Back to Journals » The Application of Clinical Genetics » Volume 13
A Novel Splice-Site Mutation in the ELN Gene Suggests an Alternative Mechanism for Vascular Elastinopathies
Authors Velandia-Piedrahita CA ![]() , Morel A
, Morel A ![]() , Fonseca-Mendoza DJ
, Fonseca-Mendoza DJ ![]() , Huertas-Quiñones VM
, Huertas-Quiñones VM ![]() , Castillo D, Bonilla JD
, Castillo D, Bonilla JD ![]() , Hernandez-Toro CJ
, Hernandez-Toro CJ ![]() , Miranda-Fernández MC
, Miranda-Fernández MC ![]() , Restrepo CM, Cabrera R
, Restrepo CM, Cabrera R
Received 16 September 2020
Accepted for publication 3 November 2020
Published 17 December 2020 Volume 2020:13 Pages 233—240
DOI https://doi.org/10.2147/TACG.S282240
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Camilo Andres Velandia-Piedrahita,1 Adrien Morel,2,* Dora Janeth Fonseca-Mendoza,2,* Victor Manuel Huertas-Quiñones,3– 5 David Castillo,6 Juan Diego Bonilla,7 Camilo José Hernandez-Toro,1 Marta Catalina Miranda-Fernández,1 Carlos Martin Restrepo,2 Rodrigo Cabrera1,2
1Laboratorio de Biología Molecular y Pruebas Diagnósticas de Alta Complejidad, Fundación Cardioinfantil‐Instituto de Cardiología, Bogotá, Colombia; 2Center for Research in Genetics and Genomics-CIGGUR, GENIUROS Research Group, School of Medicine and Health Sciences, Universidad del Rosario, Bogotá, Colombia; 3Instituto de Cardiopatías Congénitas, Fundación Cardioinfantil‐Instituto de Cardiología, Bogotá, Colombia; 4Facultad de Medicina, Universidad Nacional de Colombia, Bogotá, Colombia; 5Facultad de Medicina, Universidad del Rosario, Bogotá, Colombia; 6Servicio de Dermatología, Fundación Para la Investigación en Dermatología FUNINDERMA, Bogotá, Colombia; 7Servicio de Dermatología, Universidad El Bosque, Bogotá, Colombia
*These authors contributed equally to this work
Correspondence: Rodrigo Cabrera
Laboratorio de Biología Molecular y Pruebas Diagnósticas de Alta Complejidad, Fundación Cardioinfantil‐Instituto de Cardiología, Calle 163A # 13B-60, Bogotá CP 110131, Colombia
Tel +57 1 6672727 ext 11611
Fax +57 1 6672727 Ext 73206
Email [email protected]
Abstract: The ELN gene encodes elastin, a fundamental protein of the extracellular matrix that confers elasticity to different tissues including blood vessels. The formation of elastin fibers is a complex process involving monomer coacervation and subsequent crosslinking. Mutations in exons 1– 29 of the ELN gene have been linked to supravalvular aortic stenosis (SVAS) whereas mutations in exons 30– 33 are associated with autosomal dominant cutis laxa (ADCL). This striking segregation has led to the hypothesis that distinct molecular mechanisms underlie both diseases. SVAS is believed to arise through haploinsufficiency while ADCL is hypothesized to be caused by a dominant negative effect. Here, we describe a patient with SVAS harboring a novel splice-site mutation in the last exon of ELN. The location of this mutation is not consistent with current knowledge of SVAS, since all mutations reported in the C-terminus have been found in ADCL patients, and a thorough evaluation did not reveal significant skin involvement in this case. RT-PCR analysis of skin tissue showed that C-terminal mutations in the region can lead to the production of aberrant transcripts through intron retention and activation of cryptic splice sites and suggest that disruption of the very last exon can lead to functional haploinsufficiency potentially related to SVAS.
Keywords: SVAS, ELN, ADCL, coacervation, splicing
Introduction
Elastin is a key protein of the extracellular matrix present in connective tissue. This protein is one of two components of elastic fibers, which confer elasticity to organs and tissues including the heart, skin, lungs, and blood vessels.1 The formation of elastic fibers, or elastogenesis, is a stepwise process whereby elastin monomers bind to receptors on the membrane of fibroblasts and are subjected to coacervation and subsequent cross-linking.2–4 Elastogenesis is important during embryogenesis and shortly after birth. Mutations in the elastin gene are associated with diverse forms of elastinopathies, including supravalvular aortic stenosis (SVAS), autosomal dominant cutis laxa (ADCL) and pulmonic stenosis.1,5–11
Supravalvular aortic stenosis (SVAS) (OMIM 185500) is a congenital narrowing of the lumen of the ascending aorta with an estimated incidence of 1 in 20.000 live births6,12 This condition can present in both syndromic and nonsyndromic forms. The syndromic form is usually associated with Williams-Beuren syndrome (WBS) (OMIM 194050), which shows an autosomal dominant inheritance pattern and is caused by a deletion in 7q11.23. This syndrome is characterized by intellectual disability, distinctive facial features, hypercalcemia, connective tissue abnormalities, growth abnormalities and a gregarious personality.13 Mutations in the elastin gene (ELN) are associated with non-syndromic SVAS with compromised coronary and pulmonary artery branches, but normal descending aorta, cerebral and renal arteries.12 ELN arteriopathy is inherited in an autosomal dominant pattern, with incomplete penetrance and variable expressivity.6
Autosomal Dominant Cutis Laxa (ADCL) is an inherited connective tissue disease characterized by an excess of skin tissue with loss of elasticity and inguinal hernia, SVAS, pulmonary stenosis and emphysema in some cases.14 Histological findings in this condition show fiber fragmentation with reduced deposition, whereas in SVAS only reduced total deposition of elastin fibers without additional abnormalities can be observed.1
Even though both conditions are caused by mutations in the same gene, SVAS and ADCL are mutually exclusive in most patients, suggesting distinct underlying molecular mechanisms. Animal and in-vitro studies suggest that non-syndromic SVAS mutations lead to insufficient levels of elastin, resulting from lower mRNA levels because of degradation via the NMD pathway.10 Similarly, in-vitro studies show that ADCL mutations lead to the expression of dominant negative proteins which interfere with its function.12 Remarkably, mutations associated with one or the other show distinct clusters within the ELN gene. Analysis of ClinVar and HMGD databases showed that all frameshift (n=42) and intronic (n=14) mutations associated with SVAS are located in exons 1–29 and all frameshift (n=17) mutations associated with ADCL are found in exons 30–33 (Figure 1A).1
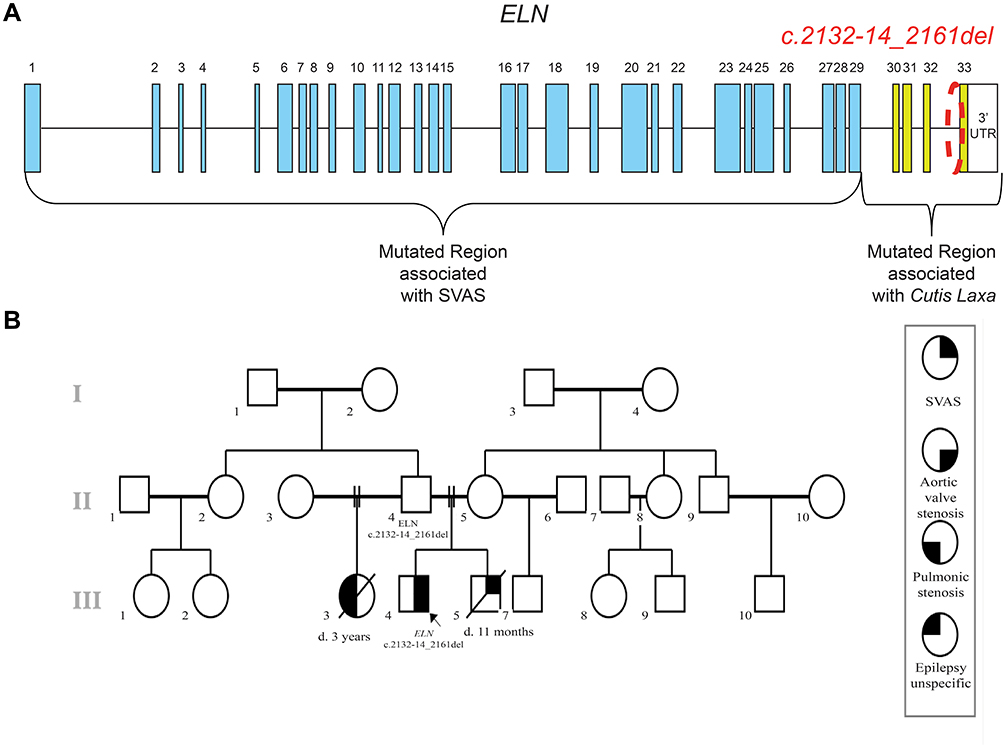 |
Figure 1 Mutations on ELN gene associated to SVAS. (A) Schematic representation of the ELN gene and the mutations associated with SVAS and Cutis Laxa showing the location of the reported mutations (B) Family pedigree. Reported case and different phenotypes in the family. Abbreviation: SVAS, supravalvular aortic stenosis. |
In this report, we present a patient with a mutation in the intron-exon boundary of the last exon of the ELN gene that leads to the production of aberrant transcripts. However, unlike all reported frameshift mutations in the C-terminus of ELN, this mutation causes SVAS and not ADCL. This finding suggests that the distinction between haploinsufficiency and dominant negative mechanisms in the pathophysiology of SVAS and ADCL is not as clear-cut as previously thought. Alternatively, this mutation suggests that there could be an additional mechanism for SVAS beyond insufficient levels of ELN.
Patient Report
The proband was born at 40w2d of gestation after an uneventful pregnancy with birth weight of 2940 g and birth length of 49 cm. When he was delivered, his mother was 23 years old and his father was 28 years old. At 4 years of age, the patient presented two syncopal episodes. In the study of these episodes, echocardiography revealed aortic valvular stenosis with a valvular gradient of 60mmHg, asymmetric thickened tri-leaflet valve and hypertrophy of the left ventricle. Balloon valvuloplasty was performed successfully. At the age of 7, a new echocardiogram and contrast cardiac computed tomography were performed and showed SVAS of 33% of the lumen with gradient of 45mmHg without coronary arteries compromise. Later, cardiac catheterization was performed, revealing evidence of the SVAS, but renal and pulmonary circulation was normal. Thus, the aorta was repaired with an autologous pericardial patch. At the age of nine, a skin biopsy was performed, which showed no significant alterations (Figure 2). Physical examination was unremarkable; the proband has neither dysmorphic characteristics nor hypermobility. The parents had another male son which died at the age of 11 months with supravalvular aortic stenosis. Additionally, the proband´s paternal half-sister died at age 3 with asymptomatic pulmonary stenosis and epilepsy (Figure 1B).
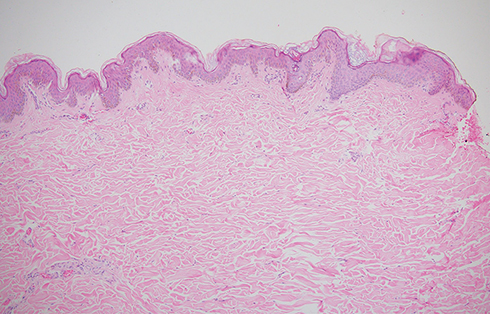 |
Figure 2 Skin biopsy, scapular region. H&E 10X. |
Materials and Methods
Whole Exome Sequencing
Total DNA from the patient was extracted from a blood sample with a QIAamp DNA mini kit (QIAGEN, Germantown, USA) according to the manufacturer’s instructions. DNA concentration was measured using a Qubit DNA assay Kit in a Qubit 2.0 Fluorometer (Life Technologies, CA, USA). A total amount of 1.0 µg genomic DNA was used for Whole Exome Sequencing (WES) as input material for library preparation. Sequencing libraries were generated using Agilent Sure Select Human All Exon Kit v6 (Agilent Technologies, CA, USA), after generation of blunt-ended 180–280 bp fragments by hydrodynamic shearing and enzyme treatment. Then, adapter oligonucleotides were ligated after adenylation at the 3´. DNA fragments with ligated adapter molecules on both ends were selectively enriched in a PCR reaction. After PCR reaction, libraries were hybridize in liquid phase with biotin-labeled probes, and captured with streptavidin-linked magnetic beads. Products were purified using the AMPure XP system (Beckman Coulter, Beverly, USA) and quantified on the Agilent Bioanalyzer 2100 system. 150 bp paired ends were sequenced on the HiSeq Illumina platform, The raw reads were aligned to the hg19 reference genome with Burrows-Wheeler Aligner (BWA) software. The variants were called with GATK. >80% of bases have a sequencing quality score >Q30 and minimum coverage of 100X. Following genomic variant detection, variants were annotated using ANNOVAR to show affected genomic regions, protein coding changes, and allele frequency reported by Genome Aggregation Database (gnomAD). Library preparation and sequencing were carried out by Novogene Inc. (Beijing-China).
The VarSeq v2.2 (Golden Helix) software was used to filter variants in the ELN gene. These variants were classified into missense, nonsense, splice site and frameshift. For our analysis, novel variants, or variants with MAF≤0.005 were selected.
Skin Biopsy and Histology
A skin sample was collected from the scapular region by punch biopsy. Half of the sample was refrigerated in trizol and the rest was fixed with formaldehyde for hematoxylin and eosin staining and histologic analysis.
RNA Extraction, RT-PCR and Cloning
To determine the effect of the splicing mutation, we performed RNA extraction using Trizol Reagent and the PureLink RNA Mini Kit (Thermo Scientific). We generated cDNA using the SuperScript™ III First-Strand Synthesis System following the manufacturer’s recommendations.
We carried out two PCR approaches to detect all possible aberrant transcripts: a first one to generate a fragment containing exon 32 and the 3´UTR (primers F3 and R3), and a second one to generate a fragment spanning exon 30 and intron 31 (primers F1 and intR1) (Figures 3A and 4A). PCR primers were generated using Primer Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). The target regions of ELN were amplified by PCR and primer sequences are available upon request.
 |
Figure 3 Deletion of splice site results in aberrant transcript formation via activation of a cryptic splice site. (A) Localization of F3-R3 primers (B) Electrophoresis Gel of F3-R3 RT-PCR on cDNA derived from skin biopsy (C) Chromatogram showing the WT and the alternative transcript (#1) with a new cryptic splice site and loss of KxxxRKRK motif (in red). |
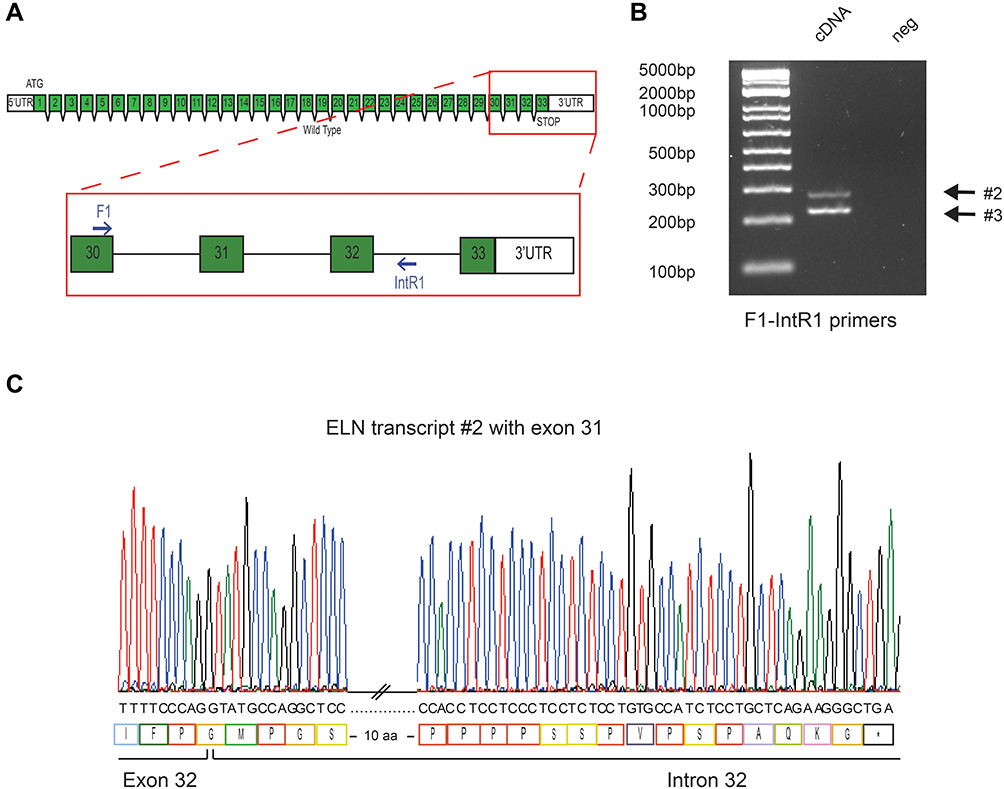 |
Figure 4 Deletion of splice site results in intron 32 retention and generates a PTC. (A) Localization of F1-IntR1 primers (B) Electrophoresis Gel of F1-IntR1 RT-PCR on cDNA derived from skin biopsy (C) Chromatogram showing the alternative transcript (#2) with intron retention and predicted stop codon in the intron 32. |
For cloning, 1uL of PCR products were cloned into pCR™4-TOPO™ Vector (Invitrogen) using TOPO™ TA Cloning™, as the manufacturer’s instructions. Ligated plasmids were inserted in One Shot TOP10 E. coli (Invitrogen) through thermic shock during 30 sec. The bacteria were incubated during 1h in SOC medium. Transformed cells were plated on LB agar plates containing 100 μg/mL ampicillin and were grown at 37°C overnight. Plasmid DNA was extracted from colonies with a Qiagen Plasmid Mini kit (Qiagen).
Sanger Confirmation
We confirmed the mutation identified by WES and the cloned RT-PCR products using sanger sequencing. The genomic DNA encompassing the variant ELN-c.2132–14_2161del was amplified by PCR using primers designed with the same tools mentioned previously (Primers available upon request). The pCR™4-TOPO™ Vector plasmids containing the PCR products of RT-PCR were sequenced with T7 and T3 primers. The obtained sequences were compared to reference sequence ENST00000252034.12 (NM_000501.3).
A detailed protocol is available at: dx.doi.org/10.17504/protocols.io.bmcnk2ve
Results
Germline sequencing of genomic DNA from the patient revealed a new splice site mutation in the ELN gene, which corresponds to a deletion of 44 bp across intron 32/exon 33 boundary (ELN-c.2132–14_2161del). This mutation has not been previously described in public databases (gnomAD) nor in the literature.
The functional effect of the mutation was determined by amplifying putative aberrant transcripts resulting from intron retention and activation of cryptic splice sites using intronic and 3ʹUTR primers (Figures 3A and 4A). Amplicons resulting from RT-PCR with the 3ʹUTR primer (F3 and R3) revealed a band smaller than expected for the wild type allele (WT), and another that was consistent with a normal allele (Figure 3B). Cloning and sequencing analysis of these PCR products identified an aberrant transcript that skipped all the coding region of exon 33 and used a new cryptic splice site in the 3´UTR (Figure 3C). This alteration, combined with a common insertion (rs34208922) in the 3´UTR, led to a predicted C-terminus containing 49 additional residues and lacking the conserved KxxxRKRK motif (r.[=, 2132_2175del]; p.G712Pext*44). (Protein #1, Figure S1).
Amplicons resulting from RT-PCR using the intronic primer (F1 and IntR1) showed two abnormal bands of 281bp and 227bp (Figure 4B). Sequencing of the 281 bp product revealed the presence of 92 bp of intronic sequence adjacent to exon 32 indicating intron 32 retention. In this case, the disruption of the splice acceptor site of intron 32 inhibits splicing and results in retention of part of intron 32 in the aberrant transcript (Figure 4C). The aberrant protein has 17 additional amino acids at the C-terminus and disrupts the KxxxRKRK motif (protein #2) (Figure S1). The 227 bp product shows an identical intron 32 retention, combined with the alternative splicing of exon 31, also observed in WT transcripts. The protein generated (protein #3) is likely to have the same functional implications as described for protein #2 (Figure S2).
Histological analysis of the skin biopsy revealed normal elastic fibers in configuration and number with adequate cellularity and without inflammatory infiltrates (Figure 2).
Discussion
SVAS is a rare disease caused by mutations in the ELN gene. The molecular mechanism proposed for the pathogenesis of SVAS is ELN haploinsufficiency, explained by premature termination codon mutations (PTCs), extensively described in affected patients.8,11,12,15 PTCs lead to insufficient expression of ELN because mutant mRNA is presumably degraded by the nonsense mediated decay (NMD) pathway.10 A reduction in ELN expression has also been found in skin fibroblasts and aortic smooth muscle cells of SVAS affected patients, thus supporting ELN haploinsufficiency as the mechanism involved in the pathogenesis of the vasculopathy.11
The location of protein truncating mutations within a gene can have a significant influence on the levels of mutant transcripts by affecting the efficiency of nonsense mediated decay. For instance, mutations that occur near the 3´end of the dystrophin gene related to Duchenne Muscular Dystrophy (DMD) result in mild phenotypes, suggesting that these truncating proteins are capable of a partial rescue of the DMD phenotype. PTCs near the 5ʹ end, however, are associated with severe forms of disease and fail to rescue the phenotype because of the NMD pathway. Furthermore, PTCs near the 3´end of gene are invisible to the NMD machinery.16 Similarly, mutations near the C-terminus of ELN can be less affected by NMD and more likely to lead to the production of dominant negative proteins as observed in ADCL.
This model has been used to explain why all frameshift and nonsense mutations in exons 1–29 lead to SVAS and similar mutations in exons 30–33 lead to ADCL. However, this model does not explain why peptides with dominant negative effects overwhelmingly affect the structure of the extracellular matrix in the skin but not the vasculature and why missense mutations can cause SVAS. Only a few missense mutations have been conclusively associated with this condition, although most seem to affect protein expression by disrupting the initial methionine or affecting splicing. However, the p.Ala707Asp mutation, which seems to affect the putative microassembly domain in exon 29 is particularly interesting, since it can potentially reduce protein expression by affecting post-transcriptional regulation.1 This suggests that most, if not all, ELN variants associated with SVAS result in reduced expression or functional haploinsufficiency.
In this study, we identified a deletion of 44 base pairs in the ELN gene situated across the intron-exon boundary of intron 32 and exon 33 in a patient with SVAS and no clinical or histological skin abnormalities. The patient had a significant family history of severe SVAS, suggesting an autosomal dominant pattern of inheritance with incomplete penetrance. cDNA analysis indicated the formation of several aberrant transcripts through the activation of cryptic splice sites in the 3´UTR (protein #1) and intron retention (proteins # 2 and 3). Since all of the transcripts generate an aberrant stop codon in the final exon, they are unlikely to trigger NMD.17,18 Since NMD is unlikely to play a role in this case of SVAS, the data suggests that an alternative mechanism can lead to SVAS.
The aberrant transcripts produced in this case are unusual because they preserve the amino acid composition of almost all of the elastin protein, with the exception of the 13 C-terminal residues. However, this region contains the highly conserved KxxxRKRK motif, known to mediate the interaction between tropoelastin and the GAGs on the cell membrane.19 We propose this disruption can lead to defects in the coacervation and/or crosslinking of tropoelastin monomers, impeding their assembly into elastin fibers. This can lead to a functional haploinsufficiency, whereby the mutant tropoelastin is not incorporated into elastin fibers but remains in its soluble form. Functional studies of the elastin fiber should be conducted to determine if these hypotheses are indeed the case.
Alternatively, Schmitz et al recently showed that intron retaining mRNAs have significantly longer 3ʹUTR sequences that are enriched for miRNA binding sites.20 miRNA sequences of 18–21 nucleotides are involved in mRNA degradation and/or inhibition of translation. In this case, the retention of intron 32 in transcripts #2 and #3 permits the formation of transcripts with longer 3ʹUTR sequences, with a greater potential of undergoing miRNA-mediated degradation or translational inhibition. This could lead to a reduced quantity of ELN protein leading to haploinsufficiency via an NMD-independent mechanism.21
Although a few other ELN mutations seem to also defy the haploinsufficiency/dominant negative model for SVAS/ADCL, careful analysis can explain why they are behaving the way they do. For example, a splicing mutation associated with ADCL was found in exon 25. However, the aberrant transcript is predicted to produce an inframe deletion of exon 25, likely avoiding the activation of NMD and producing an aberrant protein.22 This is consistent with the dominant negative model for ADCL. Similarly, a frameshift mutation affecting only the last four residues of ELN (preserving the KxxxRKRK motif) was found in a family with ADCL, suggesting that an unusual feature of the transcripts produced in this case is causing the unexpected phenotype.23
Conclusion
This study suggests that the functional consequence of extreme C-terminal ELN mutations can have more diverse effects than previously thought and must be analyzed carefully when counselling patients. Furthermore, the mechanisms proposed here can also help explain why a handful of missense mutations can cause SVAS, without likely affecting protein expression. These mutations can affect critical residues necessary for the incorporation of tropoelastin monomers into elastin fibers and can be used to map critical protein–protein interactions. Functional haploinsufficiency can play a role in several other mutations in ELN and other genes which rely on extensive protein–protein interactions for their function.
Abbreviations
ADCL, autosomal dominant cutis laxa; cDNA, complement deoxyribonucleic acid; DMD, Duchenne Muscular Dystrophy; DNA, deoxyribonucleic acid; EJC, exon junction complex; ELN, elastin; GAG, glycosaminoglycan; HGMD, human gene mutation database; mRNA, messenger ribonucleic acid; NMD, nonsense mediated decay; OMIM, Online Mendelian Inheritance in Man; PTC, premature terminal codon; RT-PCR, reverse transcription-polymerase chain reaction; SVAS, supravalvular aortic stenosis; UTR, untranslated region; WBS, Williams-Beuren syndrome; WT, wild type.
Ethical Compliance
Informed consent for genetic testing, skin biopsy and publication of clinical data and images was signed by the proband and guardian and the study protocol was approved by the institutional review board of the Fundacion CardioInfantil as a part of project “Programa para la Innovación en Cardiopatías Congénitas Humanas infrecuentes para Colombia” PINOCCHIO. (Number 21-2016).
Acknowledgments
The authors thank the family involved in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Colombian Ministry of Science, Technology and Innovation (Ministerio de Ciencia, Tecnología e Innovación - Minciencias) [contract numbers 21-2016, 726-2018 and 908-2019], Universidad del Rosario (grant ABN062) and Fundación Para la Investigación En Dermatología - FUNINDERMA.
Disclosure
Rodrigo Cabrera reports grants from Minciencias, during the conduct of the study. The authors report no other potential conflicts of interest for this work.
References
1. Duque Lasio ML, Kozel BA. Elastin-driven genetic diseases. Matrix Biol. 2018;71–72:144–160. doi:10.1016/j.matbio.2018.02.021
2. Yeo GC, Keeley FW, Weiss AS. Coacervation of tropoelastin. Adv Colloid Interface Sci. 2011;167(1–2):94–103. doi:10.1016/j.cis.2010.10.003
3. Cirulis JT, Keeley FW. Kinetics and morphology of self-assembly of an elastin-like polypeptide based on the alternating domain arrangement of human tropoelastin. Biochemistry. 2010;49(27):5726–5733. doi:10.1021/bi100468v
4. Schmelzer CEH, Hedtke T, Heinz A. Unique molecular networks: formation and role of elastin cross‐links. IUBMB Life. 2020;72(5):842–854. doi:10.1002/iub.2213
5. Hayano S, Okuno Y, Tsutsumi M, et al. Frequent intragenic microdeletions of elastin in familial supravalvular aortic stenosis. Int J Cardiol. 2019;274:290–295. doi:10.1016/j.ijcard.2018.09.032
6. Metcalfe K, Rucka AK, Smoot L, et al. Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet. 2000;8(12):955–963. doi:10.1038/sj.ejhg.5200564
7. Ewart AK, Jin W, Atkinson D, Morris CA, Keating MT. Supravalvular aortic stenosis associated with a deletion disrupting the elastin gene. J Clin Invest. 1994;93(3):1071–1077. doi:10.1172/JCI117057
8. Jelsig AM, Urban Z, Hucthagowder V, Nissen H, Ousager LB. Novel ELN mutation in a family with supravalvular aortic stenosis and intracranial aneurysm. Eur J Med Genet. 2017;60(2):110–113. doi:10.1016/j.ejmg.2016.11.004
9. Jakob A, Unger S, Arnold R, et al. A family with a new elastin gene mutation: broad clinical spectrum, including sudden cardiac death. Cardiol Young. 2011;21(1):62–65. doi:10.1017/S1047951110001563
10. Micale L, Turturo MG, Fusco C, et al. Identification and characterization of seven novel mutations of elastin gene in a cohort of patients affected by supravalvular aortic stenosis. Eur J Hum Genet. 2010;18(3):317–323. doi:10.1038/ejhg.2009.181
11. Park S, Seo EJ, Yoo HW, Kim Y. Novel mutations in the human elastin gene (ELN) causing isolated supravalvular aortic stenosis. Int J Mol Med. 2006;18(2):329–332. doi:10.3892/ijmm.18.2.329
12. Merla G, Brunetti-Pierri N, Piccolo P, Micale L, Loviglio MN. Supravalvular aortic stenosis: elastin arteriopathy. Circ Cardiovasc Genet. 2012;5(6):692–696. doi:10.1161/CIRCGENETICS.112.962860
13. Wilson M, Carter IB. Williams Syndrome. StatPearls Publishing; 2020. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31334998.
14. Hbibi M, Abourazzak S, Idrissi M, Chaouki S, Atmani S, Hida M. Cutis laxa syndrome: a case report. Pan Afr Med J. 2015;20. doi:10.11604/pamj.2015.20.3.5878
15. Lindsay ME, Dietz HC. The genetic basis of aortic aneurysm. Cold Spring Harb Perspect Med. 2014;4(9):a015909. doi:10.1101/cshperspect.a015909
16. Kerr TP, Sewry CA, Robb SA, Roberts RG. Long mutant dystrophins and variable phenotypes: evasion of nonsense-mediated decay? Hum Genet. 2001;109(4):402–407. doi:10.1007/s004390100598
17. Nickless A, Bailis JM, You Z. Control of gene expression through the nonsense-mediated RNA decay pathway. Cell Biosci. 2017;7(1):26. doi:10.1186/s13578-017-0153-7
18. Kurosaki T, Popp MW, Maquat LE. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol. 2019;20(7):406–420. doi:10.1038/s41580-019-0126-2
19. Broekelmann TJ, Kozel BA, Ishibashi H, et al. Tropoelastin interacts with cell-surface glycosaminoglycans via its COOH-terminal domain. J Biol Chem. 2005;280(49):40939–40947. doi:10.1074/jbc.M507309200
20. Schmitz U, Pinello N, Jia F, et al. Intron retention enhances gene regulatory complexity in vertebrates. Genome Biol. 2017;18(1). doi:10.1186/s13059-017-1339-3
21. Monteuuis G, Wong JJL, Bailey CG, Schmitz U, Rasko JEJ. The changing paradigm of intron retention: regulation, ramifications and recipes. Nucleic Acids Res. 2019;47(22):1149711513. doi:10.1093/nar/gkz1068
22. Graul-Neumann LM, Hausser I, Essayie M, Rauch A, Kraus C. Highly variable cutis laxa resulting from a dominant splicing mutation of the elastin gene. Am J Med Genet A. 2008;146(8):977–983. doi:10.1002/ajmg.a.32242
23. Hadj-Rabia S, Callewaert BL, Bourrat E, et al. Twenty patients including 7 probands with autosomal dominant cutis laxa confirm clinical and molecular homogeneity. Orphanet J Rare Dis. 2013;8(1):36. doi:10.1186/1750-1172-8-36
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.