Back to Journals » The Application of Clinical Genetics » Volume 15
A Novel POGZ Variant in a Patient with Intellectual Disability and Obesity
Authors Giraldo-Ocampo S, Pacheco-Orozco RA, Pachajoa H ![]()
Received 5 April 2022
Accepted for publication 8 June 2022
Published 6 July 2022 Volume 2022:15 Pages 63—68
DOI https://doi.org/10.2147/TACG.S369483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Sebastian Giraldo-Ocampo,1 Rafael Adrian Pacheco-Orozco,2 Harry Pachajoa2,3
1Universidad del Valle, Cali, Colombia; 2Genetics Division, Fundación Valle del Lili, Cali, Colombia; 3Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras (CIACER), Cali, Colombia
Correspondence: Harry Pachajoa, Genetics Division, Fundación Valle del Lili, Carrera 98 # 18-49, Cali, Colombia, Tel +57 5552334 ext 7653, Email [email protected]
Abstract: White–Sutton syndrome is a rare type of autosomal dominant neurodevelopmental disorder caused by mutations, mostly de novo, in the POGZ gene. No more than 120 patients have been described so far in the literature. Common clinical manifestations include intellectual disability, developmental delay, autism spectrum disorder, other behavioral abnormalities, sleeping problems, hyperactivity and visual problems. We describe a 20-year-old male patient from Colombia who presented with delayed psychomotor development, intellectual disability, obesity, sleep difficulties, hypotonia, hypogonadism, gynecomastia, visual abnormalities and several facial dysmorphisms. Genetic testing showed a novel heterozygous frameshift variant (c.3308del; p.Leu1103Profs*19) in the POGZ gene (NM_015100.3). This is the first report of a diagnosed patient with WHSUS in Colombia.
Keywords: WHSUS, neurodevelopmental disorder, Colombia, case report
Introduction
White–Sutton syndrome (WHSUS, OMIM:616364), also known as POGZ-Related Intellectual Disability Syndrome, is a monogenic, autosomal dominant neurodevelopmental disorder (NDD) identified for the first time by the Deciphering Developmental Disorders Study (2015) in two out of 1133 children with severe, undiagnosed developmental disorders analyzed using exome sequencing and detection of chromosome rearrangement.1 WHSUS is caused by mutations in the pogo transposable element derived with ZNF domain (POGZ) gene,2 which encoded a 1410 aminoacids protein. A growing line of evidence suggests that POGZ protein is involved in transcriptional dysregulation,3 chromosome segregation during mitosis and meiosis4 and also binding to different isotypes of human heterochromatin protein 1 (H1P), which in turn, is involved in transcriptional silencing and modulation of chromatin arrangement.5 Although it is not fully understood what the role of POGZ protein is in a physiological and pathological context, pogz knockout mice exhibit abnormal brain development, smaller absolute brain, altered neurogenesis in the embryo and adult mice, growth delay and abnormal motor, cognitive and social behavior.3
Currently, there are no established diagnostic criteria for White–Sutton syndrome (WHSUS) and of cases published so far, a discrete phenotype for this disorder has not yet been identified.6 Furthermore, WHSUS is heterogeneous at both phenotypic and genotypic levels which is why the only manner of properly diagnosing this condition is by looking for mutations in the POGZ gene that may lead to a lack or malfunction of POGZ protein and are associated with a set of neurodevelopmental abnormalities.1
Patients with WHSUS have intellectual disability varying from borderline to severe, developmental delay with speech and language more affected than motor development, autism spectrum disorder (ASD), other behavioral abnormalities such as anxiety and limited social interactions, sleeping problems, hyperactivity and visual problems. Some other variables may be present in some patients, such as brachycephaly, microcephaly, hypertelorism, midface hypoplasia, small mouth with thin upper lip hypotonia, feeding problems, obesity or other gastrointestinal manifestations, seizures, strabismus, hearing loss, genital abnormalities, urinary tract anomalies, adducted thumb, and peripheral polyneuropathy.6–9 Furthermore, a novel and distinctive phenotypes have been recently described, consisting of paroxysmal non-epileptic events, EEG abnormalities without seizures, or the coexistence of both in the same patient.10 Overall, the expressivity of WHSUS is variable, with limited knowledge about the genotype-phenotype correlation in this disorder. However, individuals with the same variant in the POGZ gene tend to have the same clinical features.6 Since its discovery and up to 2021, nearly 117 patients with WHSUS and 72 different disease-causing variants in the POGZ gene have been reported.6 Among these patients, most have frameshift (41%) and nonsense (40%) variants and to a lesser extent, missense (8.5%), splice site (7%), deletions of several exons or the whole gene (2.5%) and small in-frame deletions (1%) variants.6 Here we report the first Colombian patient with a novel heterozygous frameshift variant in the POGZ gene causing a clinical phenotype compatible with White–Sutton syndrome.
Case Presentation
A 20-year-old Colombian male was born to healthy parents and delivered at 40 weeks of gestation without complications. His mother and father were non-consanguineous and aged 27 and 35 years, respectively, at the time of birth. Birth weight was 3600 g (10th-25th percentile) and height was 51 cm (50th-75th percentile). No facial, phenotypic or neurological abnormalities were noticed at birth. The patient crawled and walked at 6 and 14 months, respectively. His family history was unremarkable.
At the age of 10 years, the patient was diagnosed with obesity. A neuropsychological test was performed at 14 years old, revealing an Intelligence Quotient (IQ) of 40, suggestive of moderately cognitive impairment. Between ages 15 and 16 years, Multiplex Ligation-dependent Probe Amplification (MLPA) for the 15q11.2 region and Array CGH KaryoNIM genetic testing were made with no abnormal results. Clinical exome sequencing (Illumina NGS technology) performed at 18 years old showed the heterozygous variant c.3308del (p.Leu1103Profs*19) in the POGZ gene (NM_015100.3), Subsequent Sanger sequencing confirmed, classified as likely pathogenic (criteria: PVS1, PM2, according to the College of Medical Genetics and Genomics ACMG)11 and was neither described in the literature to date nor reported in databases such as Clinvar, ExAC, 1000 genomes and HGMD. This mutation causes a frameshift with a premature stop codon, truncating the POGZ protein at aminoacid residue 1122. Analysis of translation-dependent Nonsense-Mediated Decay (NMD) using the NMDEscPredictor computational tool (https://nmdpredictions.shinyapps.io/shiny/)12 revealed that this variant escapes NMD (a manner in which the cells destroy aberrant RNA whose translation may lead to altered proteins), therefore is not likely a loss-of-function variant or null allele.
Physical examination at the age of 20 years showed delayed psychomotor development, intellectual disability, sleep difficulties, hypotonia, central obesity, genital features compatible with Prader stage III and testicular volume of 10mL, gynecomastia, acanthosis nigricans in the neck, visual abnormalities, strabismus, astigmatism and myopia. Dysmorphic features included hypotonic facies, epicanthus and bitemporal narrowing, curved eyebrows, midface hypoplasia, flat nasal bridge, broad nasal tip, open mouth, short philtrum, downturned corners of the mouth, long malar ridge, flat malar ridge, pointed chin, prognathism, high-arched palate, short neck and the presence of a mild brachycephaly cannot be excluded (Figures 1 and 2). Obsessive, hoarding behavior was observed. Cranial computed tomography, echocardiogram, audiometry and electroencephalogram were normal. No gastrointestinal, skeletal, cardiovascular, self-injurious behavior or severe autistic features were noticed. Weight was 111 kg (>97th percentile), height was 181 cm (50–75th percentiles) and head circumference was 55.6 cm (64th percentile). Paraclinical tests showed that LDL cholesterol (97 mg/dL), total cholesterol (156 mg/dL), Thyroid-stimulating hormone (1.4 mIU/L), free thyroxine (0.9 mcg/dl), total bilirubin (0.7 mg/dL), glycosylated hemoglobin (5.3%), triglycerides (101 mg/dL) and aspartate aminotransferase (20 U/L) were normal. Alanine aminotransferase (44 U/L) and free insulin (20 mcU/mL) were elevated.
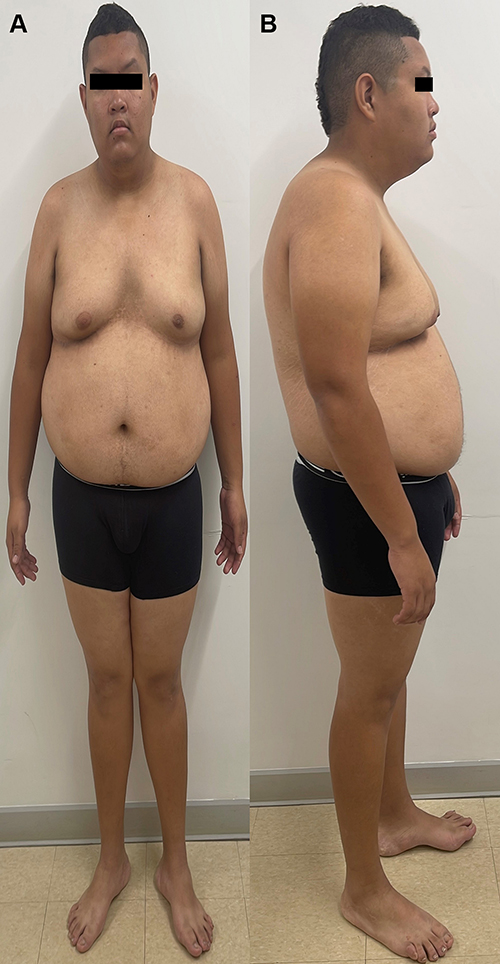 |
Figure 1 (A) frontal and (B) lateral view of the patient. The pictures were taken when the patient was 20 years old. |
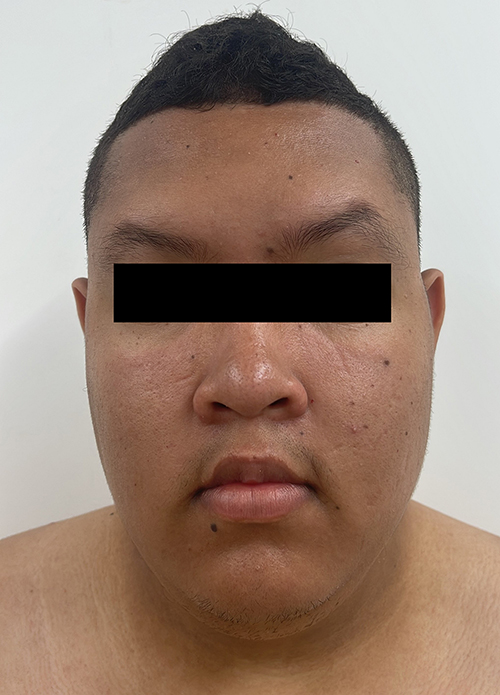 |
Figure 2 Frontal view of patient’s face. The picture was taken when the patient was 20 years old. |
Discussion
Neurodevelopmental disorders (NDD) are a group of pathologies with onset in the developmental period and include a wide variety of conditions such as intellectual disability, communication disorders, autism spectrum disorder (ASD), attention-deficit/hyperactivity disorder (ADHD), neurodevelopmental motor disorders and specific learning disorders.13 Latin America has the highest estimated prevalence of NDD, 33.4 (95% CI=28.9–38.0) per 1000 people, among low and middle-income countries.14 One particular and often problematic feature for the diagnosis of NDD is their considerable heterogeneous but also overlapping phenotypes, which is why the genetic diagnosis of NDD provides important information. The patient in this case had delayed psychomotor development, intellectual disability, sleep difficulties, hypotonia, central obesity, hypogonadism, visual abnormalities and several face dysmorphic features. He also had a finding of a likely pathogenic variant in the POGZ gene, classified as such because has not been reported in the literature so far and there is no experimental evidence of causality.
72 different variants, almost all de novo, have been reported in the POGZ gene of White–Sutton syndrome (WHSUS) patients6 spanning almost all the coding sequences of the gene and with 38 corresponding to frameshift variants. However, the most severe phenotypes seem to be associated with frameshift, nonsense and missense variants that escape NMD and are located in the proline-rich domain of POGZ protein.6 The frameshift variant found in our patient is located in the interdomain region between the Centromere protein (CENP)-B-DNA-binding domain and the DDE domain; it removes almost 288 aminoacid residues from the POGZ protein, although it escapes NMD and therefore can be functional to some extent. Taking together all symptoms and the lack of severe behavioral abnormalities, congenital malformations, seizures, brain architecture alteration, hearing problems and central nervous system abnormality, this patient had a mild WHSUS phenotype that could contribute to the delay in diagnosis compared to the median of reported genetic diagnosis (18 vs 6 years old, respectively).6
Only one patient has been reported to have a frameshift variant (c.3312del; p.Phe1104Leufs*18) in the same protein domain as our patient (c.3308del; p.Leu1103Profs*19). This 7-year-old male patient had mild intellectual disability (TIQ score 50–69), global developmental delay, gross motor developmental delay, speech delay, hypotonic facies, obstructive sleep apnea, vision problems, obesity and dysmorphic features such as brachycephaly and microcephaly, flat nasal bridge and overfield ears.15 Similar conditions were found in our patient except for brachycephaly, microcephaly, global developmental delay and gross motor developmental delay; which may be associated with the similar type of alteration in the POGZ gene in this and our patient. Furthermore, although monogenic defects leading to NDD are the minority of cases,13 some patients will benefit from genetic testing as this can influence the disease management, monitor possible complications and guide treatment. This case highlights the importance of genomic evaluation in such patients. Finally, one important limitation in this study is that no genetic testing has been performed on the patient’s parents, and therefore it is not possible to confirm whether this variant arose the novo in the proband.
Conclusion
Here we report a variant in the POGZ gene leading to White–Sutton syndrome. As far as we know, this is the first patient with WHSUS diagnosed in Colombia as well as the first time the variant c.3312del; p.Phe1104Leufs*18 in the POGZ gene has been reported. The characteristics of our patient are compatible with those described previously for patients with WHSUS and, as expected, they overlap with conditions of other NDD but also contribute to the phenotypic characterization of these patients. In this case, genetic testing allowed us to identify this rare condition in the patient that otherwise, would have been difficult to suspect or diagnose.
Abbreviations
WHSUS, White–Sutton syndrome; POGZ, pogo transposable element derived with ZNF domain; NDD, neurodevelopmental disorders; NMD, nonsense-mediated decay; ASD, autism spectrum disorder.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
This study was approved by the Ethics Committee of Fundación Valle del Lili, Colombia (human study protocol #1504) and performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from the patient. Information revealing the subject’s identity was not included in the manuscript. The patient was identified by number and not by his real name.
Consent for Publication
Written informed consent for publication of clinical details and images/photographs was obtained from the patient.
Acknowledgments
The authors would like to thank the patient for agreeing to the publication of this report. We also thank the people who have contributed to the development and execution of this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the manuscript; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to declare.
Disclosure
The authors report no conflicts of interest in relation to this work.
References
1. Fitzgerald TW, Gerety SS, Jones WD, et al. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi:10.1038/nature14135
2. Ye Y, Cho MT, Retterer K, et al. De novo POGZ mutations are associated with neurodevelopmental disorders and microcephaly. Mol Case Stud. 2015;1:a000455. doi:10.1101/mcs.a000455
3. Suliman-Lavie R, Title B, Cohen Y, et al. Pogz deficiency leads to transcription dysregulation and impaired cerebellar activity underlying autism-like behavior in mice. Nat Commun. 2020:11. doi:10.1038/s41467-020-19577-0.
4. Nozawa RS, Nagao K, Masuda HT, et al. Human POGZ modulates dissociation of HP1α from mitotic chromosome arms through Aurora B activation. Nat Cell Biol. 2010;12(7):719–727. doi:10.1038/ncb2075
5. Jones DO, Cowell IG, Singh PB. Mammalian chromodomain proteins: their role in genome organisation and expression. BioEssays. 2000;22(2):124–137. doi:10.1002/(SICI)1521-1878(200002)22:2<124::AID-BIES4>3.0.CO;2-E
6. Nagy D, Verheyen S, Wigby KM, et al. Genotype-phenotype comparison in POGZ-related neurodevelopmental disorders by using clinical scoring. Genes. 2022;13(1):154. doi:10.3390/genes13010154
7. White J, Beck CR, Harel T, et al. POGZ truncating alleles cause syndromic intellectual disability. Genome Med. 2016;8(1):1–11. doi:10.1186/s13073-015-0253-0
8. Stessman HAF, Willemsen MH, Fenckova M, et al. Disruption of POGZ is associated with intellectual disability and autism spectrum disorders. Am J Hum Genet. 2016;98(3):541–552. doi:10.1016/j.ajhg.2016.02.004
9. Trimarchi G, Caraffi SG, Radio FC, et al. Adducted thumb and peripheral polyneuropathy: diagnostic supports in suspecting white–sutton syndrome: case report and review of the literature. Genes. 2021:12. doi:10.3390/genes12070950.
10. Donnarumma B, Riccio MP, Terrone G, Palma M, Strisciuglio P, Scala I. Expanding the neurological and behavioral phenotype of White-Sutton syndrome: a case report. Ital J Pediatr. 2021;47(1):1–6. doi:10.1186/s13052-021-01101-9
11. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
12. Coban-Akdemir Z, White JJ, Song X, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet. 2018;103(2):171–187. doi:10.1016/j.ajhg.2018.06.009
13. Morris-Rosendahl DJ, Crocq MA. Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin Neurosci. 2020;22(1):65–72. doi:10.31887/DCNS.2020.22.1/macrocq
14. Bitta M, Kariuki SM, Abubakar A, Newton CRJ. Burden of neurodevelopmental disorders in low and middle-income countries: a systematic review and meta-analysis. Wellcome Open Res. 2017;2:121. doi:10.12688/wellcomeopenres.13540.1
15. Garde A, Cornaton J, Sorlin A, et al. Neuropsychological study in 19 French patients with White-Sutton syndrome and POGZ mutations. Clin Genet. 2021;99:407–417. doi:10.1111/cge.13894
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.