Back to Journals » International Journal of Nanomedicine » Volume 16
A Novel pH-Sensitive Multifunctional DNA Nanomedicine: An Enhanced and Harmless GD2 Aptamer-Mediated Strategy for Guiding Neuroblastoma Antitumor Therapy
Authors Zhang L, Wang M, Zhu Z, Ding C, Chen S, Wu H, Yang Y, Che F, Li Q, Li H
Received 19 January 2021
Accepted for publication 9 March 2021
Published 10 May 2021 Volume 2021:16 Pages 3217—3240
DOI https://doi.org/10.2147/IJN.S302450
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Liyu Zhang,1,2 Meng Wang,3 Zeen Zhu,4 Chenxi Ding,1 Shengquan Chen,1 Haibin Wu,2 Ying Yang,2 Fengyu Che,2 Qiao Li,5 Hui Li1,6
1Department of Neonatology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China; 2Shaanxi Institute of Pediatric Diseases, Affiliated Children’s Hospital of Xi’an Jiaotong University, Xi’an, People’s Republic of China; 3Department of Emergency Surgery, Shaanxi Provincial People’s Hospital, Xi’an, Shaanxi, People’s Republic of China; 4Department of Hepatobiliary Surgery, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi 710061, People’s Republic of China; 5Department of Clinical Laboratory, Affiliated Children’s Hospital of Xi’an Jiaotong University, Xi’an, People’s Republic of China; 6Department of Neonatology, Affiliated Children’s Hospital of Xi’an Jiaotong University, Xi’an, People’s Republic of China
Correspondence: Hui Li
Department of Neonatology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China
Email [email protected]
Background: GD2 is a mainstream biomarker for neuroblastoma (NB)-targeted therapy. Current anti-GD2 therapeutics exhibit several side effects since GD2 is also expressed at low levels on normal cells. Thus, current anti-GD2 therapeutics can be compromised by the coexistence of the target receptor on both cancer cells and normal cells.
Propose: Aptamers are promising and invaluable molecular tools. Because of the pH difference between tumor and normal cells, in this study, we constructed a pH-sensitive aptamer-mediated drug delivery system (IGD-Targeted).
Methods: In vivo Systematic Evolution of Ligands by Exponential Enrichment (SELEX) was used to generate a novel GD2 aptamer. Flow cytometry and molecular docking were applied to assess the binding specificities, affinities abilities of the aptamers. Confocal microscope, CCK8 assay, and BrdU assay were utilized to evaluate whether IGD-Targeted could only bind with GD2 at acidic environment. To evaluate whether IGD-Targeted could inhibit GD2-positive tumor and protect normal cells, in vivo living imaging, histomorphological staining, blood test, and RNA-sequencing were observed in animal model.
Results: GD2 aptamer termed as DB67 could bind with GD2-positive cells with high specificity, while has minimal cross-reactivities to other negative cells. It has been validated that the i-motif in IGD-Targeted facilitates the binding specificity and affinity of the GD2 aptamer to GD2-positive NB tumor cells but does not interfere with GD2-positive normal cells at the pH of the cellular microenvironment. In addition, IGD-Targeted is capable of delivering Dox to only GD2-positive NB tumor cells and not to normal cells in vivo and in vitro, resulting in precise inhibition of tumor cells and protection of normal cells.
Conclusion: This study suggests that IGD-Targeted as a promising platform for NB therapy which could show greater tumor inhibition and fewer side effects to normal cells, regardless of the existence of the same receptor on the target and nontarget cells.
Keywords: neuroblastoma, GD2 aptamer, pH-sensitive, i-motif, antitumor therapy
Introduction
GD2, a disialoganglioside, is one of the most ideal targets for targeted neuroblastoma (NB) therapy.1 GD2 is an acidic glycolipid that is biosynthesized from its precursor gangliosides GD3/GM3 by β-1,4-N-acetylgalactosaminyl transferase (GD2 synthase).2 Although the normal biological function of GD2 is not well understood, it is thought to play a role in neural differentiation, repair, invasion and motility enhancement, and immunosuppressive effects on effector cells.3 Generally, GD2 is expressed on the outer cell membrane of neural and mesenchymal stem cells.4 The expression of GD2 on the neuroblastoma tumor cell membrane is ubiquitous, with an estimated 5×106 molecules/cell.5 In addition, GD2 is highly expressed in other embryonal cancers, such as retinoblastoma, brain tumors, Ewing’s sarcoma, rhabdomyosarcoma, osteosarcoma, and even neural crest-derived cancers in adults.6 Thus, based on multiple criteria, GD2 is ranked 12th on a list of prioritized cancer antigens by the National Cancer Institute program. Moreover, unlike other tumor antigens, the expression of GD2 will not be influenced by therapy and can persist on the neuroblastoma cell membrane.7 Furthermore, circulating GD2 will not interfere with the binding between GD2-specific targeting molecules and the GD2 expressed on cells. Therefore, these properties make GD2 a promising target for NB-targeted therapy.8 To date, GD2 has been treated as an attractive antigen for NB drug development and has great potential to achieve substantial clinical benefits in NB therapy. Several anti-GD2 antibodies have been evaluated.8–10 Among them, dinutuximab, the first GD2 antibody approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for neuroblastoma treatment, has shown excellent effects on the OS rates of patients in several clinical trials.8,11,12 However, dinutuximab may also cause several side effects, with the most major adverse events including pain, fever, hypertension, and urticarial reactions.13–15 These symptoms can generally be controlled but sometimes they may not. In rare cases, side effects such as motor peripheral neuropathy, transverse myelitis, and unique toxicity due to the manifestation of internal ophthalmoplegia (mydriasis and loss of accommodation) have also been reported.16 The root of these side effects is that although GD2 is highly expressed on NB tumor cells, it is unfortunately also expressed on peripheral neurons, in the central nervous system, and on skin melanocytes among normal tissues.17 Current GD2 monoclonal antibodies are incapable of distinguishing between GD2 expressed on normal cells from that expressed on the tumor cell membrane; thus, GD2 antibodies could not only damage tumor cells but also cause cytotoxicity to normal cells. Therefore, to improve the safety and effectiveness of anti-GD2 therapeutics, a novel GD2-targeting strategy is urgently needed so that the GD2 expressed on NB tumor cells is exclusively affected.
The growth rate of tumor cells is much faster than that of normal cells, resulting in insufficient nutrients and blood oxygen for tumor growth. Thus, for adequate growth, tumor cells have to survive through anaerobic glycolysis. Based on this mode of metabolism, tumor cells will produce large amounts of lactic acid and adenosine triphosphate (ATP) hydrolysates, as well as excess carbon dioxide and protons in the cytoplasm.18 To avoid the toxicity of the acidic environment and to maintain a normal pH, tumor cells will excrete more H+ outside of the cells, resulting in an acidic extracellular environment and an alkaline intracellular environment.19 In general, the extracellular pH values of normal tissues are approximately 7.2–7.4, while those of tumor tissues and tumor cells are approximately 6.5–6.9.20 Due to the abnormal pH in tumor cells, which is conducive to tumor proliferation, invasion, and adhesion, this characteristic presents a chance to design endogenous tumor-specific intelligent targeting agents. To make therapeutic agents recognize and function in only tumor cells, it is necessary to design these agents in an “off” state in the normal tissue environment that turns “on” after being ingested by tumor cells.21 It has been predicted that this approach has higher therapeutic sensitivity and selectivity for tumor cells as well as fewer side effects to normal tissues.
Several studies have explored molecular probes and therapeutics with pH stimuli-responsive properties for cell-specific imaging and biomedical applications. In particular, the i-motif, which is a quadruplex structure formed by the chimerism of cytosine-cytosine base pairs in an antiparallel manner, has been explored.22 Since the i-motif structure is stabilized by the formation of hydrogen bonds between protonated cytosine and non-protonated cytosine, its spatial structure is greatly affected by the pH value.23 The i-motif structure is mostly formed and stabilized under acidic conditions but not under neutral or alkaline conditions. Thus, the i-motif has been extensively used in DNA nanotechnology for molecular sensing and therapeutic applications.24 Kan et al developed a bimolecular i-motif-mediated FRET strategy for the accurate and dynamic in situ imaging of Met homodimerization on a living tumor cell surface.25 Lee et al constructed i-motif-based pH-responsive hyaluronic acid/polyethylenimine complexes for selective drug delivery.26 This complex demonstrated potential for pH-responsive anticancer drug delivery systems. Thus, the i-motif has been validated as an attractive and powerful tool for tumor-specific therapeutic strategies.27 Therefore, in this study, we tried to explore the potential of i-motif-functionalized targeting molecules that are able to distinguish between GD2 expressed on tumor cells from GD2 expressed on normal cells.
Targeted therapeutics have undergone a revolution with the entry of monoclonal antibodies into the medical toolkit. In addition to antibodies, aptamers, which are targeting DNA or RNA molecules synthesized via straightforward phosphoramidite chemistry, have been rapidly developed in recent years.28,29 Aptamers can display several defined secondary motifs and further form complex three-dimensional structures to confer these molecules the ability to recognize and bind to targets with high affinity and specificity.28 Consequently, they are also known and function as “chemical antibodies”. When compared with antibodies, aptamers have unique advantages as targeting molecules due to their high affinity for binding to target molecules, limited synthetic cost, small sizes that allow them to penetrate solid tumors and even the blood-brain barrier, non-immunogenicity that facilitates long-term therapeutic efficacy and safety, and geometrical conformational flexibility and synthetic dynamics for easy synthesis and chemical modification for various therapeutic applications.30–32 Thus, so far, however, there has been no literature reported about GD2 aptamer and GD2 aptamer-mediated drug delivery system. Generally, a typical SELEX process usually treat a pure protein or whole cells as targets. To remove non-specific sequences, bovine serum albumin (BSA) protein that usually treated as negative protein or control cells without targets expressed were applied for negative selection. However, some aptamers may only recognize their targets in vitro and may not function well in vivo. Aptamers selected in vitro may be challenged in vivo: ① the spatial structure of targets in the physiological environment may be different when compared with recombinant proteins as well as in cells in vitro; ② Aptamers will face a more complex environment, including nuclease digestion and temperature. Aptamer may be destroyed quickly in vivo. All these factors are challenges for aptamers to identify targets in vivo. Only aptamers still applicable in vivo are promising for targeting therapeutics. Therefore, in this study, we have successfully selected a novel GD2 aptamer via the in vivo SELEX technique. This GD2 aptamer was termed DB67 and could recognize and bind to GD2-positive NB cells with high specificity and affinity. However, DB67 could not distinguish between the GD2 expressed on normal cells from that expressed on NB tumor cells. Since the extracellular pH of tumor cells is acidic and the i-motif can play a role in acidic environments, we tried to explore whether the combination of DB67 and the i-motif has the potential to turn the GD2 aptamer DB67 “on” or “off” to distinguish between the GD2 expressed on NB cells from that on normal cells according to the different pH values.
We first explored the core sequence of DB67 for GD2 recognition and binding as noted above. Utilizing a molecular docking technique, the core region of DB67 that binds to GD2 was predicted. According to the prediction results, DB67 was truncated into sequences of different lengths to determine which sequence exhibited the best binding specificity and affinity. Flow cytometry was applied to evaluate the binding specificity of these truncated GD2 aptamers. It was demonstrated that the core sequence of DB67 was 27 nt in length, and this sequence was termed as DB67-active. Further, DB67-active was designed into an i-motif-modified aptamer containing both DB67-active and the i-motif structure. As shown in Figure 1, a split i-motif was integrated into the aptamer DB67-active. To avoid interference caused by steric hindrance between DB67-active and the i-motif, a linker zone was designed between these two moieties. Thus, this modified aptamer, termed IG-Targeted, consists of three functional regions that maintain both the binding ability of DB67-active and the pH-sensitive features of the i-motif. These regions are the GD2 recognition domain, the linker zone, and the split i-motif. Additionally, doxorubicin (Dox), a typical NB chemotherapeutic agent, can forcefully kill cancer cells but also brings many unexpected side effects (eg, tissue necrosis) due to its nonspecific distribution. To avoid its cytotoxicity to normal cells, dox should only be released around tumor cells and in presence with GD2. Since Dox can intercalate into C/G base pairs and the i-motif has an abundance of C bases, the i-motif sequence in IG-Targeted was paired with its complementary strand, resulting in an abundance of C/G base pairs for Dox loading. IG-Targeted loaded with Dox was termed as IGD-Targeted. As shown in Figure 1, at physiological pH, the i-motif is unstructured, resulting in a disrupted DB67 active structure that prevents the GD2 recognition domain from forming the correct three-dimensional structure for GD2 recognition. Dox is still retained in IG-Targeted and cannot be released. However, in acidic environments, the i-motif can fold into the correct three-dimensional structure, causing the structure of the GD2 recognition domain to be unrestricted, further enabling restoration of the targeting ability of IGD-Targeted. After the i-motif is produced, the dsDNA of the i-motif and complementary strand denatures, causing the dissociation and accumulation of Dox around NB tumor cells, causing further damage. We explored the binding specificity and affinity of IG-Targeted at different pH values, and IG-Targeted can recognize and bind to the GD2 structure in an acidic environment but not in a physiological pH environment. Additionally, IGD-Targeted effectively inhibited GD2-positive NB tumor cells but did not generate severe side effects in vitro and in vivo. Overall, these results indicated the potential of utilizing the i-motif to enhance the targeting specificity toward tumor cells in acidic environments while eliminating damage to normal cells that express the target antigens.
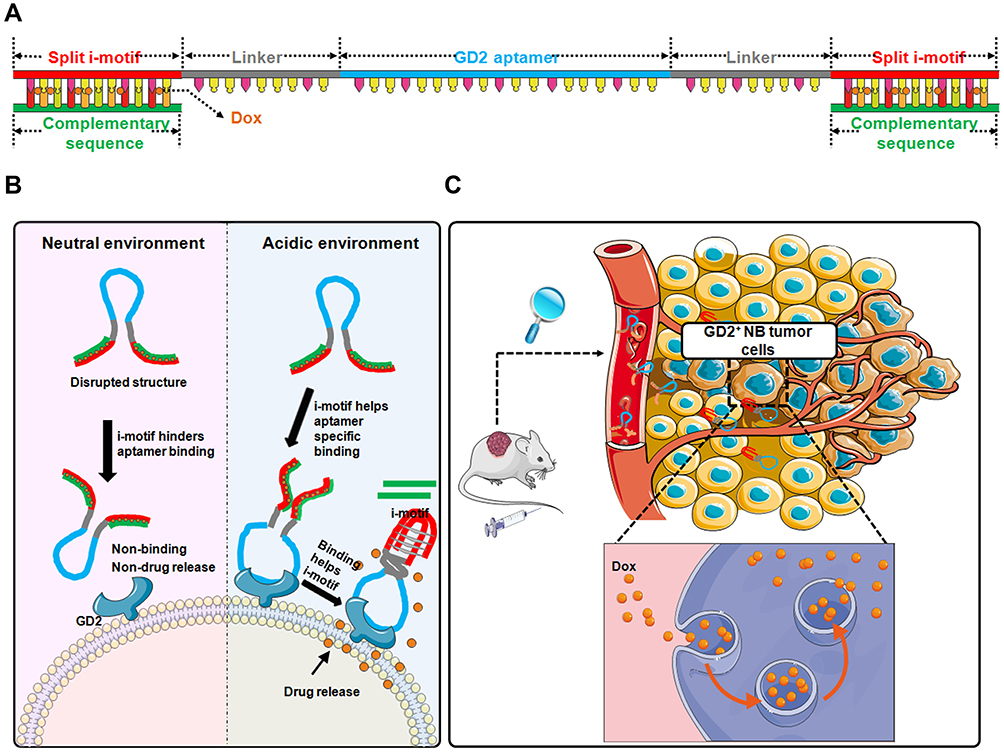 |
Figure 1 Schematic illustration of pH-sensitive multifunctional DNA nanomedicine for guiding neuroblastoma anti-tumor therapy. (A) Construction of IGD-Targeted. IGD-Targeted contained a split i-motif, GD2 aptamer, and linker. i-motif sequences could pair with complementary sequences and further for Dox loading. (B) pH-sensitive IGD-Targeted resulting in different targeting ability to GD2. When in physiological pH, i-motif was unstructured, Dox could not be released, resulting in disrupted GD2 aptamer structure and further preventing GD2 recognition domain to form the correct three-dimensional structure for GD2 recognition. When in acidic environment, the structure of i-motif was changed and the structure of GD2 recognition domain was not restricted and further enabling the restoration of its targeting ability. The three-dimensional formation of GD2 aptamer further helped correct construction of i-motif, the complementary pairing was destroyed and Dox was released. (C) Anti-cancer of IGD-Targeted in vivo. When IGD-Targeted was injected in tumor-bearing mice, since the pH value around tumor tissues was acidic, i-motif structure could form, IGD-Targeted could further recognize GD2, and Dox could be released into tumor cells. Normal tissues could be protected due to neutral pH value. |
Materials and Methods
Materials and Reagents
All ssDNA, modified ssDNA, and primers were synthesized by Sangon Biotech (Shanghai China). Bovine serum albumin (BSA, Sigma-Aldrich, Japan, Catalogue#: V900933), trypsin (Sigma-Aldrich, Japan, Catalogue#: T2600000), IgG (Sigma-Aldrich, US, Catalogue#: 14506), MTS cell proliferation colorimetric assay Kit (Abcam, UK, Catalogue#: ab197010), cell counting kit 8 (CCK8, Sigma-Aldrich, US, Catalogue#: 96992), BrdU cell proliferation ELISA Kit (Abcam, UK, Catalogue#: ab126556), Doxorubicin (Dox, Sangon Biotech, Shanghai, China, Catologue#: A603456), DMEM (Gibco®, Life Technologies, Carlsbad, CA), fetal bovine serum (FBS, Gibco®, Life Technologies, Carlsbad, CA), bovine serum albumin (BSA, Fisher Scientific), yeast tRNA (Sigma-Aldrich, St. Louis, MO, USA), MTS CellTiter reagent (Promega, USA).
Cell Lines and Cell Culture
GD2 positive Human neuroblastoma cell line IMR32[American Type Culture Collection (ATCC) no. ATCC® CCL-127™] and GD2 negative epidermoid carcinoma cell line A431 [American Type Culture Collection (ATCC) no. ATCC® CRL-1555™] were both purchased from ATCC (Manassas, VA, USA). Healthy cell lines including human normal pulmonary epithelial cells BEAS2B (BFN6080086), human hepatocytes cell line L-02 (BFN608006124), human hepatocytes cell line LX-2 (BFN608006123), human hepatocytes cell line THLE-3 (BFN608006122), human retinal epithelial cells RPE (BFN60807591), and human prostatic epithelial cells RWPE-1 (BFN60800802) were obtained from BLUEFBIO (Shanghai, China). Adhesion cells were cultured with DMEM with 10% fetal bovine serum and a mixture of penicillin/streptomycin. Cells were seeded on culture plates at a density of 2×105 cells/mL and cultured at 37°C in a humidified atmosphere with 5% CO2. All experiments were performed on cells in the exponential growth phase.
GM2/GD2 Synthase Knock Down of IMR32 Cell Line and Activation of A431 Cells
Cells were seeded in 6-well culture plate with density/well of 1.5×105-2.5×105 cells in 3 mL of antibiotic-free standard growth medium 24 h before transfection.
For IMR32 cells: the siRNA/miRNA oligo solution was added to 100 μL Opti-MEM at room temperature for 5 min. Two microliters siRNA-Mate was added immediately. After incubating for 5 min, this complex was added into cells drop by drop. Cells were incubated for 24–96 hours. IMR32 cell that knock down GM2/GD2 synthase was termed as KD-IMR32.
For A431 cells: prepare a mixture of complete medium with Polybrene® (sc-134220) at a final concentration of 5 µg/mL. Remove media from plate wells and replace with 3 mL of this Polybrene® media mixture per well (for 6-well plate). Remove the culture medium and replace with 3 mL of complete medium (without Polybrene®). To select stable activated clones, split cells 1:3 to 1:5, depending on the cell type, and continue incubating for 24–48 hours in complete medium Select activated clones via Puromycin dihydrochloride (sc-108071), hygromycin B (sc-29067) and blasticidin S HCl (sc-495389) selection. Use an amount sufficient to kill the non-transduced cells. Replace medium with fresh selective antibiotic-containing medium every 3–4 days. Pick stable target gene activation colonies and termed as GD2-positive A431.
In vivo SELEX and Evaluation
The protocol of the animal study in this paper was reviewed and approved by the Ethics Committee of Xi’an Jiaotong University Affiliated Children’s Hospital (Xi’an Children’s Hospital, Xi’an, China), no. C2018004. Six- to eight-week-old female immunodeficient mice were purchased from the Xi’an Jiaotong University Lab Animal Centre (Xi’an) and raised under pathogen-free conditions. For targeting ability evaluation assay, mice were anesthetized with 2% vaporized isoflurane, and 1.5×106 GD2-positive cells IMR32 were subcutaneously inoculated. After 14 days, mice were used for SELEX. In total, 80 mice were prepared for SELEX. The random DNA pool thiophosphorylated by 5ʹ-monothiophosphate substitutions of all dA was synthesized. 20 nmol of DNA aptamer pool in 200 μL saline was administered through tail vein injection. For each round, seven mice were used for selection. Mice were sacrificed and tumor tissues were isolated. DNA was extracted and amplified with forward primer (5ʹ-TGCGTGTGTAGTGTGTCTG-3ʹ) and biotin-labeled reverse primer (5ʹ-CCGCCCAAATCCCTAAGAG-3ʹ). DNA pool was enriched by PCR amplification. PCR products were assessed by flow cytometry by incubating with GD2-coated beads. When there was a strong signal of binding, sequences were clones. Briefly, the selected ssDNA pool was PCR-amplified using unmodified primers and cloned into Escherichia coli with TA cloning kit (Cat# CT101) for DNA sequencing. The binding specificities of each clone were detected by flow cytometry. Separated FAM-ssDNA was incubated with IMR32 cells or A431 cells at 37°C for 30 min. Cells were washed and evaluated.
The Active Sites Prediction and Dynamics Simulation of Aptamer to Target by Molecular Docking
With the three-dimensional structure of DNA short hairpin as the ligand and the target protein as the receptor, the conformation of the interaction between the ligand and the receptor was searched by NPDock program, and the relevant parameters of NPDock were set for docking. The detailed parameters of NPDock were shown as follows: clustering model: 20000; Clustering the best score model: 100; RMSD cut-off for clustering [Ångström]: 0.5 angstrom; RMSD: 0.9 Å; Steps of simulation: 1000; Temperature of the first step of simulation [in Kelvin]: 15000 K; Temperature of the last step of simulation [in Kelvin]: 295 K. In the complex structure model of ligand and target protein generated in each docking, the result with the best score and reasonable conformation was selected according to the statistical analysis of the most dominant conformation. In view of the rough degree of homologous modeling results, a certain molecular dynamics simulation analysis is needed, and the modeling results are optimized to obtain a relatively reliable three-dimensional structure of protein. The number of molecular kinetic simulations, for the sake of dynamic reliability, it is necessary to repeat a molecular dynamics simulation experiment in order to achieve the most and least controversial protein structure. After structural overlap and completion, the conformational proteins were optimized by molecular dynamics (6 ns) until the structure was balanced. After the molecular dynamics’ equilibrium, the average structure was selected for later analysis and docking.
Binding Specificity and Affinity of Truncated GD2 Aptamer
According to the result of molecular docking, GD2 aptamer was truncated into different fragments (Table 1). These fragments were synthesized by Sangon and were modified with FAM at 5ʹ end. These modified truncated aptamers were incubated with 1×106 IMR32. Random DNA was treated as negative control. Cells were washed by PBS twice and FAM fluorescence was determined with a FACS caliber cytometer (BD).
 |
Table 1 Sequences of Truncated Aptamers |
To evaluate binding affinities of truncated GD2 aptamer, various concentrations of aptamer were incubated with IMR32 cells at 37°C in PBS for 30 min. Cells were washed by PBS twice and subjected to flow cytometric analysis. The mean fluorescence intensity of target labeled by aptamers was used to quantify specific binding by subtracting the mean fluorescence intensity of non-specific binding to negative cells. The dissociation constants (Kd) of the aptamer-cell interaction were calculated by the equation Y = BmaxX/(Kd + X), where Y represented the reciprocal of the average fluorescence intensity, X represented the reciprocal of GD2 aptamer’s concentration, and Bmax represented the maximum binding capacity of aptamer bound to GD2-positive cells.33
Preparation of Buffers at Different pH
The basic binding buffer with neutral pH (pH 7.4) was prepared from Dulbecco’s Phosphate-Buffered Saline (PBS) with 5 mM MgCl2, 2 g/mL BSA, and 100 mg/L yeast tRNA. To adjust to the required pH values, 100 mM HCl and/or NaOH was applied and the pH was measured using a calibrated pH meter (pH 6.5).
Selection of i-Motif Sequences
Biotin-labeled GD2 molecule was fixed on streptavidin-coated magnetic beads. Several combination sequences containing GD2 aptamer and i-motif were listed in Supplementary Figure S1B. Sequences ①, ②,③, ④, ⑤, ⑥ (5 mM) were labeled with TARMA and were mixed with complementary sequence of i-motif at equal concentration at 37°C for 30 min, respectively. Dox (5 mM) were added and intercalated. Next, the constructed sequences were mixed with GD2-coated beads or null-GD2 beads at either pH 7.4 buffer or pH 6.5 buffer. Bull-GD2 beads treated as control were beads without GD2 coating. Sequences were incubated with beads at 37°C for 30 min, and beads were separated under magnetic field. To assess the GD2 binding specificity, beads were detected by flow cytometry; To assess the i-motif formation and Dox releasing, supernatant fluid were evaluated under Synergy4 analyzer (UK) (Ex=488 nm, Em=590 nm). The data were calculated and analyzed by fluorescence value ratio of GD2 group/control group.
Construction of IG-Targeted
ssDNA strands containing GD2 recognition domain, linker zone, and split i-motif were synthesized (see Table 2 for detailed sequences). Sequences of complementary sequences for split i-motif were presented in Table 2. ssDNA strands and complementary sequences were mixed at proper ratio in annealing buffer (10 mM Tris-HCL, 1 mM ethylenediaminetetraacetic acid, 150 mM sodium chloride, pH 8.0). The mixed solution was heated at 95°C for 5 min and cooled on ice immediately. IG-Control, which was constructed with control aptamer (could not recognize GD2) and i-motif, was treated as control group to test GD2 binding specificity of IGD-Targeted; C-Targeted, which was constructed with GD2 aptamer and non-i-motif sequence, was treated as control group to test the i-motif structure formation of IGD-Targeted (Table 2).
 |
Table 2 Sequences and Modifications of Truncated Aptamers |
Evaluation of IG-Targeted Specificity
ssDNA strands with different length and sequences were all modified with fluorescent groups and quenching groups, briefly, with TAMRA modified at 5ʹ end. Modified ssDNA strands were mixed with complementary sequences to construct IG-Targeted. Then, IG-Targeted was incubated with IMR32 or A431 in prepared buffers at different pH (6.5 and 7.4) at 37°C for 30 min. Cells were washed by PBS with equivalent pH and evaluated by flow cytometry. C-Targeted and IG-control were treated as control group.
Evaluation of IG-Targeted Formation Mechanism
IG-Targeted was modified with fluorescent groups and quenching groups, briefly, with TAMRA modified at 5ʹ end and BHQ-2 modified at 3ʹ end. IG-Targeted was treated in various pH environments (pH 6.5–7.5) in the presence of GD2 and the fluorescence was monitored by fluorescence spectrometry by Synergy4 analyzer (UK) (Ex=560 nm, Em=580 nm). IG-control (i-motif with control aptamer that could not bind with GD2) was used as negative control.
Drug Loading for IGD-Targeted
IG-Targeted with a concentration gradient was incubated in an aqueous solution of Dox (5 nM) for 30 min in a black 96-well plate at a range of aptamer/dox molar ratios in the presence of GD2 molecules (10 μM). The fluorescence of Dox was monitored by Synergy4 analyzer (UK) (Ex=488 nm, Em=590 nm). The same method was used for Dox loading of C-Targeted, which termed as CD-Targeted.
Cytotoxicity Study of IGD-Targeted in vitro
Targeting Drug delivery study assessed by confocal microscope: IMR32, KD-IMR32, A431, and GD2-positive A431 cells were seeded on 6-well plate and incubated with IGD-Targeted at 37°C for 1 h at either pH 7.4 buffer or pH 6.5 buffer, respectively. Dox in cells were scanned with a Leica TCS SP5 confocal microscope (Leica Microsystems).
IMR32 cells and A431 cells were seeded in 96-well plate (5×104 cells per well). Both cells were treated with various concentration of IGD-Targeted (10 nM, 20 nM, 50 nM, 100 nM, 200 nM, 500 nM) at 37°C for 6 h at either pH 7.4 buffer or pH 6.5 buffer. Then, after removing supernatant, cells were washed with PBS buffer and cultured for a further 96 h. Cytotoxicity study assessed by MTS: 20 µL MTS reagent was added into each well for 4 h incubation. The absorbance (490 nm) of each well was determined to evaluate the cell viability per standard protocol outlined by the manufacturer's instruction. Cytotoxicity study assessed by CCK8: 10 µL CCK8 reagent was added into each well for 4 h incubation. The absorbance (450 nm) of each well was determined to evaluate the cell viability per standard protocol outlined by the manufacturer's instruction.
Cell proliferation assessed by BrdU ELISA: IMR32 cells, A431 cells, and 14 kinds of healthy cell lines (BEAS2B, CCD-8L, 6T-CEM, L-02, LX-2, THLE-3, RPE, D407, CRL-2713, RWPE-1, CRL-7601, GP-293, hFOB 1.19, and HMy2.CIR) were seeded in 96-well plate (5×104 cells per well). All cells were treated with various concentration of IGD-Targeted (10 nM, 20 nM, 50 nM, 100 nM, 200 nM, 500 nM) at 37°C for 1 h at either pH 7.4 buffer or pH 6.5 buffer. Then, after removing supernatant, cells were washed with PBS buffer and cultured for a further 96 h. BrdU buffer was added and incubated for 24 h. Then, remove the incubation buffer and added 100 μL fixed buffer for 30 min. Remove supernatant fluid and added 100 μL BrdU antibody buffer for 1 h. 300 μL washing buffer was added for three times. HRP-conjugated goat anti-mouse antibody was added and incubated for 1 h. Cells were washed three times and finally added with 100 μL TMB. After 15 min, 50 μL terminating buffer was added and the absorbance (450 nm) of each well was determined.
Targeting and Inhibition Evaluation of IGD-Targeted in vivo
The protocol of the animal study in this paper was reviewed and approved by the Ethics Committee of Xi’an Jiaotong University Affiliated Children’s Hospital (Xi’an Children’s Hospital, Xi’an, China), no. C2018004. Four- to six-week-old female BALB/c (nu/nu) athymic nude mice were purchased from the Xi’an Jiaotong University Lab Animal Centre (Xi’an) and raised under pathogen-free conditions. 1.5×106 IMR32 cells were administered into the flank by subcutaneous injection. After 14 days of cell injection, mice were subjected to further experiments. All animal procedures were performed according to the standards of the Institutional Animal Care and Use Committee. Firstly, to evaluate whether IGD-Targeted could only deliver Dox to GD2-positive tumor cells, IGD-Targeted was injected in tumor-bearing mice. 30 min later, the fluorescence signal of Dox was detected by the IVIS 200 Imaging System to image whole body. Further, to assess whether IGD-Targeted could prolong survival rate and inhibit tumor growth, tumor-bearing mice were randomly divided into four groups, with six mice in each group: ① treated with saline (10 μL); ② treated with IGD-Targeted (2 mg/kg); ③ treated with CD-Targeted (C-Targeted loaded with Dox, 2 mg/kg); ④ treated with free Dox (2 mg/kg). The Dox loading ability of IGD-Targeted and CD-Targeted were similar, so they were injected with same volume. Mice were injected with agents by s.c. every two days for 10 days, and followed by observation until all mice died. Live mice number was recorded every day and the survival rate in each group was calculated using the following equation: Survival rate=the number of live mice/the number of total mice. The mice were weighed every 3 days. After mice were died, tumor tissues were separated and imaged. To further assess the anti-cancer and specificity ability of IGD-Targeted, tumor tissues in saline, CD-Targeted, and IGD-Targeted group were submitted to RT-PCR (Caspase-9, Bcl-2, RPS27L, PPMID, SESN1, FAS, and β-actin) and transcriptomic analysis using the Novaseq 6000 (Illumina, US). Transcriptomic data were analyzed using Heatmap software. RT-PCR primers: Caspase-9 (Forward,5ʹ-GGCTGTCTACGGCACAGATGGA-3ʹ;Reverse,5ʹ-CTGGCTCGGGGTTACTGCCAG-3ʹ);Bcl2(Forward,5ʹ-CTGAGTACCTGAACCGGCACC-3ʹ;Reverse,5ʹ-GAGCAGAGTCTTCAGAGACAG-3ʹ);RPS27L(Forward,5ʹ-TGCCGAGCGCAGATCGCTTG-3ʹ;Reverse,5ʹ-TGGCCTTTCCTCCTGTAGGCTGG-3);SESN1(Forward,5ʹ-CTTCTGGAGGCAGTTCAAGC-3ʹ;Reverse,5ʹ-TGAATGGCAGCCTGTCTTCAC-3);PPM1D(Forward,5ʹ-TTCTCGCTTGTCACCTTGCC-3ʹ;Reverse,5ʹ-CCAAACTACACGATTCACCCC-3);FAS(Forward,5ʹ-GTGGGTAAAGAAGCCAAACG-3ʹ;Reverse,5ʹ-TCAAAAGCACCAGTTCACAGA-3ʹ);β-actin(Forward,5ʹ-CAGCCTTCCTTCTTGGGTAT-3ʹ; Reverse, 5ʹ-TGGCATAG AGGTCTTTACGG-3ʹ). β-actin was treated as internal reference gene.
Assessment of IGD-Targeted’s Normal Cells Protection Ability
To assess the targeting ability of IGD-Targeted to protect normal cells, tumor-bearing mice were randomly divided into four groups, with six mice in each group: ① treated with saline (10 μL); ② treated with IGD-Targeted (2 mg/kg); ③ treated with CD-Targeted (C-Targeted loaded with Dox, 2 mg/kg); ④ treated with free Dox (2 mg/kg). Mice were injected with agents by s.c. every two days for 9 days, and followed by observation for 5 days. Tumor volume values were calculated every three days. Subsequently, the mice were sacrificed, and the tumors and vital organs (lung, liver, spleen, kidney, and heart) were collected for analysis. The organ tissue and tumor tissues were divided into two parts: one part was grinded for Dox evaluation. The grinded products were dissolved in saline. The Dox concentration of these products and mice urine were assessed by Synergy4 analyzer (UK) (Ex=488 nm, Em=590 nm); The other part of tissues were collected and subjected to hematoxylin and eosin (H&E) staining to evaluate elimination ability of any adverse effects of IGD-Targeted on normal tissues. Additionally, whole blood was collected in BD Microtainer® blood collection tubes with K2EDTA (Becton, Dickinson and Company, USA). Blood samples were subjected to blood routine examination and to measure serum markers of organ damage [aspartate aminotransferase (AST) for liver, alanine aminotransferase (ALT) for liver, creatinine for kidney, and blood urea nitrogen (BUN) for kidney]. Blood routine examination was detected by automatic hematology analyzer (BC-2800vet, Mindray). There were 15 parameters evaluated in blood routine examination: white blood cell count (WBC), red blood cell count (RBC), granulocyte count (Gran#), mononuclear cell count (Mon#), platelet count (PLT), red blood cell specific volume (HCT), the level of hemoglobin (HGB), lymphocyte count (Lymph#), the level of mean corpuscular hemoglobin (MCH), lymphocyte percentage (Lymph%), monocytes percentage (Mon%), granulocyte percentage (Gran%), mean red blood cell volume (MCV), red blood cell distribution width (RDW), and mean platelet volume (MPV). Additionally, blood samples were also subjected to serum markers of organ damage [creatinine, aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatinine, and blood urea nitrogen (BUN)]. Evaluations were operated according to assay kit instruments.
To evaluate the application potential of IGD-Targeted, the anti-cancer ability was compared with GD2 antibody Dinutuximab. 1.5×106 IMR32 cells were administered into the flank by subcutaneous injection to BALB/c (nu/nu) athymic nude mice (Four- to six-week-old). Mice were kept with regular circadian rhythm light. After 14 days of cell injection, mice were subjected to further experiments. Mice were divided into three groups: ① treated with saline; ② treated with IGD-Targeted (2 mg/kg); ③ treated with GD2 antibody Dinutuximab (15 µg/mouse). Agents were injected via tail vein every 2 days for 8 days and further observed for 7 days. Tumor volume values were calculated every three days to assess the anti-cancer ability of IGD-Targeted compared with Dinutuximab. Additionally, to further confirm IGD-Targeted’s anti-cancer ability and protection to normal tissues, four- to six-week-old female BALB/c (nu/nu) athymic nude mice were kept with regular circadian rhythm light. Mice were divided into three groups: ① treated with saline; ② treated with IGD-Targeted (low dose: 1 mg/kg, mediate dose: 2 mg/kg; and high dose: 20 mg/kg); ③ treated with GD2 antibody Dinutuximab (low dose: 5 µg/mouse; mediate dose: 15 µg/mouse; and high dose: 25 µg/mouse). The self-harm behavior of mice was recorded. Generally, self-harm generally starts from biting toes. Scores were calculated as followed: if one or more toes were bite, added 1 point; the distal end of each foot is bitten off plus one point, and the proximal end is bitten off plus another point.
Statistical Analysis
SPSS11.0 software was applied from Xi’an Jiaotong University. All numerical data were expressed as the mean±SD. Differences between the groups were examined with Student’s two-tailed t-test. A p value of <0.05 was identified as statistical difference.
Results
Aptamer Selection by in vivo SELEX and Aptamer Binding Specificity Evaluation
The process of in vivo SELEX is presented in Figure 2A. Firstly, thiophosphorylated random DNA pool was performed on tumor-bearing mice whereby nude mice bearing a previously implanted human xenograft through tail vein injection. Unbounded sequences may circulate and eliminated by kidney and liver, whereas binding clones could target and bind with tumor tissues. Then, mice were sacrificed and tumor tissues were harvested. DNA was extracted and amplified. To monitor selection effect of each round, each round DNA pool was amplified and labeled by FAM. Modified DNA was incubated with GD2. As shown in Figure 2B, with the process of SELEX, there was more DNA that could bind with GD2. When in 9th round, the fluorescence signal reached the most. DNA sequences were cloned and assessed further for binding specificities. Ninety-nine clones were performed for further function evaluations. Aptamers were incubated with GD2-positive cell line IMR32 and GD2-negative cell line A431. As shown in Figure 2C, when compared with random DNA pool, DB67 generated a relatively stronger signal in GD2-positive cell lines, whereas showed little binding to GD2-negative cells, indicating a relatively strong binding specificity to GD2.
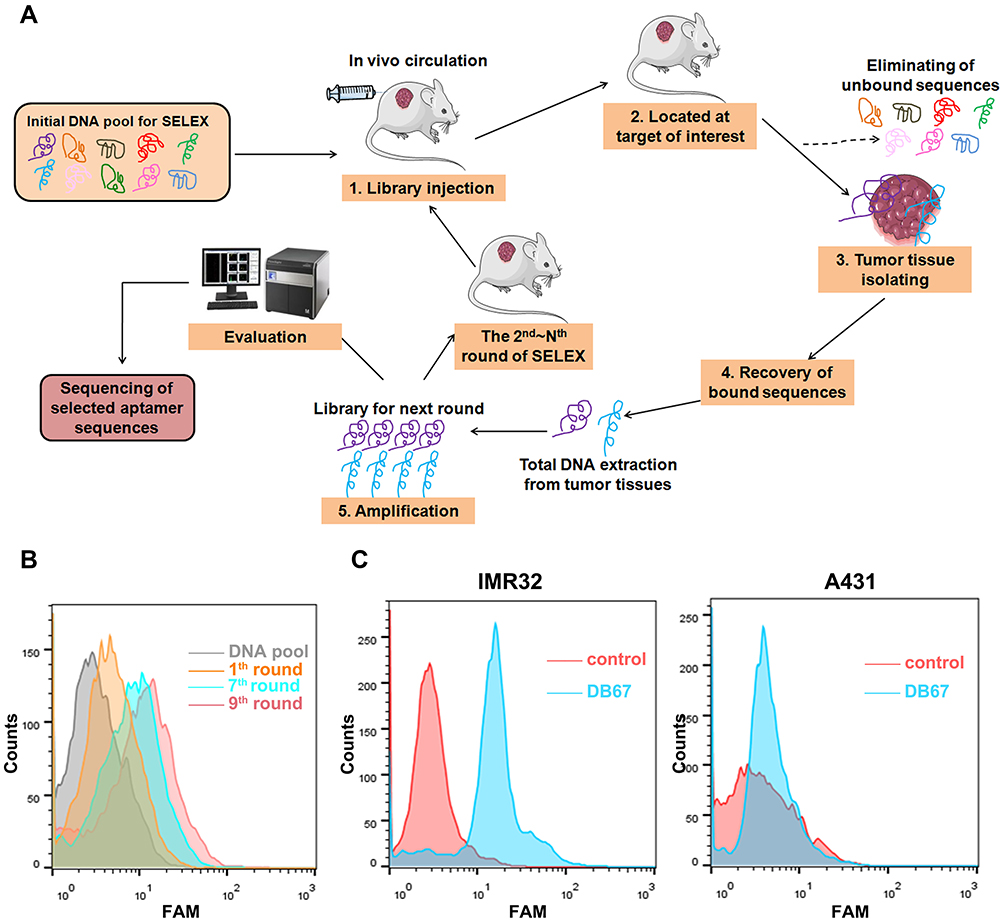 |
Figure 2 Selection and evaluation of GD2 aptamer. (A) In vivo SELEX process. DNA pool was administered through tail vein injection to GD2+ tumor-bearing mice. DNA pool could distribute rapidly ssDNA were extracted and amplified by PCR. PCR products were preceded to next selection part. (B) Flow cytometry monitoring of selection effect. Selected pool of each round was amplified and labeled by FAM, and further incubated with GD2-coated beads. Fluorescence signal was assessed. (C) Flow cytometry assessment of aptamer for GD2 binding specificities. FAM-DB67 was incubated with GD2+ cells IMR32 and GD2– cells A431, respectively. Fluorescence signal was assessed. |
The Core Region of DB67 Can Bind to GD2 with High Specificity and Affinity
Although aptamers generally consist of nearly 60–100 nucleotides, only a few of these exhibit main roles in target recognition and binding. When aptamers fold into their three-dimensional structures, only the key nucleotides form a unique central tertiary structure from which their function emerges, as the other residues are not necessary for target recognition. To explore the key region of DB67 that recognizes GD2, molecular docking technology was utilized to simulate the interaction between GD2 and DB67. The three-dimensional structure of DB67 is presented in Figure 3A, and it was predicted that DB67 was mainly composed of several stem-ring structures. Upon docking with GD2, the docking position result indicated that the binding was related to the structure of the nucleic acids and had a certain consistency (Figure 3B). Furthermore, RMSD was 0.9 Å, it was found that a balance in the system was reached on the left and right sides after 0.5 ns when the whole system tended to be stable (Figure 3C). In addition, the active binding site of DB67 was assessed, and C11, C12, C13, C32, C34, A35, and C36 could form polar interactions, which are very significant for target recognition (Figure 3D and E). Furthermore, it was predicted that C12, C13, C44, and A45 could form an intimate local space that makes a greater contribution to target recognition. Next, the GD2 aptamer was truncated according to the molecular docking results, and the truncated sequences were designed and synthesized. The truncated sequences are shown in Table 1. All sequences were modified with FAM at the 5ʹ end and incubated with IMR32 or A431 cells. The fluorescence signals were assessed by flow cytometry. As presented in Figure 3F, when compared with other sequences (L-DB67 and R-DB67), core-DB67 and DB67 exhibited relatively stronger binding to GD2-positive cells. After removing C11, C12, and C13 in R-DB67 or C32, C34, A35, and C36 in L-DB67, the fluorescence signal decreased significantly. This result indicated that consistent with molecular docking, C11, C12, C13, C32, C34, A35, and C36 were critical for GD2 binding. Interestingly, after removing the unnecessary nucleotides, the binding affinity of core-DB67 was slightly stronger than that of DB67. This may be due to the removal of unnecessary steric hindrance. Thus, core-DB67, termed DB67-active, was applied for further evaluation.
 |
Figure 3 Evaluation of DB67 core region for GD2 recognition and binding. (A) The three-dimensional of DB67. DB67 was mainly composed of several stem-ring structures. (B) Black box docking position of DB67. (C) Recognition of system balance between DB67 and GD2 molecules. (D and E) Molecular docking. The C11th, C12th, C13th, C32th, C34th, A35th, and C36th could form polar interaction. (F) Flow cytometry assessment of truncated DB67 for GD2 binding specificities. DB67 was truncated and truncated sequence L-DB67, R-DB67, DB67-active, and DB67 were incubated with IMR32 cells. Fluorescence signals were detected. |
Selection and Characterization of i-Motif
An ideal combination of GD2 aptamer and i-motif should meet two requirements: ① Sequences should not bind with GD2-positive cells even in pH 7.4 environment. Since once binding, it may be internalized by cells and Dox will damage cells; ② i-motif should be only formed and Dox should be released only in the presence with GD2, otherwise i-motif should be formed and Dox will be released in acidic environment such as gastric juice. Thus, to solve this issue, several i-motif sequences were taken into account (Supplementary Figure S1B). In brief, sequences①, ②, ③, ④, ⑤, ⑥ were labeled with TARMA and constructed with complementary sequence and Dox. The constructed sequences were mixed with GD2-coated beads or null-GD2 beads at either pH 7.4 buffer or pH 6.5 buffer. Further, beads were detected by flow cytometry and supernatant fluid were evaluated for Dox releasing. The sequence which could only bind with GD2 at acidic environment and Dox could be released in the presence of GD2 should be utilized for further study (Supplementary Figure S1A). The GD2 binding data was presented in Supplementary Figure S1C and Dox releasing was presented in Supplementary Figure S1D. For sequence ①, no matter in pH 7.4 or 6.5, the TRAMA signal in GD2-beads was much higher than control-beads (pH 7.4: 3.4-fold; pH 6.5: 4.3-fold), and there was no difference between pH 7.4 and 6.5, indicating a GD2 recognition and binding regardless of pH value; Additionally, Dox releasing of sequence ① was quite little and there was no difference between pH 7.4 and pH 6.5, indicating a failure of i-motif formation and Dox releasing. This may be due to a strong influence caused by GD2 aptamer steric hindrance and i-motif formation energy was in-sufficient. For sequence ②, no matter in pH 7.4 or 6.5, the TRAMA signal in GD2-beads was much higher than control-beads (pH 7.4: 8.2-fold; pH 6.5: 9.6-fold), and there was no difference between pH 7.4 and 6.5, indicating a GD2 recognition and binding regardless of pH value; It is worth noting that when in pH 7.4, there was little Dox release at either GD2-beads or control-beads, the ratio was nearly 1; When in pH 6.5, there was a much stronger drug release in GD2-beads when compared with control beads; It could predicted that sequence ② could only release Dox at acidic environment and in presence with GD2. This may because with a longer length, i-motif has enough energy to overcome aptamer steric hindrance, but still did not have sufficient energy to influence aptamer formation. For sequence ③, there generated a significant difference in TRAMA signal ratio between pH 7.4 and pH 6.5. The TRAMA signal ratio (GD2-beads/control-beads) at pH 6.5 was much higher pH 7.4 (p<0.01), indicating ③ could only recognize GD2 at acidic environment; Meanwhile, Dox releasing ratio of sequence ③ at pH 6.5 was much stronger when compared with pH 7.4 (p<0.01), indicating a successful i-motif formation and Dox releasing. It should be predicted that sequence ③ matched a balance between GD2 aptamer and i-motif. When in acidic environment, a longer i-motif sequence generated a stronger change in three-dimensional structure and help aptamer to form correct structure to bind with GD2, and after aptamer binding with GD2, aptamer structure may further facilitate i-motif formation. When in pH 7.4, i-motif structure could not form and further hinder GD2 aptamer recognition. For sequence ④, ⑤, ⑥, there generated a significant difference in TRAMA signal ratio between pH 7.4 and pH 6.5. The TRAMA signal ratio in GD2-beads at pH 6.5 was much higher pH 7.4 (p<0.01), indicating ④, ⑤, ⑥ could only recognize GD2 at acidic environment; However, with a longer sequence of i-motif, the formation of i-motif was not influenced by GD2 aptamer anymore, when in pH 7.4, there was no i-motif formation and Dox release in both GD2-beads and control-beads, thus the ratio was approximately equal to 1; When in pH 6.5, i-motif was formed regardless of GD2, dox was released and the ratio in GD2-beads and control-beads was also approximately equal to 1. It should be predicted that with the longer sequence of i-motif, the formation energy was much stronger and enough to get rid of GD2 aptamer. Therefore, only ③ was suitable for further study.
Construction and Characterization of IG-Targeted
According to the hypothesis, IG-Targeted consisted of a GD2 recognition domain, a linker zone, and a split i-motif element. When IG-Targeted was in a neutral environment, the i-motif was extended and could not form the correct three-dimensional structure, which influenced the DB67-active structure for targeted binding. Consequently, DB67-active could not recognize GD2 and failed to recognize the target. However, in an acidic environment, the i-motif could successfully form and help the DB67-active structure to enhance the binding with GD2. Therefore, we assessed how to incorporate an i-motif structure into IG-Targeted to facilitate both recognition and binding in accordance with microenvironmental stimuli. First, the influence of pH on the binding of DB67-active was investigated. As presented in Figure 4A, there was no obvious difference in the binding of DB67-active to GD2 at the two different pH values tested, suggesting that such a small pH range could not influence the ability of DB67-active to bind to GD2. Further, this i-motif is sensitive in the pH 6.5–7.2 range with a midpoint pH of 6.9, which has been validated to be responsive to the different extracellular pH values of tumor cells and normal cells. In addition, to further enhance the balance between aptamer and i-motif, a linker zone was designed and inserted between DB67-active and the i-motif. To determine the most suitable length of the linker zone for IG-Targeted, three IG-Targeted molecules with different linker lengths were designed, synthesized, and modified with TAMRA modified at the 5ʹ end (Table 2). To mimic both normal GD2-positive cells at physiological pH and tumor cells in an acidic tumor microenvironment, GD2-positive IMR32 cells were seeded in culture medium at pH 6.5 and pH 7.4, respectively. Untreated cells were treated as control. To estimate which IG-Targeted sequence was most effective, three modified sequences were incubated with the cells in different pH environments, and the fluorescence signals were detected by flow cytometry. As presented in Figure 4B and Supplementary Figure S4, all three sequences exhibited binding to the target cells at pH 6.5 but showed only a small signal at pH 7.4, indicating the successful formation of the three-dimensional structure of DB-active and construction of the i-motif structure (p<0.05). Interestingly, sequences with intermediate length linkers (IG-5) exhibited better binding affinity than sequences with shorter and longer length linkers (IG-3 and IG-7, respectively). When compared with pH 7.4, IG-5 generated a most significant fluorescence signal difference at pH 6.5 (p<0.01). The possible reason for this result is that longer linkers allowed enough space for both DB-active and the i-motif to perform their biological functions. Therefore, IG-5 was chosen for further investigation.
 |
Figure 4 Construction and characterization of IGD-Targeted. (A) Evaluation of pH-sensitive ability of DB67-active. DB67-active was incubated with IMR32 cells at pH 6.5 or 7.4. Fluorescence intensity of cells were assessed by flow cytometry. (B) Binding specificities evaluation. ssDNA strands with different length and sequences were all modified with fluorescent groups and quenching groups. Sequences were incubated with IMR32 in prepared buffers at different pH (6.5 and 7.4) at 37°C for 30 min. Cells were washed by PBS with equivalent pH and evaluated by flow cytometry. (C) Evaluation of IG-Targeted formation mechanism. IG-Targeted was modified with fluorescent groups and quenching groups, and treated in various pH environments (pH 6.5–10) and the fluorescence was monitored by fluorescence spectrometry (The difference compared with pH 10 was labeled with *, *p < 0.05, **p < 0.01). (D) Fluorescence histogram of doxorubicin solution (5 nM) with increasing molar ratios of the IG-Targeted. The fluorescence quenching indicates Dox loading into IG-Targeted (The difference compared with Free Dox was labeled with *, *p < 0.05, **p < 0.01, ***p < 0.001). |
IG-Targeted Formation Mechanism
To explore the proposed mechanism of the response of IG-Targeted to pH, forster resonance energy transfer (FRET) was employed to reveal the formation of IG-Targeted at different pH values. To generate a FRET signal, IG-5 was modified with fluorescent groups and quenching groups; with fluorophore TAMRA modification at the 5ʹ end and quencher BHQ-2 modification at the 3ʹ end. A sequence termed C-targeted, which consisted of DB67-active and poly T of the same length (which could not form an i-motif structure), acted as a control. Both sequences were treated in various pH environments (pH 6.5, 6.8, 7, 7.5, 8, 8.5, 9, 9.5, 10), and their fluorescence was monitored by fluorescence spectrometry with a Synergy 4 analyzer at 580 nm. As shown in Figure 4C, during the pH between 10~7, the fluorescence changed slightly since i-motif could not form in these pH values, thus the TARMA could not be quenched; When under pH 7, there was an obvious fluorescence decreasing at pH 6.8 since i-motif structure was beginning to form (p<0.05); When pH value reached at 6.5, there was an almost 7-fold decrease in the fluorescence at 580 nm (p<0.01), indicating strong fluorescence quenching with decreasing pH value. However, there was no significant difference in the fluorescence of C-targeted (Supplementary Figure S2). These data indicated the successful formation of a closed i-motif architecture and the occurrence of FRET in IG-Targeted.
Construction of IGD-Targeted
Since the chemotherapeutic agent Dox is widely used in clinical treatment and could intercalate into cytosine-guanosine base pairs, IG-Targeted was designed as a drug loading region that is abundant in cytosine and guanosine. Free Dox can emit fluorescence that can be quenched after intercalation into DNA. To assess the most Dox loading capacity of IG-Targeted, different concentrations of IG-Targeted were incubated in an aqueous solution of Dox (5 nM) for 30 min in a black 96-well plate for a range of aptamer/dox molar ratios. The fluorescence of Dox was monitored by a Synergy 4 analyzer. As shown in Figure 4D, as the ratio of DNA/free Dox increased, there was a sequential decrease in fluorescence when compared with free Dox, suggesting the loading of Dox into IG-Targeted. When the DNA/Dox molar ratio was above 10,000:1, the Dox fluorescence reached its lowest value, indicating Dox saturation into IG-Targeted. IG-Targeted loaded with Dox was termed IGD-Targeted. The construction of CD-Targeted was the same with IGD-Targeted and the data was presented in Supplementary Figure S3. When the DNA/Dox molar ratio was above 10,000:1, the Dox fluorescence reached its lowest value, indicating Dox saturation into CD-Targeted.
IGD-Targeted Can Selectively Transport Dox to GD2-Positive Cells in Acidic Environments in vitro
Generally, free Dox can be taken up by both tumor cells and normal cells, resulting in damage to normal tissues and adverse effects. According to our design, it was assumed that when IGD-Targeted was in an acidic tumor microenvironment and in the presence with GD2, the i-motif would form, and the double-stranded DNA structure of the linker region would be disrupted. Dox could then be released around the tumor cells to subsequently selectively damage tumor cells. However, in neutral environments, the i-motif does not form, and Dox remains intercalated in the loading region, resulting in protection of normal cells. To test this hypothesis, IGD-Targeted was incubated with IMR32, KD-IMR32, A431, or GD2-positive A431 cells at 37°C for 1 h in either pH 7.4 buffer or pH 6.5 buffer. The distribution of Dox in the cells was analyzed by confocal microscopy. As shown in Figure 5A, for GD2 expressing cells (IMR32, KD-IMR32, and GD2-positive A431), there is obvious Dox fluorescence signal at pH 6.5, but not at pH 7.4. For GD2-negative cells A431, there was no Dox release either at pH 7.4 or pH 6.5. This result indicated again that GD2 aptamer and i-motif may help each other and IGD-Targeted could only release Dox at acidic environment and in the presence of GD2.
 |
Figure 5 IGD-Targeted could transport Dox to GD-positive cells and inhibit cell proliferation in acidic environment selectively in vitro. (A) IGD-Targeted could release Dox to GD2+ cells in acidic environment. IGD-Targeted was incubated with IMR32 or A431 cells in pH 7.4 or pH 6.5, respectively. Cells were observed under confocal microscope. (B) Cell Viability inhibition ability of IGD-Targeted to GD2+ cells. Different concentration of IGD-Targeted was incubated with IMR32 or A431 cells in pH 7.4 or pH 6.5, cell proliferations were detected by MTS assay. (C) Cell Proliferation inhibition of IGD-Targeted to GD2+ cells. Different concentration of IGD-Targeted was incubated with IMR32 or A431 cells in pH 7.4 or pH 6.5, cell proliferations were detected by CCK8 assay (*p<0.05, **p<0.01, ***p<0.001). |
Further, IGD-Targeted was incubated with IMR32 and A431 cells at 37°C for 6 h in either pH 7.4 buffer or pH 6.5 buffer, after 96 h, cell viability and proliferation were assessed. MTS assay showed that when compared with untreated cells, there was no significant change of cell viability; Only when IMR32 was at pH 6.5 environment, IGD-Targeted generated with a significant change compared with untreated cells. The difference was becoming more and more significant with the increasing concentration of IGD-Targeted (20 nM: p<0.05; 50 nM: p<0.05; 100 nM: p<0.05; 200 nM: p<0.01; 500 nM: p<0.001) (Figure 5B). Additionally, CCK8 assay indicated for IMR32 at pH 7.4 and A431 at both pH 7.4 and 6.5, when compared with untreated cells, there was no significant change of cell viability; Only when IMR32 was at pH 6.5 environment, IGD-Targeted generated with a significant change compared with untreated cells. The difference was becoming more and more significant with the increasing concentration of IGD-Targeted (20 nM: p<0.05; 50 nM: p<0.05; 100 nM: p<0.01; 200 nM: p<0.01; 500 nM: p<0.001) (Figure 5C). BrdU assay showed that when in IMR32 cells, IGD-Targeted generated a stronger cell proliferation inhibition at pH 6.5, whereas little damage at pH 7.4 (p<0.05); Additionally, IGD-Targeted did not cause obvious inhibition to tumor cell A431 and 14 kinds of healthy cell lines either at pH 7.4 or pH 6.5 (Supplementary Figure S5). These data indicated that IGD-Targeted could selectively damage GD2-positive cells in acidic environments but not normal cells or GD2-positive cells at neutral pH values. Taken together, IGD-Targeted could selectively transport Dox to GD2-positive cells in acidic environments.
IGD-Targeted Can Inhibit GD2-Positive NB Tumor Growth and Protect Normal Tissue in vivo
To further evaluate whether IGD-Targeted could inhibit tumor growth and reduce cytotoxicity to normal tissues in vivo, we first determined whether IGD-Targeted could release Dox only to tumor cells. IGD-Targeted was injected into IMR32 tumor-bearing mice, and Dox fluorescence was observed with an IVIS 200 Imaging System. CD-targeted was used as a control. As presented in Figure 6A, mice injected with CD-targeted did not exhibit obvious Dox fluorescence at the tumor site. However, mice treated with IGD-Targeted showed a strong fluorescence signal at the tumor site, indicating selective Dox delivery to the target. Second, to assess whether IGD-Targeted could prolong mice survival and inhibit tumor growth, tumor-bearing mice were randomly divided into four groups and treated with saline, IGD-Targeted, CD-targeted, or free Dox. As illustrated in Figure 6B, after injection with IGD-Targeted or free Dox, the tumor volumes visibly decreased compared with that of the saline group, indicating an effective treatment of IG-Targeted. CD-Targeted showed a similar result with saline group, which was consistent with the previous result that CD-Targeted did not possess targeting Dox release ability. In addition, the mice in the IGD-Targeted and free Dox groups survived longer than those in the CD-targeted group and saline group. When compared with saline group, CD-Targeted did not extend survival obviously, whereas IGD-Targeted (p<0.01) and free Dox (p<0.05) induced significant longer surviving time. In addition, when compared with Free Dox, IGD-Targeted showed a meaningful longer survival rate (p<0.05), indicating successful antitumor effects of IGD-Targeted (Figure 6C). The body weights of the mice are shown in Figure 6D. Mice treated with IGD-Targeted and free Dox presented weights that remained relatively stable during the first 10 days, and then the weights in the Dox group declined rapidly due the cytotoxicity of Dox to normal cells (p<0.05). Mouse weights in the saline and CD-targeted group increased rapidly due to rapid tumor growth. When compared with Free Dox group at 24 days, IGD-targeted generated a much slower weight loss (p<0.05). As shown in Figure 6E, when compared with saline group, both IGD-Targeted and Free Dox group presented a much slower increasing of tumor volume (p<0.01), indicating an effective Dox delivery ability of IGD-Targeted; Since CD-Targeted possessed no drug targeted delivery ability, CD-Targeted group showed little tumor inhibition effect. Additionally, to assess whether IGD-Targeted reduced the Dox release in normal cells, Dox concentrations in organ tissues, tumor tissues, and urine were detected. As shown in Figure 6F, when compared with saline group, Dox concentrations in tissues were much higher and in average (p<0.05), indicating Dox had no cell selectivity; When compared with Free Dox group, CD-Targeted group showed a higher concentration in urine (p<0.01), suggesting that CD-Targeted was rapidly excreted due to its non GD2 targeting ability; When compared with saline group, IGD-Targeted group showed a much higher concentration in tumor tissues (p<0.01). When compared with CD-Targeted group, IGD-Targeted group also showed a higher concentration in tumor tissues (p<0.05). These data indicated the effectiveness of IGD-Targeted as a novel strategy for the targeted therapy of GD2-positive NB tumors. Further, to assess whether IGD-Targeted could protect normal tissues, hematoxylin-eosin staining of organs and organ damage markers were evaluated. As presented in Figure 7A, when compared with saline group, hematoxylin-eosin staining revealed injury to the organs of the mice in the free Dox group, whereas no obvious damage was observed in the IGD-Targeted group, indicating an effective targeting Drug delivery ability of IGD-Targeted. Meanwhile, CD-Targeted group showed little obvious damage in vital organ, reflecting a successful formation design of IGD-Targeted for recognizing and Dox delivery ability. Moreover, the blood routine examination results were shown in Figure 7B. When compared with saline group, there was no significant change in IGD-Targeted and CD-Targeted group, suggesting there was no obvious damage to blood cells caused by IGD-Targeted, indicating a great targeting ability of IGD-Targeted. Since CD-Targeted owned no targeting and Dox release ability, it should be excreted fast without cells damage. However, there was an obvious difference in Free Dox group. The counts of white blood cell (WBC), granulocyte (Gran#), and platelets (PLT) were significantly higher than saline group (p<0.05). This may be caused by non-selective delivery to blood cells and resulting in cytotoxicity effect. It should be noticed that other parameters like red blood cell (RBC), percentage of monocytes (Mon#), percentage of granulocyte (Gran%) in free Dox group was a little higher than saline group, but have not reached the statistical difference, it may be due to an insufficient agent administration time. It may significantly change with a longer medication time. Organ damage bio-marker levels are presented in Figure 7C. When compared with saline group, AST, ALT, creatinine, and BUN were significantly increased in the free Dox group (p<0.05), indicating an organ damage caused by Dox. However, there was no significant change in IGD-Targeted group, indicating a targeted Dox delivery ability of IGD-Targeted. The levels in CD-Targeted also stayed in a normal range, reflecting non-targeting and delivery ability as we designed. To assess RNA expression changing after IGD-Targeted administration, tumor tissues in saline, CD-Targeted group and IGD-Targeted group were extracted and subjected to RNA sequencing. Since it has been reported that after Dox was internalized by cells, it may influence several gene expressions. Thus, in this study, these genes were evaluated from RNA sequencing data and presented as ratio: expression level of IGD-Targeted group or CD-Targeted group/expression level of saline group. Ratios were presented as heat map. As shown in Figure 8A, there were 30 genes up-regulated (1.2~1.8-fold) in IGD-Targeted when compared with CD-Targeted. Of which, PRR13, KPN2, EDEM2, PCDHA7, SESN1, PPM1D, RPS27L, ISG20L1, TOB1, TNFRSF108, ARID3A, IER5, CDKN1A, ACTA2, SPATA18, DRAM, BTG2, PHLDA3, STEAP3, GADD45A, CDKN1A, CYF1P2, and RN7SK were reported as Dox-reactive genes; BAD, Caspase-3, Bax, and Caspase-9 have been validated that were associated with cell apoptosis induced by Dox. Additionally, three genes were found down-regulated in IGD-Targeted group: Bcl-2, PCNA, and Ki67. Bcl-2 gene was associated with anti-apoptosis, PCNA and Ki67 were associated with cell proliferation. The expression level change in IGD-Targeted group indicated its effective Dox delivery and cell inhibition ability IGD-Targeted. Further, to confirm gene expression changing, six vital genes were evaluated by RT-PCR. As shown in Figure 8A, Dox-reactive gene RPS27L, PPMID, and SESN1 were significantly increased in IGD-Targeted group, indicating a positive Dox uptake compared with control group (p<0.05); Further, caspase-9 and FAS gene were also up-regulated in IGD-Targeted group (p<0.05), indicating cells apoptosis induced by Dox. In the contrary, Bcl-2 gene was down-regulated in IGD-Targeted group, further suggesting cell apoptosis caused by Dox. These data were consistent with other published reports: Dox may up-regulated nearly 1.5-fold of Dox-reactive genes, 1.3-fold of Dox-related apoptosis genes, and down-regulated nearly 0.6-fold of Dox-related cells proliferation genes.34–38 These data identified a specific Dox delivery and tumor inhibition of IGD-Targeted to GD2-positive cells.
 |
Figure 6 IGD-Targeted could inhibit GD2-positive NB tumor growth selectively in vivo. (A) Specific targeting ability of IGD-Targeted to GD2+ tumor site in mouse model detected by IVIS 200 imaging. (B) IGD-Targeted could inhibit tumor growth in model mice. Mice were divided into three groups and administrated with Saline, CD-Targeted, IGD-Targeted, and free Dox (N=6). Mice and tumor tissues were photographed. One mouse was died due to severe side effect in free Dox group. (C) Survival rate (%) of each group (The difference compared with saline group was labeled with *, *p < 0.05, **p < 0.01; The difference compared with Free Dox group was labeled with #, #p < 0.05). (D) Weights of mice in each group (The difference compared with saline group was labeled with *, *p < 0.05; The difference compared with Free Dox group was labeled with #, #p < 0.05). (E) Tumor volume of mice in each group. (The difference compared with saline group was labeled with *, **p < 0.05). (F) Dox fluorescence intensity evaluation of tissues in each group (The difference compared with saline group was labeled with *, *p < 0.05, **p < 0.05; The difference compared with Free Dox group was labeled with #, #p < 0.05, ##p < 0.05; The difference compared with CD-Targeted group was labeled with &, &p < 0.05). |
 |
Figure 7 Potential reduced side effects of IGD-Targeted in vivo. (A) Histochemistry of mice organs. After initial treatment, mice were euthanized and the heart, liver, kidney, spleen, and lung tissues were observed by H&E staining. (B) Blood routine examination of mice peripheral blood (The difference compared with saline group was labeled with *, *p < 0.05,). (C) Serology assessment. Each bar represents means with SD of six replicates (The difference compared with saline group was labeled with *, *p < 0.05). |
 |
Figure 8 Transcriptomic analysis and comparison between IGD-Targeted and dinutuximab. (A) Transcriptomic analysis of the CD-Targeted treated tumor samples, IGD-Targeted treated tumor samples, and saline-treated tumor samples were compared with using Illumina system and Heatmap software. The value was presented as ratio of CD-Targeted/saline group or IGD-Targeted/saline group. RT-PCR analysis of Caspase-9, Bcl-2, RPS27L, PPMID, SESN1, and FAS was evaluated. (The difference compared with saline group was labeled with *, *p < 0.05). (B) Anti-cancer evaluation of IGD-Targeted and dinutuximab. (C) Tumor volume of mice in each group. (The difference compared with saline group was labeled with *, *p < 0.05). (D) Self-harm behavior scores of mice. Mice were injected with three concentration level of IGD-Targeted or GD2 antibody, respectively. The scores were calculated (The difference compared with saline group was labeled with *, *p<0.05; The difference compared with IGD-Targeted group was labeled with #, #<0.05). |
Additionally, to evaluate the application potential of IGD-Targeted, the inhibition ability and side effects of IGD-Targeted were compared with GD2 antibody Dinutuximab. Dinutuximab, a kind of GD2 antibody, has shown promising therapeutic effect clinically. Dinutuximab binds to GD2, and induces antibody-dependent cell-mediated cytotoxicity (ADCC) and compliant-dependent cytotoxicity (CDC). First, tumor-bearing mice were injected with saline, IGD-Targeted, and GD2 antibody Dinutuximab, respectively. After 15 days, mice were sacrificed and tumor tissue volume values were evaluated. As shown in Figure 8B and C, when compared with saline group, IGD-Targeted and Dinutuximab significantly decreased tumor growth (p<0.01), and there was no obvious difference between IGD-Targeted and Dinutuximab, indicating an effective inhibition ability of IGD-Targeted. Several reports have reminded severe pain after Dinutuximab administration. The clinical instruction of Dinutuximab also indicated severe pain to normal nerve cells. The main reason for this was due to the GD2 expression of normal nerve cells. Dinutuximab may also damage these normal cells and induce severe pain. Thus, to evaluate whether IGD-targeted could avoid this side effect, four- to six-week-old female BALB/c (nu/nu) athymic nude mice were treated with saline, IGD-Targeted (low dose, medium dose, and high dose) and dinutuximab (low dose, medium dose, and high dose). If nerve cells were damaged, they may cause pains. For mice, a self-harm of biting toes was started at the beginning of nerve damage and pain. Therefore, the number and degree of bitten toe were recorded. As presented in Figure 8D, the score in the GD2 antibody group was much higher than that in the IGD-Targeted group and saline group at both mediate dose and high dose (p<0.05), and the score in the IGD-Targeted group did not show significant change when compared with saline group, indicating an obvious alleviation of pain in IGD-Targeted group. These results indicated the ability of IGD-Targeted to protect normal nerve cells. Overall, these data demonstrated the effectiveness and safety of IGD-Targeted as a novel strategy for GD2-positive NB tumor-targeting therapy.
Discussion
In this study, we successfully designed a pH-sensitive drug delivery system termed IGD-Targeted to target GD2-positive NB tumor cells. IGD-Targeted consists of the GD2 aptamer DB67-active, an i-motif element, and a linker (Figure 1). The structure of IGD-Targeted switches depending on the pH environment. Due to the i-motif element, IGD-Targeted is capable of binding to GD2 molecules expressed on cell membranes and releasing Dox around tumor cells; however, it does not recognize GD2 and liberate Dox in neutral media (Figures 2–4). Our in vitro and in vivo data indicated that IGD-Targeted could selectively recognize GD2 in a simulated acidic tumor microenvironment but not GD2 in neutral media (Figure 5). Additionally, IGD-Targeted precisely delivered Dox to tumor cells, prolonged survival, and protected normal tissues (Figures 6–8).
Gangliosides, including GM3, GM2, GM1, and GD1, etc., are a large category of molecules that are widely expressed on many normal human tissues and mediate multiple biological functions.39 GD2 has been considered an attractive tumor antigen and target for NB cancer therapy.40 GD2 is abundantly expressed on a wide range of tumors, such as almost all neuroblastomas, most melanomas and retinoblastomas, and gliomas.41 In addition, it has been validated that GD2 plays a role in cell proliferation, migration, and invasion. Scientists have found that high circulating GD2 levels correspond to patients showing rapid disease progression, low response to therapy, or low survival. Thus, the US National Cancer Institute ranked GD2 twelfth among 75 potential targets for anticancer therapy in 2009, suggesting that GD2 is a valuable and ideal target for cancer therapy.42 Previous studies have demonstrated the applicability of GD2 in targeted NB therapy.43 Research and development of NB tumor therapy has focused on GD2, including GD2 antibodies and GD2 CAR-T cells.44 Many of these studies have exhibited powerful anticancer abilities in animal models. Among them, the anti-GD2 monoclonal antibody dinutuximab (ch14.18) has been approved by the US Food and Drug Administration, and dinutuximab β (ch14.18/CHO) has been approved by the European Medicines Agency for high-risk neuroblastoma treatment.8 Nevertheless, GD2 antibodies can cause severe side effects, such as capillary leak syndrome, hypersensitivity reactions, intense neuropathic pain, infection, and fever. The root of these side effects is that although GD2 is over-expressed on NB tumor cells, it is also expressed in low amounts on peripheral nerves, peripheral sensory nerve fibers, mesenchymal stem cells, brain parenchyma, etc.45 Current GD2 antibodies are incapable of distinguishing between GD2 expressed on tumor cells or normal cells, resulting in cytotoxicity, such as neurotoxicity, to normal cells. To solve this obstacle, it is necessary and urgent to develop a novel strategy to damage only GD2-positive tumor cells and not GD2-expressing normal cells. Aptamers, a promising kind of target molecule, are generated via a ligand phylogenetic technique with exponential enrichment in vitro. Structured oligonucleotide sequences of RNA or DNA have strict recognition ability and high affinity for their corresponding target molecules (proteins, viruses, bacteria, cells, heavy metal ions, etc.).46 Aptamers can fold into unique three-dimensional structures to recognize and bind to their targets. Interestingly, in addition to their well-known structure, aptamers also have more complex structures, such as the i-motif and A-motif. The i-motif is a pH-sensitive tetramer structure. In acidic environments, C:C+ base pairs are arranged, and the interphase intercalates.47 Generally, the i-motif structure can produce a fast and reversible response to pH values between 5.0 and 7.0. It is noteworthy that the pH in the tumor microenvironment is acidic due to rapid metabolism while the pH in normal tissues remains neutral. To date, the i-motif has been widely used as a biosensor and in targeted therapy. Generally, nanoparticles containing aptamers and the i-motif are the most common, and the chemotherapeutic agent Dox is loaded in nanoparticles.48 When the environmental pH decreases from neutral (~7.4) to acidic, the i-motif can switch from a single-stranded structure to a C-tetrad, further releasing Dox only to tumor cells. Notably, Tan et al first distinguished targets at different pH values by aptamers utilizing an i-motif.27 Thus, in this study, to make it safe to GD2-positive normal cells, such as some nervous system cell, the novel targeting drug delivery system should only release Dox to GD2-positive tumor cells. Therefore, it should meet two conditions: first, it should only recognize GD2 in acidic environment, since extracellular pH of normal cells was neutral; Second, Dox could only be released in the presence with GD2, since some physiological environment such as gastric juice was acidic. Thus, how to make a balance between aptamer and i-motif was quite important. Here, we designed a smart system termed IGD-Target that could differentiate between GD2 expressed on tumor cells from that expressed on normal cells, exhibiting an anticancer ability against only GD2-positive tumor cells. IGD-Target contains a GD2 aptamer, a linker region, and an i-motif element. Our data indicated the effectiveness, safety, and specificity of IGD-Target in GD2-positive NB targeted therapy. Compared with nanoparticles, IGD-Target exhibits certain advantages. First, the easily designed and programmed IGD-Target can be assembled by only complementary DNA pairing and Dox self-loading. This strategy is helpful for scale-up production. Second, IGD-Target is biodegradable and will not lead to chronic accumulation in tissues. The aforementioned findings provide a simple and smart strategy for NB-targeted therapy. IGD-Target recognizes and damages only NB tumor cells irrespective of the coexistence of identical receptors on normal cells. Future molecular and structure analysis of IGD-Targeted should be processed to clarify the interaction of aptamer and i-motif and clear how they influence each other and reach an ideal balance. The structural stability and chemical stability of IGD-Targeted and pharmacokinetic should be our main focus work for the translation of IGD-Targeted from bench to bedside in the future.
Conclusions
In summary, in this study, we constructed a pH-sensitive switchable drug delivery system termed IGD-Target based on the GD2 aptamer and i-motif element, which cold distinguish targeted expressed on tumor or normal cells. IGD-Target recognizes GD2 expressed on tumor cells but not the same antigen expressed on normal cells due to the allosterically regulated i-motif. Our data demonstrated that IGD-Target could bind with GD2 only in an acidic tumor microenvironment in vitro and further release Dox to only GD2-positive tumor cells in vivo. Additionally, IGD-Target inhibits the growth of tumor cells in vivo while protecting normal cells. Thus, when compared with typical aptamer and drug delivery systems, IGD-Target could be more practical to solve the problem of having identical GD2 target antigens on both NB tumors and healthy cells. Collectively, these features make IGD-Target an attractive compound for NB-targeted therapy. It is apparent that the combination of an aptamer and i-motif is a simple and efficient strategy for tumor therapy in which the target is also expressed on normal cells, suggesting great clinical potential for various tumor therapy applications.
Ethics Approval
All mouse studies were approved by Affiliated Children’s Hospital (Xi’an, China). All animal studies followed the international guidelines on animal welfare and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. In addition, all animal studies complied with the Animal Research: Reporting of In Vivo Experiments guidelines and the 2013 American Veterinary Medical Association euthanasia guidelines.
Acknowledgments
The authors would like to thank Ms Yafei Zhou and Ms Sha Yin for their assistance with the cell experiments at the Affiliated Children’s Hospital of Xi’an Jiaotong University.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Shaanxi Provincial Program (grant no. 2019SF-207 and 2017SF-280).
Disclosure
Mrs Fengyu Che reports that she was supported by some research fees sponsored by the affiliated Children's Hospital of Xi'an Jiaotong University during the conduct of the study. The authors declare that they have no other potential conflicts of interest for this work.
References
1. Park JA, Cheung N-KV. Targets and antibody formats for immunotherapy of neuroblastoma. J Clin Oncol. 2020;38(16):1836–1848. doi:10.1200/JCO.19.01410
2. Matthay KK. Interleukin 2 plus anti-GD2 immunotherapy: helpful or harmful? Lancet Oncol. 2018;19(12):1549–1551. doi:10.1016/S1470-2045(18)30627-2
3. Nguyen R, Moustaki A, Norrie JL, et al. Interleukin-15 enhances Anti-GD2 antibody-mediated cytotoxicity in an orthotopic PDX model of neuroblastoma. Clin Cancer Res. 2019;25(24):7554–7564. doi:10.1158/1078-0432.CCR-19-1045
4. Wienke J, Dierselhuis MP, Tytgat GAM, et al. The immune landscape of neuroblastoma: challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur J Cancer. 2021;144:123–150.
5. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol. 2020;10:1000. doi:10.3389/fonc.2020.01000
6. Schengrund CL. Gangliosides and Neuroblastomas. Int J Mol Sci. 2020;21:15. doi:10.3390/ijms21155313
7. Mount CW, Majzner RG, Sundaresh S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat Med. 2018;24(5):572–579. doi:10.1038/s41591-018-0006-x
8. Zobel MJ, Zamora AK, Wu H-W, et al. Initiation of immunotherapy with activated natural killer cells and anti-GD2 antibody dinutuximab prior to resection of primary neuroblastoma prolongs survival in mice. J Immunother Cancer. 2020;8(2):2. doi:10.1136/jitc-2020-001560
9. Wienke J, Dierselhuis MP, Tytgat GAM, et al. The immune landscape of neuroblastoma: challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur J Cancer. 2020;144:123–150.
10. Sujjitjoon J, Sayour E, Tsao S-T, et al. GD2-specific chimeric antigen receptor-modified T cells targeting retinoblastoma- assessing tumor and T cell interaction. Transl Oncol. 2020;14(2):100971.
11. Cicek F, Troschke-Meurer S, Ceylan K, et al. Impact of IL-2 on treatment tolerance in patients with high-risk neuroblastoma treated with dinutuximab beta-based immunotherapy. Front Pediatr. 2020;8:582820. doi:10.3389/fped.2020.582820
12. Park JA, Cheung N-KV. GD2 or HER2 targeting T cell engaging bispecific antibodies to treat osteosarcoma. J Hematol Oncol. 2020;13(1):172. doi:10.1186/s13045-020-01012-y
13. Levy G, Bonnevalle M, Rocourt N, et al. Necrotizing enterocolitis as an adverse effect of recombinant interleukin-2 and Ch14.18 in maintenance therapy for high-risk neuroblastoma. J Pediatr Hematol Oncol. 2015;37(4):e250–2. doi:10.1097/MPH.0000000000000304
14. Anghelescu DL, Goldberg JL, Faughnan LG, et al. Comparison of pain outcomes between two anti-GD2 antibodies in patients with neuroblastoma. Pediatr Blood Cancer. 2015;62(2):224–228. doi:10.1002/pbc.25280
15. Gorges M, West N, Deyell R, et al. Dexmedetomidine and hydromorphone: a novel pain management strategy for the oncology ward setting during anti-GD2 immunotherapy for high-risk neuroblastoma in children. Pediatr Blood Cancer. 2015;62(1):29–34. doi:10.1002/pbc.25197
16. Terme M, Dorvillius M, Cochonneau D, et al. Chimeric antibody c.8B6 to O-acetyl-GD2 mediates the same efficient anti-neuroblastoma effects as therapeutic ch14.18 antibody to GD2 without antibody induced allodynia. PLoS One. 2014;9(2):e87210. doi:10.1371/journal.pone.0087210
17. Yu J, Hung J-T, Wang S-H, et al. Targeting glycosphingolipids for cancer immunotherapy. FEBS Lett. 2020;594(22):3602–3618. doi:10.1002/1873-3468.13917
18. Hulikova A, Harris AL, Vaughan-Jones RD, et al. Regulation of intracellular pH in cancer cell lines under normoxia and hypoxia. J Cell Physiol. 2013;228(4):743–752. doi:10.1002/jcp.24221
19. Lim SC, Lee T-B, Kang B-S, et al. Extracellular acidity-mediated expression of cPLA2gamma confers resistance in gastric cancer cells. Anticancer Res. 2021;41(1):211–218. doi:10.21873/anticanres.14767
20. Swietach P, Vaughan-Jones RD, Harris AL, et al. The chemistry, physiology and pathology of pH in cancer. Philos Trans R Soc Lond B Biol Sci. 2014;369(1638):20130099. doi:10.1098/rstb.2013.0099
21. Miao D, Yu Y, Chen Y, et al. Facile construction of i-Motif DNA-conjugated gold nanostars as near-infrared and pH dual-responsive targeted drug delivery systems for combined cancer therapy. Mol Pharm. 2020;17(4):1127–1138. doi:10.1021/acs.molpharmaceut.9b01159
22. Turaev AV, Isaakova EA, Severov VV, et al. Genomic DNA i-motifs as fast sensors responsive to near-physiological pH microchanges. Biosens Bioelectron. 2021;175:112864. doi:10.1016/j.bios.2020.112864
23. Spence P, Fielden J, Waller Z. Beyond solvent exclusion: i-Motif detecting capability and an alternative DNA light-switching mechanism in a Ruthenium (II) polypyridyl complex. J Am Chem Soc. 2020;142(32):13856–13866. doi:10.1021/jacs.0c04789
24. Wei H, Zhao Z, Wang Y, et al. One-step self-assembly of multifunctional DNA nanohydrogels: an enhanced and harmless strategy for guiding combined antitumor therapy. ACS Appl Mater Interfaces. 2019;11(50):46479–46489. doi:10.1021/acsami.9b15874
25. Kan A, Liu X, Xu X, et al. A bimolecular i-motif mediated FRET strategy for imaging protein homodimerization on a living tumor cell surface. Chem Commun (Camb). 2020;56(87):13405–13408. doi:10.1039/D0CC05607C
26. Lee GJ, Kim T-I. pH-responsive i-motif conjugated hyaluronic acid/polyethylenimine complexes for drug delivery systems. Pharmaceutics. 2019;11(5):5. doi:10.3390/pharmaceutics11050247
27. Li L, Jiang Y, Cui C, et al. Modulating aptamer specificity with pH-responsive DNA bonds. J Am Chem Soc. 2018;140(41):13335–13339. doi:10.1021/jacs.8b08047
28. Rangel AE, Hariri AA, Eisenstein M, et al. Engineering aptamer switches for multifunctional stimulus-responsive nanosystems. Adv Mater. 2020;32(50):e2003704. doi:10.1002/adma.202003704
29. Tran P, Xiang D, Tran TTD, et al. Exosomes and nanoengineering: a match made for precision therapeutics. Adv Mater. 2020;32(18):e1904040. doi:10.1002/adma.201904040
30. Wu H, Wang M, Dai B, et al. Novel CD123-aptamer-originated targeted drug trains for selectively delivering cytotoxic agent to tumor cells in acute myeloid leukemia theranostics. Drug Deliv. 2017;24(1):1216–1229. doi:10.1080/10717544.2017.1367976
31. Zhao J, Wang M, Yang Y, et al. CD123 thioaptamer protects against sepsis via the blockade between IL-3/CD123 in a cecal ligation and puncture rat model. Nucleosides Nucleotides Nucleic Acids. 2021;40(1):16–31. doi:10.1080/15257770.2020.1815770
32. Wang M, Wu H, Li Q, et al. Novel aptamer-functionalized nanoparticles enhances bone defect repair by improving stem cell recruitment. Int J Nanomedicine. 2019;14:8707–8724. doi:10.2147/IJN.S223164
33. Davis KA, Abrams B, Lin Y, et al. Use of a high affinity DNA ligand in flow cytometry. Nucleic Acids Res. 1996;24(4):702–706. doi:10.1093/nar/24.4.702
34. Mao F, Zhang J, Cheng X, et al. miR-149 inhibits cell proliferation and enhances chemosensitivity by targeting CDC42 and BCL2 in neuroblastoma. Cancer Cell Int. 2019;19(1):357. doi:10.1186/s12935-019-1082-9
35. Abd EN, et al. Sesamol upregulates death receptors and acts as a chemosensitizer in solid ehrlich carcinoma model in mice. Nutr Cancer. 2021;1–15.
36. Horwacik I, Durbas M, Boratyn E, et al. Analysis of genes involved in response to doxorubicin and a GD2 ganglioside-specific 14G2a monoclonal antibody in IMR-32 human neuroblastoma cells. Acta Biochim Pol. 2015;62(3):423–433. doi:10.18388/abp.2015_1035
37. Chen S, Yuan X, Xu H, et al. WNT974 inhibits proliferation, induces apoptosis, and enhances chemosensitivity to doxorubicin in lymphoma cells by Inhibiting Wnt/β-catenin signaling. Med Sci Monit. 2020;26:e923799. doi:10.12659/MSM.923799
38. Hua Z, Zhan Y, Zhang S, et al. P53/PUMA are potential targets that mediate the protection of brain-derived neurotrophic factor (BDNF)/TrkB from etoposide-induced cell death in neuroblastoma (NB). Apoptosis. 2018;23(7–8):408–419. doi:10.1007/s10495-018-1467-6
39. Cumin C, Huang Y-L, Everest-Dass A, et al. Deciphering the importance of glycosphingolipids on cellular and molecular mechanisms associated with epithelial-to-mesenchymal transition in cancer. Biomolecules. 2021;11(1):1. doi:10.3390/biom11010062
40. Chen Y, Sun C, Landoni E, et al. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and Interleukin-15. Clin Cancer Res. 2019;25(9):2915–2924. doi:10.1158/1078-0432.CCR-18-1811
41. Ho W-L, Hsu W-M, Huang M-C, et al. Protein glycosylation in cancers and its potential therapeutic applications in neuroblastoma. J Hematol Oncol. 2016;9(1):100. doi:10.1186/s13045-016-0334-6
42. Cheung IY, et al. Survival impact of anti-GD2 antibody response in a phase II ganglioside vaccine trial among patients with high-risk neuroblastoma with prior disease progression. J Clin Oncol. 2020;JCO2001892.
43. Forlenza CJ, Boudreau JE, Zheng J, et al. KIR3DL1 allelic polymorphism and HLA-B epitopes modulate response to anti-GD2 monoclonal antibody in patients with neuroblastoma. J Clin Oncol. 2016;34(21):2443–2451. doi:10.1200/JCO.2015.64.9558
44. Sujjitjoon J, Sayour E, Tsao S-T, et al. GD2-specific chimeric antigen receptor-modified T cells targeting retinoblastoma – assessing tumor and T cell interaction. Transl Oncol. 2021;14(2):100971.
45. Diccianni MB, Kempińska K, Gangoti JA, et al. Anti-GD2 induced allodynia in rats can be reduced by pretreatment with DFMO. PLoS One. 2020;15(7):e0236115. doi:10.1371/journal.pone.0236115
46. Moghadam F, LeGraw R, Velazquez JJ, et al. Synthetic immunomodulation with a CRISPR super-repressor in vivo. Nat Cell Biol. 2020;22(9):1143–1154. doi:10.1038/s41556-020-0563-3
47. Chen B, Wang Y, Ma W, et al. A Mimosa-inspired cell-surface-anchored ratiometric DNA nanosensor for high-resolution and sensitive response of target tumor extracellular pH. Anal Chem. 2020;92(22):15104–15111. doi:10.1021/acs.analchem.0c03250
48. Li C, Chen Z, Zhang Y, et al. Guanine-lighting-up fluorescence biosensing of silver nanoclusters populated in functional DNA constructs by a pH-triggered switch. Anal Chem. 2020;92(19):13369–13377. doi:10.1021/acs.analchem.0c02744
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.