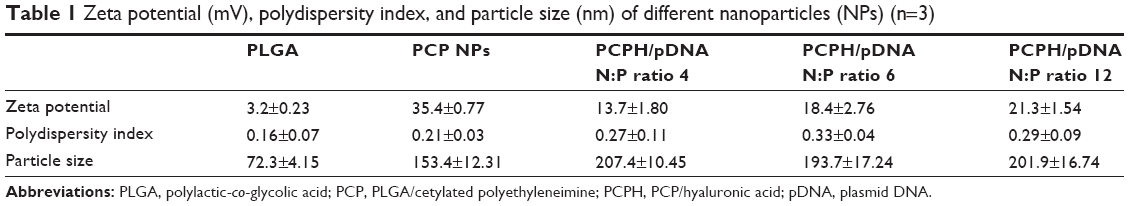 | Table 1 Zeta potential (mV), polydispersity index, and particle size (nm) of different nanoparticles (NPs) (n=3) |
Furthermore, PLGA NPs, PCP NPs (6 mg PEI-cetyl/50 mg PLGA), and PCPH/pDNA (N:P ratio of 12) nanocomplexes were visualized using transmission electron microscopy. As shown in Figure 2A, the pure PLGA NPs had a smooth, spherical morphology with a diameter of 70 nm. Compared to pure PLGA NPs, the introduction of PEI-cetyl increased the size of PCP NPs slightly, but did not alter the shape; moreover, the core–shell structure can be observed (Figure 3B). The PCPH/pDNA NPs also showed sphericity, and mean size was about 150 nm, suggesting that DNA and HA did not change the structural properties of microspheres.
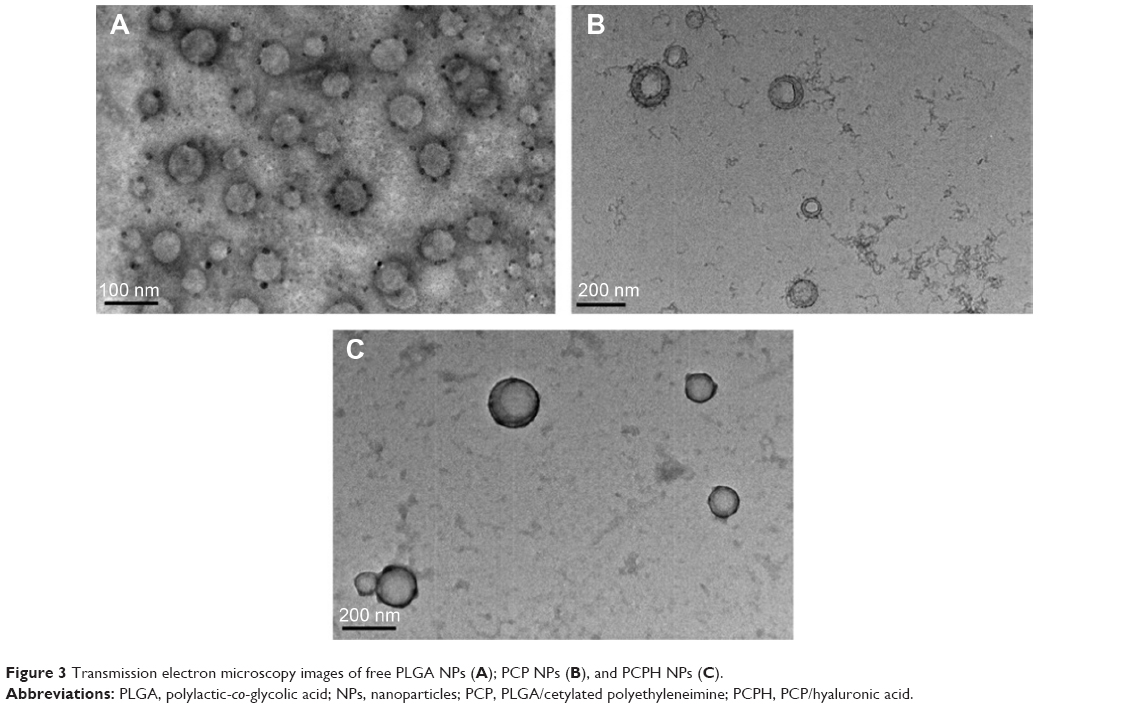 | Figure 3 Transmission electron microscopy images of free PLGA NPs (A); PCP NPs (B), and PCPH NPs (C). |
Loading efficiency of PCPH NPs
The formation of the NP/pDNA complex was assessed by agarose gel electrophoresis. As Figure 3A shows, with N:P ratio increasing DNA gel retardation became more apparent, and DNA was fully retarded when the N:P ratio was 6:1 or higher, indicating PCPH/DNA was formed through an integrated self-assembly. Moreover, protecting genes from agent degradation, such as nucleases and destructive enzymes, within the endolysosome is critical in gene delivery. An electrophoretic band was always observed in PCP/pDNA with all N:P ratios (Figure 4B). Meanwhile, with the increase of N:P ratio, the electrophoretic band of PCPH/pDNA become more and more weak, and the band was mostly invisible when PCPH/pDNA had an N:P ratio of 10:1. This confirmed HA successfully attached to the surface by electrostatic forces when the N:P ratio was at or above 10:1. In addition, the attached HA effectively protected pDNA from staining by ethidium bromide. Therefore, the result suggests that HA may offer an effective protection to encapsulated DNA from nuclease attack.
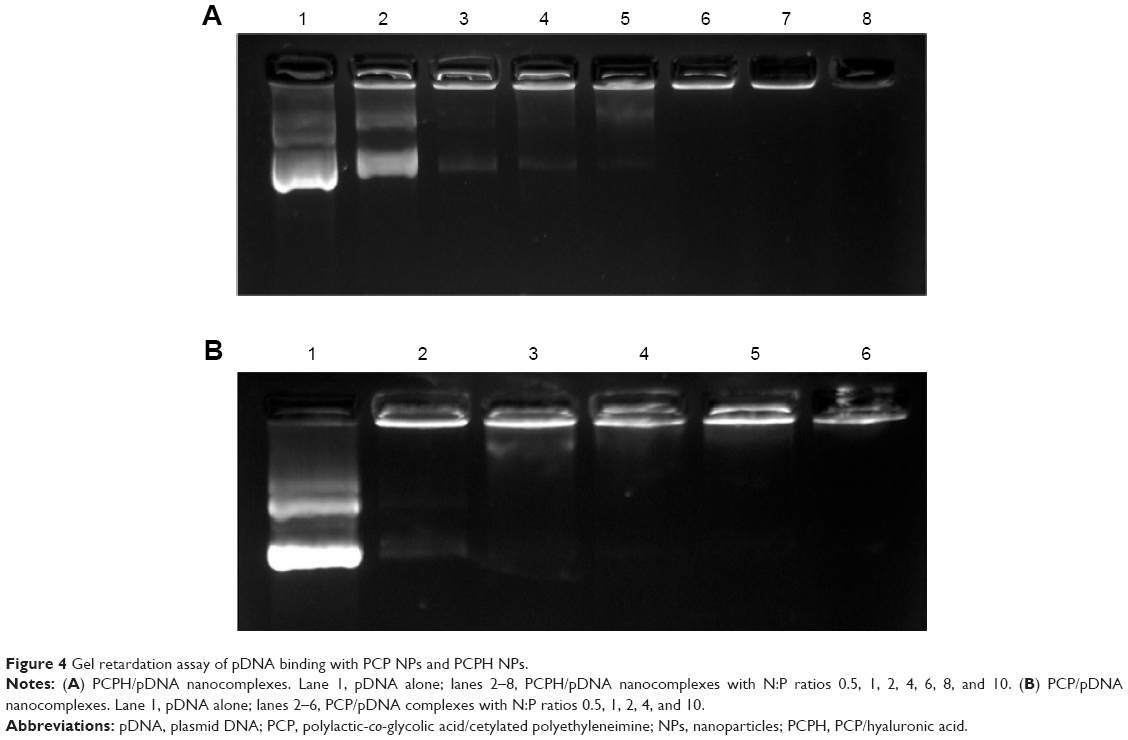 | Figure 4 Gel retardation assay of pDNA binding with PCP NPs and PCPH NPs. |
Cytotoxicity of PCPH NPs
MTT assay was used to investigate the cytotoxicity of the NPs on HepG2 cells. HepG2 cells were incubated with PEI/pDNA, PCP/pDNA, or PCPH/pDNA. As expected, Figure 5 shows a dose-dependent cytotoxicity of NPs on HepG2 cells, and this is in agreement with previous research on other cationic polymers.26 No significant cytotoxicity was found among the NPs at low concentrations. Nevertheless, as the concentration of NPs increased, the cytotoxicity of different NPs increased in various degrees: the cytotoxicity of PEI/pDNA increased the fastest. This confirmed that high-MW PEI mostly destroyed membranous structures of cells and organelles, inducing lysosomal breakdown and mitochondrial damage, thereby affecting cell viability.18 Under the same nitrogen concentration in culture media, the PCPH/pDNA nanocomplexes exhibited markedly lower cytotoxicity than PEI/DNA and PCP/pDNA, especially when the medium had higher nitrogen. This result demonstrates that the incorporation of HA into the NPs reduced their toxicity profile, and HA coating on the NP surface may provide shielding between PEI and cell membranes, effectively suppressing the toxicity of PEI as well. Earlier work has found that PEI direct contact with cell membranes can cause toxicity, as early necrotic-like changes resulting from compromised membrane integrity, assessed by considerable lactate dehydrogenase release and phosphatidylserine translocation from the inner plasma membrane to the outer cell surface, HA constituted the outermost layer of the atmosphere prevent PEI from interacting with cell membrane, so HA can decrease the cytotoxicity of NPs.18
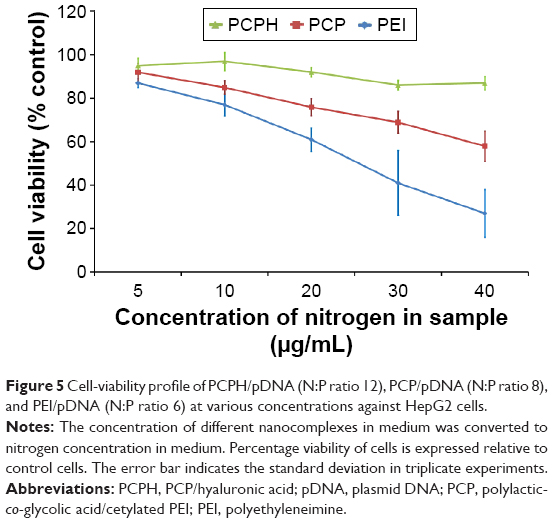 | Figure 5 Cell-viability profile of PCPH/pDNA (N:P ratio 12), PCP/pDNA (N:P ratio 8), and PEI/pDNA (N:P ratio 6) at various concentrations against HepG2 cells. |
In vitro transfection efficiency
To assess the transfection efficiency mediated by PCPH NPs, we performed in vitro transfection-activity assays using CD44-positive HepG2 cells and CD44-negative HEK293 cells. To obtain the best transfection result of PEI/pDNA, PCP/pDNA, or PCPH/pDNA, HepG2 and HEK293 cells were transfected by these three delivery vectors with the same amount of pDNA (2 μg) at different N:P ratios (range 1–15). The transfection efficiency of various NP/DNA complexes was visualized by fluorescence microscopy and quantified by flow cytometry. The highest transfection efficiency of PEI/DNA, PCP/pDNA, and PCPH/pDNA was obtained at N:P ratios of 6, 8, and 12, respectively (Figures 6 and 7). When the N:P ratio level was below optimum, NPs displayed low levels of transfection efficiency, suggesting that the zeta potential with low N:P ratio of nanocomplexes was too low to combine with negatively charged cell surfaces tightly, resulting in lower EGFP expression. Interestingly, transfection efficiency decreased with further increasing N:P ratio after having reached optimum N:P ratio. The possible reasons for this might be the cytotoxic effect. Interestingly, as shown in Figure 7, in their respective optimum conditions, transfection efficiency with PCP/pDNA complexes yielded comparable transfection efficiency with PCPH/pDNA complexes in HEK293 cells. On the other hand, the expression of EGFP in HepG2 cells transfected with PCPH/pDNA complexes was higher than the group transfected with PEI/pDNA or PCP/pDNA complexes, indicating that a better performance in transfection efficiency in HepG2 cells might be attributed to HA/CD44-mediated endocytosis.
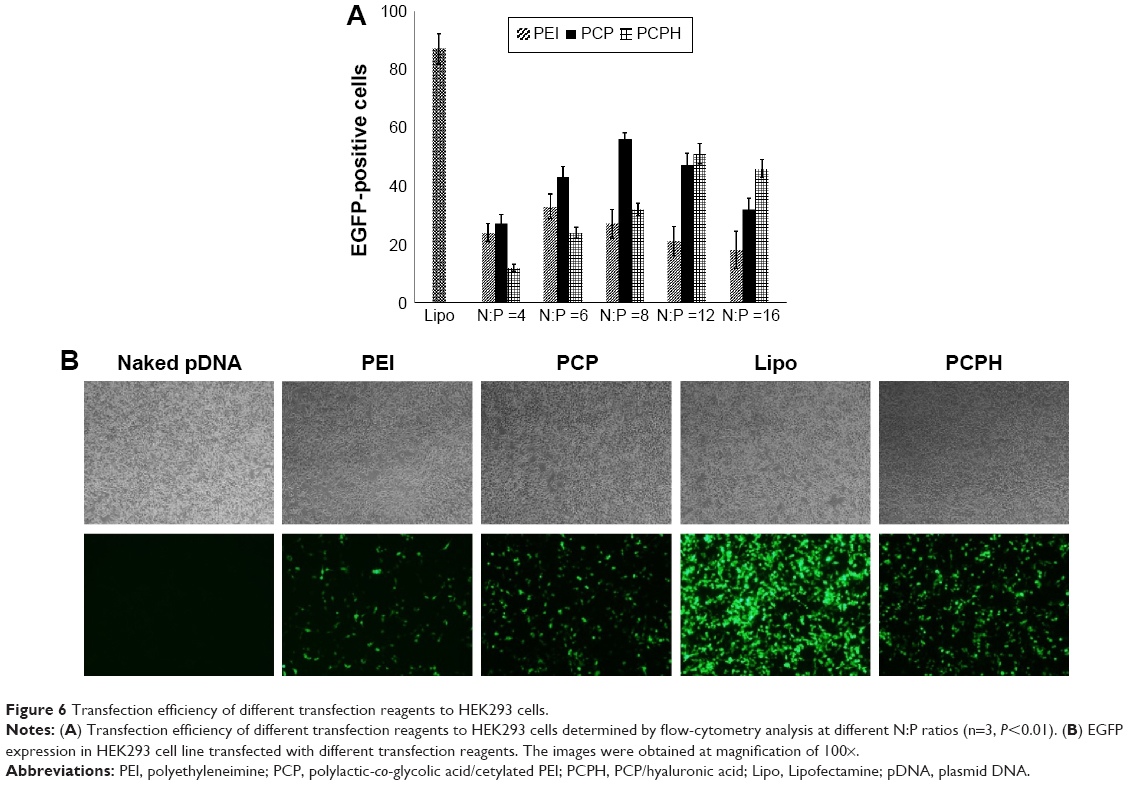 | Figure 6 Transfection efficiency of different transfection reagents to HEK293 cells. |
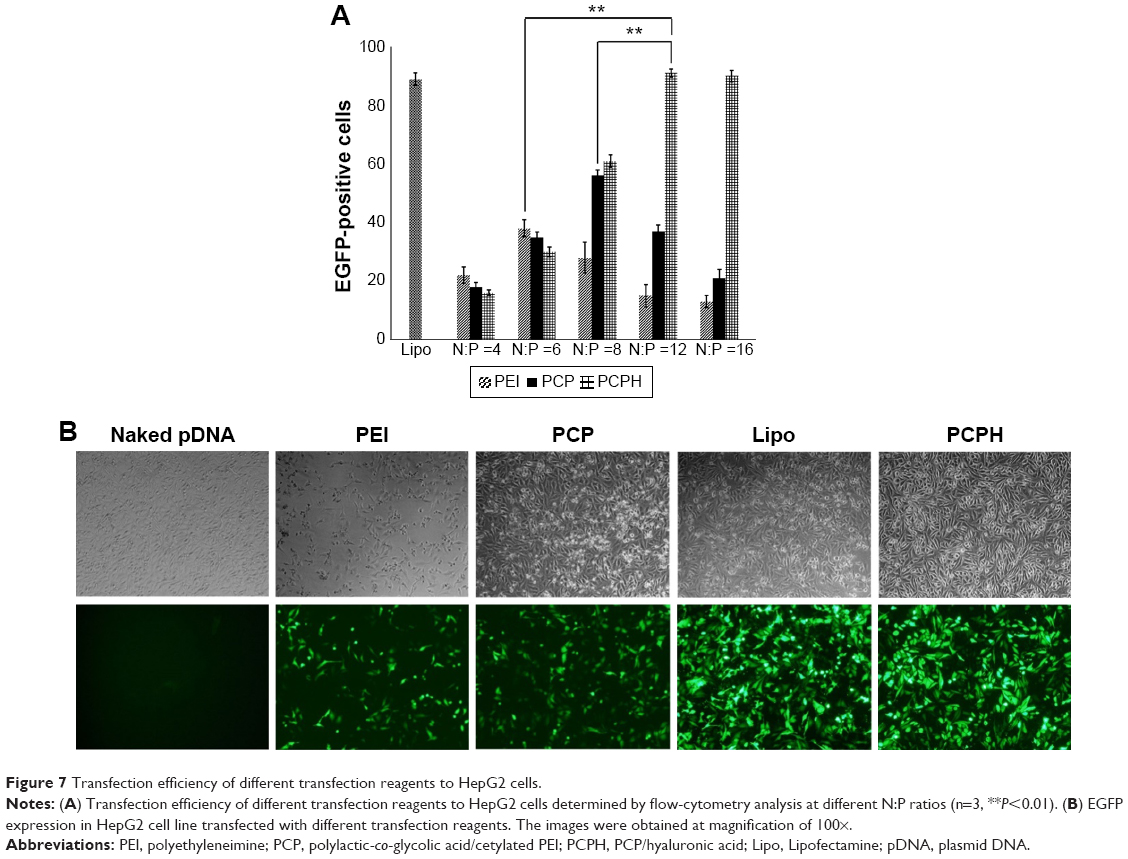 | Figure 7 Transfection efficiency of different transfection reagents to HepG2 cells. |
To investigate further whether the higher transfection efficiency of PCPH NPs was attributed to HA/CD44-mediated endocytosis, we first prepared HepG2 cells in which CD44 expression was knocked down using siRNA. Then, we transfected EGFP pDNA with various formations for untreated HepG2 and CD44-suppressed HepG2. Flow-cytometry analysis demonstrated that there were no significant differences in the transfection efficiency of PEI and PCP NPs between untreated HepG2 cells and CD44-suppressed HepG2 cells, whereas the fluorescence intensity of CD44 HepG2 cells transfected with PCPH NPs declined dramatically compared to the control group (Figure 8). Taken together, these results indicate PCPH NPs are an effective gene vector and can target CD44-expressing tumors through the CD44-mediated pathway.
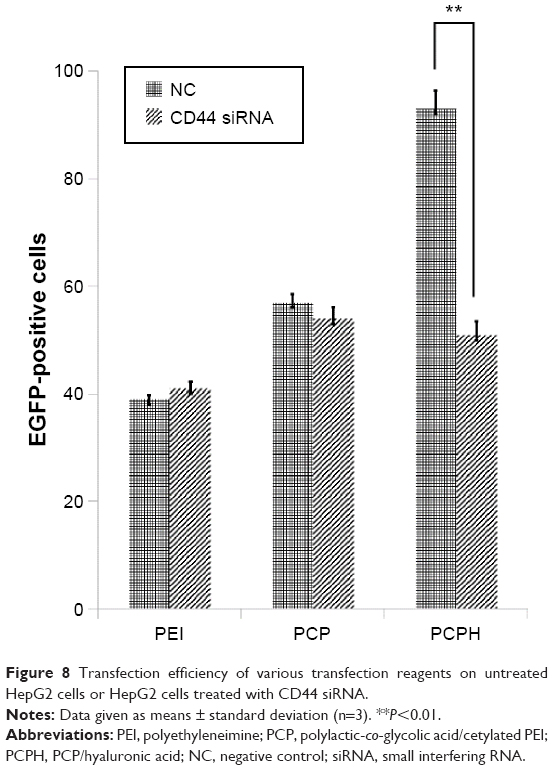 | Figure 8 Transfection efficiency of various transfection reagents on untreated HepG2 cells or HepG2 cells treated with CD44 siRNA. |
Transfected PCPH/anti-miR-221 into HepG2 cells
We further investigated whether PCPH NPs are also capable of driving oligonucleotides into targeted cells efficiently, especially to HA receptor-rich cancer cells, such as HepG2. PCPH NPs were used as anti-miR-221 oligonucleotide carriers (according to the result of the former experiment, the N:P ratio was set at 12:1) to transfect HepG2 cells in vitro.
To assess the capacity of PCPH NPs for anti-miRNA loading and subsequent cellular internalization, we carried out confocal laser scanning microscopy imaging with FAM-labeled anti-miR-221. NP-mediated uptake in HepG2 is shown in Figure 9, where FAM-labeled anti-miR-221 fluoresced green and cellular nuclei fluoresced blue (Hoechst 33342). Within cells that were untreated or treated only with FAM-labeled anti-miR-221, there was no detectable green fluorescence, indicating no oligonucleotide internalization, as expected. In contrast, there was clear gene carrier-mediated intracellular accumulation of anti-miRNA in all imaged cells. By comparing the fluorescence of cell samples transfected with different carriers, the fluorescence in cells transfected with PCP or PCPH was diffuse, indicating that cytosolic rather than endosomal localization of anti-miRNA had been achieved. In addition, cells transfected with PCPH/FAM-oligonucleotides showed stronger fluorescence intensity than the fluorescence intensity displayed on cells transfected with PCP/FAM oligonucleotides. This demonstrated that because of the HA coated on the PCPH NP surface, PCPH can enter into cells easier than PCP. Instead, the green fluorescence signal was porhyritic rather than dispersive in cells transfected with PEI, and demonstrated that PEI as a gene carrier forms clumps easily. This makes the carried gene hard to release to cytoplasm. Therefore, the visible results demonstrated PCPH NPs can ferry anit-miR-221 into HepG2 cells.
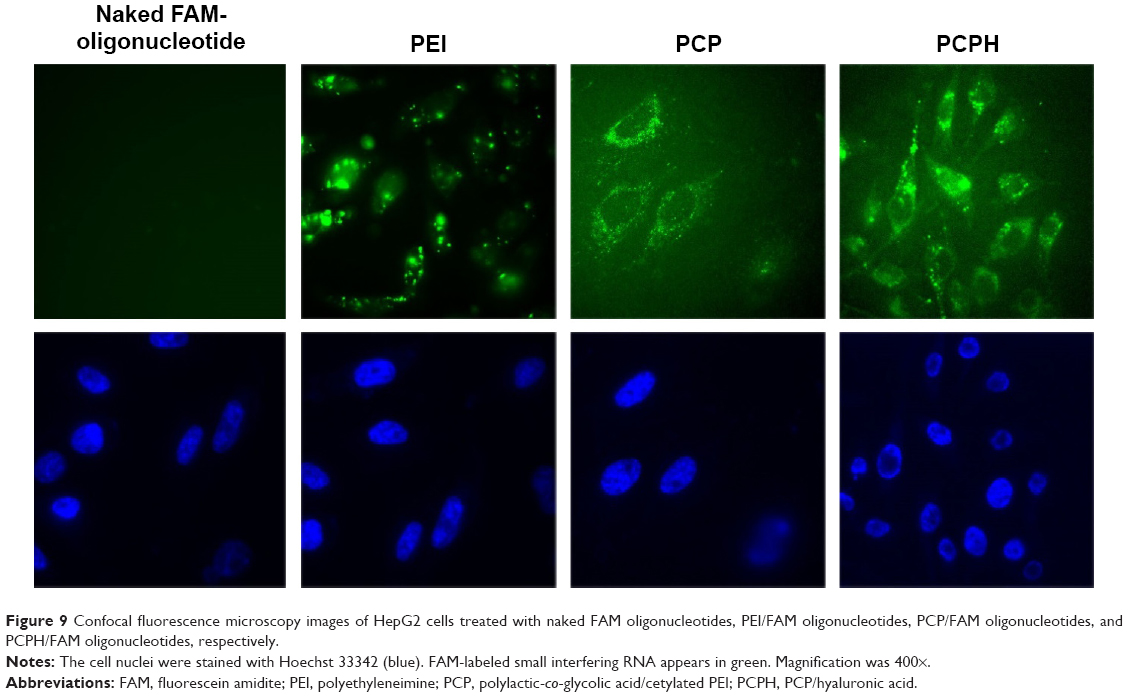 | Figure 9 Confocal fluorescence microscopy images of HepG2 cells treated with naked FAM oligonucleotides, PEI/FAM oligonucleotides, PCP/FAM oligonucleotides, and PCPH/FAM oligonucleotides, respectively. |
In another study, the internalizations of PCPH/anti-miR-221 were also monitored by qRT-PCR. Previous research has shown that the apparent expression levels of miRNA detected by qRT-PCR will be reduced by anti-miRNA AMOs, because stable formation of base-pairing between mature miRNA and anti-miRNA molecules prevent the binding of the miRNA-specific looped RT primer and subsequently inhibit reverse transcription.27 As illustrated in Figure 10A, when the cells were transfected with PCPH/anti-miR-221, the endogenous miR-221 was slightly reduced by 5 nM of anti-miR-221, while 90% of miR-221 was silenced with 50 nM of anti-miR-221, indicating the detected levels of miR-221 declined in a dose-dependent manner. These results suggested that PCPH NPs could efficiently deliver anti-miR-221 into the cytoplasm, and specifically silenced the miR-221 in HepG2 cells.
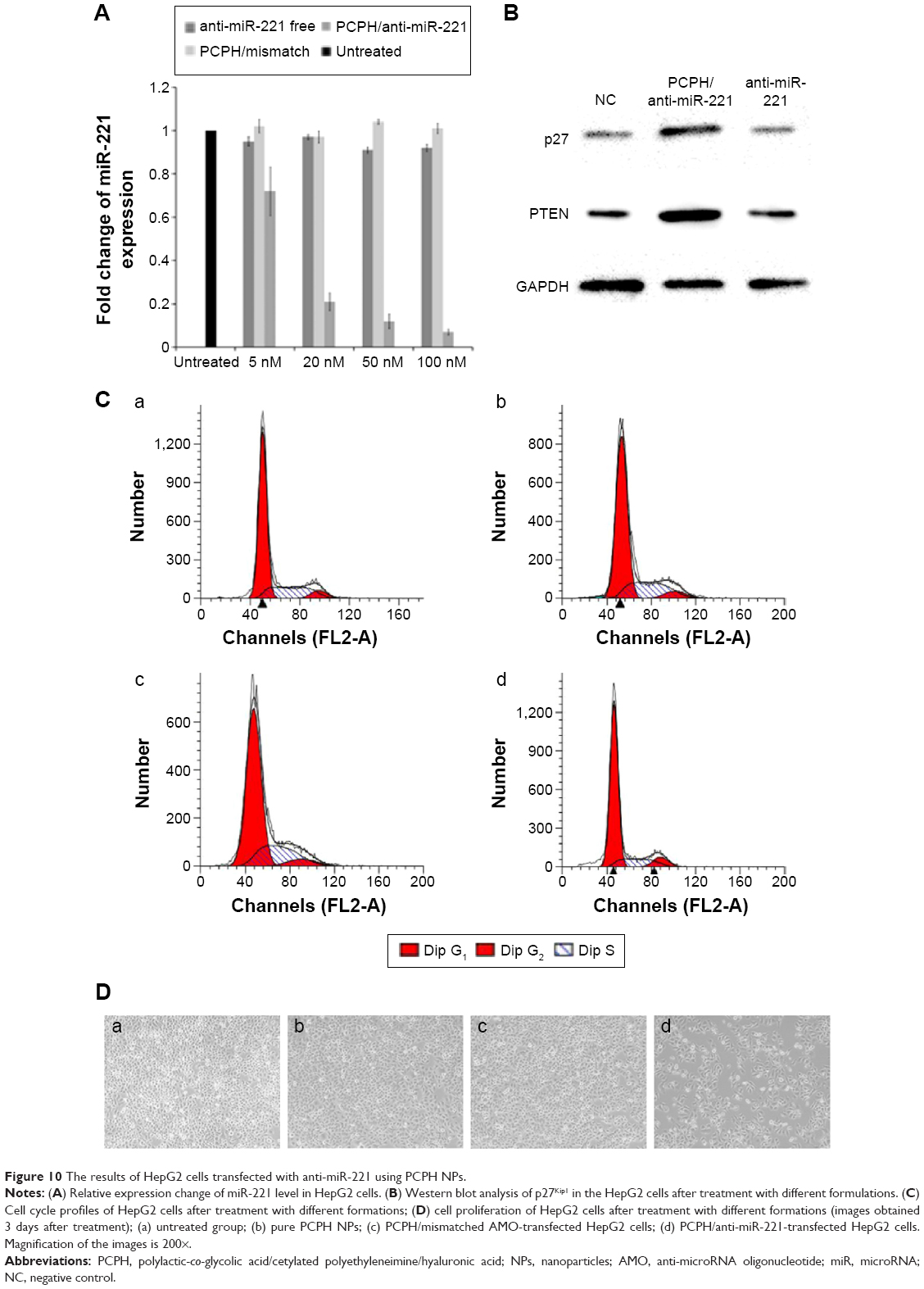 | Figure 10 The results of HepG2 cells transfected with anti-miR-221 using PCPH NPs. |
To confirm the biological function induced by PCPH/anti-miR-221 in HepG2 cells, we evaluated the protein-expression levels of the downstream targets of miR-221, such as p27Kip1 and PTEN, respectively. As anticipated, an increase in p27 and PTEN protein expression was observed in HepG2 cells treated with PCPH/anti-miR-221 compared to untreated cells, whereas the free anti-miR-221 had no effect (Figure 10B).
It has been proposed that miR-221 is oncogenic, based on its upregulation in tumor cells and the suppressive effect on PTEN and p27Kip1.28,29 p27Kip1 is a member of the Cip/Kip family of cyclin-dependent kinase inhibitors, and these are known to prevent cell-cycle progression from G1- to S-phase expression, permitting increased cell proliferation.30 Therefore, to evaluate further the activity of PCPH/anti-miR-221, we evaluated HepG2 cell apoptosis and cell cycle by MTT and flow-cytometry analysis, respectively, at 48 hours posttransfection. Results showed the HepG2 cells in the S phase decreased after being transfected with anti-miR-221 by PCPH NPs (Figure 10C, Table 2), and proliferation ability was obviously inhibited (Figure 10D). Finally, the aforementioned could be more proof that PCPH NPs can transfect anti-miR-221 into HepG2 cells to function without any damage.
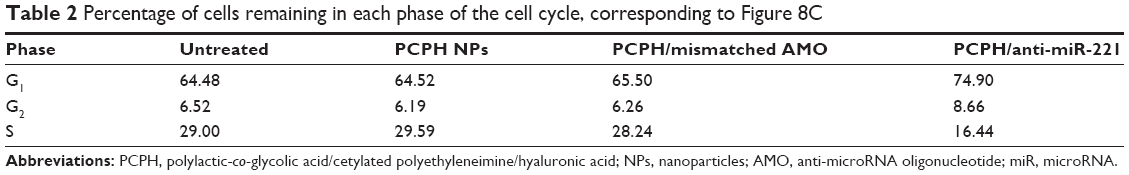 | Table 2 Percentage of cells remaining in each phase of the cell cycle, corresponding to Figure 8C |
Conclusion
In this study, we have reported a novel type of NPs for targeted gene delivery. We introduced amphiphilic PEI-cet to modified PLGA NPs in order to increase the loading dose of genes and the cellular uptake capacity. To reduce cytotoxicity and increase tumor cell-targeting ability, HA was coated on the surface of NPs. With a negligible cytotoxicity effect, the resulting NPs successfully transferred pDNA or AMOs into HepG2 cells. Therefore, PCPH could be a promising nonviral delivery system for gene therapy. Further studies are necessary to investigate the detailed uptake mechanism and its clinical safety properties.
Acknowledgments
This work was financially supported by the NSFC (20875014, 61071047) and the 973 Program (2013CB932900).
Disclosure
The authors report no conflicts of interest in this work.
References
Verma IM, Weitzman MD. Gene therapy: twenty-first century medicine. Annu Rev Biochem. 2005;74:711–738. | ||
Mountain A. Gene therapy: the first decade. Trends Biotechnol. 2000;18(3):119–128. | ||
Somia N, Verma IM. Gene therapy: trials and tribulations. Nat Rev Genet. 2000;1(2):91–99. | ||
Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10(2):126–139. | ||
Shimoni Y, Friedlander G, Hetzroni G, et al. Regulation of gene expression by small non-coding RNAs: a quantitative view. Mol Syst Biol. 2007;3:138. | ||
Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9(3):219–230. | ||
Rossbach M. Small non-coding RNAs as novel therapeutics. Curr Mol Med. 2010;10(4):361–368. | ||
Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346–358. | ||
Pouton CW, Seymour LW. Key issues in non-viral gene delivery. Adv Drug Deliv Rev. 2001;46(1–3):187–203. | ||
Morille M, Passirani C, Vonarbourg A, Clavreul A, Benoit JP. Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials. 2008;29(24–25):3477–3496. | ||
Chen S, Cheng SX, Zhuo RX. Self-assembly strategy for the preparation of polymer-based nanoparticles for drug and gene delivery. Macromol Biosci. 2011;11(5):576–589. | ||
Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(d,l-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16(10):1217–1226. | ||
Perez C, Sanchez A, Putnam D, Ting D, Langer R, Alonso MJ. Poly(lactic acid)-poly(ethylene glycol) nanoparticles as new carriers for the delivery of plasmid DNA. J Control Release. 2001;75(1–2):211–224. | ||
Su WP, Cheng FY, Shieh DB, Yeh CS, Su WC. PLGA nanoparticles codeliver paclitaxel and Stat3 siRNA to overcome cellular resistance in lung cancer cells. Int J Nanomedicine. 2012;7:4269–4283. | ||
Benfer M, Kissel T. Cellular uptake mechanism and knockdown activity of siRNA-loaded biodegradable DEAPA-PVA-g-PLGA nanoparticles. Eur J Pharm Biopharm. 2012;80(2):247–256. | ||
Han XP, Li ZB, Sun J, et al. Stealth CD44-targeted hyaluronic acid supramolecular nanoassemblies for doxorubicin delivery: probing the effect of uncovalent pegylation degree on cellular uptake and blood long circulation. J Control Release. 2015;197:29–40. | ||
Sun Q, Kang ZS, Xue LJ, et al. A collaborative assembly strategy for tumor-targeted siRNA delivery. J Am Chem Soc. 2015;137(18):6000–6010. | ||
Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther. 2005;11(6):990–995. | ||
Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6(5):376–385. | ||
Lin SB, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15(6):321–333. | ||
Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132(21):4653–4662. | ||
Marilena VI, Carlo MC. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. EMBO Mol Med. 2012;4(3):143–159. | ||
Kumar M, Bakowsky U, Lehr CM. Preparation and characterization of cationic PLGA nanospheres as DNA carriers. Biomaterials. 2004;25(10):1771–1777. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. | ||
Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int J Pharm. 2002;244(1–2):105–115. | ||
Jin S, Ye KM. Nanoparticle-mediated drug delivery and gene therapy. Biotechnol Prog. 2007;23(1):32–41. | ||
Scherr M, Venturini L, Battmer K, et al. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007;35(22):e149. | ||
Visone R, Russo L, Pallante P, et al. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr Relat Cancer. 2007;14(3):791–798. | ||
le Sage C, Nagel R, Egan DA, et al. Regulation of the p27Kip1 tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26(15):3699–3708. | ||
Toyoshima H, Hunter T. P27, a novel inhibitor of G1 cyclin-Cdk protein-kinase activity, is related to p21. Cell. 1994;78(1):67–74. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.