Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
A novel nanobody specific for respiratory surfactant protein A has potential for lung targeting
Authors Wang S, He X, Li N, Yu F, Hu Y, Wang L, Zhang P, Du Y, Du S, Yin Z, Wei Y, Mulet X, Coia G, Weng D, He J, Wu M ![]() , Li H
, Li H
Received 10 November 2014
Accepted for publication 23 January 2015
Published 13 April 2015 Volume 2015:10(1) Pages 2857—2869
DOI https://doi.org/10.2147/IJN.S77268
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Shan-Mei Wang,1,* Xian He,2,* Nan Li,1,* Feng Yu,3 Yang Hu,1 Liu-Sheng Wang,1 Peng Zhang,4 Yu-Kui Du,1 Shan-Shan Du,1 Zhao-Fang Yin,1 Ya-Ru Wei,1 Xavier Mulet,5 Greg Coia,6 Dong Weng,1 Jian-Hua He,3 Min Wu,7 Hui-Ping Li1
1Department of Respiratory Medicine, Shanghai Pulmonary Hospital, School of Medicine, Tongji University, Shanghai, 2School of Medicine, Suzhou University, SuZhou, 3Shanghai Institute of Applied Physics, Chinese Academy of Sciences, 4Department of Chest Surgery, Shanghai Pulmonary Hospital, School of Medicine, Tongji University, Shanghai, People’s Republic of China; 5CSIRO (Commonwealth Scientific and Industrial Research) Materials Science and Engineering, Clayton, 6CSIRO Materials Science and Engineering, Parkville, Melbourne, VIC, Australia; 7Department of Basic Sciences, University of North Dakota, Grand Forks, ND, USA
*These authors contributed equally to this work
Abstract: Lung-targeting drugs are thought to be potential therapies of refractory lung diseases by maximizing local drug concentrations in the lung to avoid systemic circulation. However, a major limitation in developing lung-targeted drugs is the acquirement of lung-specific ligands. Pulmonary surfactant protein A (SPA) is predominantly synthesized by type II alveolar epithelial cells, and may serve as a potential lung-targeting ligand. Here, we generated recombinant rat pulmonary SPA (rSPA) as an antigen and immunized an alpaca to produce two nanobodies (the smallest naturally occurring antibodies) specific for rSPA, designated Nb6 and Nb17. To assess these nanobodies’ potential for lung targeting, we evaluated their specificity to lung tissue and toxicity in mice. Using immunohistochemistry, we demonstrated that these anti-rSPA nanobodies selectively bound to rat lungs with high affinity. Furthermore, we intravenously injected fluorescein isothiocyanate-Nb17 in nude mice and observed its preferential accumulation in the lung to other tissues, suggesting high affinity of the nanobody for the lung. Studying acute and chronic toxicity of Nb17 revealed its safety in rats without causing apparent histological alterations. Collectively, we have generated and characterized lung-specific nanobodies, which may be applicable for lung drug delivery.
Keywords: nanobodies, rSPA, phage-nanobody library, VHH, lung-targeting drugs
Introduction
Development of lung-targeting ligands has been an important focus of drug delivery for the treatment of respiratory diseases to increase drug concentrations in the lung and minimizing toxicity. Currently, two major forms of lung-targeting drugs have been developed: 1) inhaled drugs, which are directly delivered to the respiratory tract, thereby improving pharmacological effects of therapy and minimizing unwanted systemic side effects (such as inhaled broncholytics, inhaled anti-inflammatory drugs, and inhaled vasodilators)1–3 and 2) passive lung-targeting drugs, which are likely localized in pulmonary capillary mechanical retention (eg, levofloxacin liposome).4–6 However, inhaled drugs have shown only limited efficacy against airway diseases, due to the barriers of respiratory structures and cooperation of patients.7 Furthermore, clinical application of passive lung-targeting drugs is also hampered by the high retention of the reticuloendothelial system, such as the liver and spleen. Therefore, there is an urgent need to develop more effective lung-targeting drugs or delivery ligands.
An antibody may recognize its antigen in vivo with high affinity and specificity. Based on this principle, active targeting drugs that consist of antibody ligands and nanoparticles via chemical conjugation have increased drug concentrations in the targeting tissue and decreased distribution in unrelated organs (especially in the liver and spleen) compared to their nontargeting counterparts.8–10 Because certain cancer cells overexpress growth-factor receptors or other molecules that can be easily targeted, recent studies have largely focused on tumor-targeting drugs.11–13 However, to date, the development of active lung-targeting drugs has been hampered by the lack of specific ligands for lung tissue.
Studies have shown that pulmonary surfactant protein A (SPA) is predominantly produced by alveolar epithelial type II cells and rarely expressed elsewhere.14 In recent years, SPA has become a commonly used marker of the lung for diagnosis of lung diseases.15,16 Therefore, SPA may serve as an attractive lung-targeting molecule for the development of active lung-targeting drugs. Particularly, targeting SPA by an antibody may be a viable approach. We have successfully developed dexamethasone liposomes with active lung-targeting distribution by linking anti-SPA polyclonal antibodies (SPA-poly-ant) with a dexamethasone (DXM) nanoliposome (NLP) surface (SPA-DXM-NLP).17 The concentration of the SPA-DXM-NLP complex in the lung is approximately fivefold greater than the free DXM control initially and reached 40-fold greater at 12 hours postinjection. Also, administration of SPA-DXM-NLP significantly attenuates lung injury in animal models. However, using full-size antibodies like anti-SPA-poly-ant for ligands is not ideal, because of their high molecular weight, strong immunogenicity, and low tissue penetration.18
To overcome this problem, recent studies have illustrated that the serum of Camelidae contains some special antibodies that are naturally devoid of light chains, termed heavy-chain antibodies (HcAbs).19 HcAbs can recognize their cognate antigens by a single variable-domain, referred to as VHH. Due to the nanometer-scale size of VHH, Ablynx (a company focusing on therapeutic applications of camelid antibodies) renamed VHHs Nanobodies® (Nbs).20,21 Nanobodies are the smallest naturally occurring intact antigen-binding domains of antibodies identified to date. In addition, benefiting from their small size, nanobodies are of higher affinity, more soluble, and more stable than other antibody fragments, such as F(ab’)2 or single-chain antibodies.22 Nanobodies also have better tissue penetration and less immunogenicity,23 which may be promising for application in the antitumor and anti-infection fields.24–26
In this study, we report the development of novel anti-rSPA nanobodies and the assessment of in vitro and in vivo targeting ability and toxicity.
Materials and methods
Materials
GenScript constructed the recombinant plasmid pGEX-4T-1-rSPA containing the rSPA gene sequence (Nanjing, People’s Republic of China [PRC]). The plasmid (pET-44a, pET-30a, pCANTAB5E), Escherichia coli BL21, Ex Taq deoxyribonucleic acid (DNA) polymerase, deoxynucleotide triphosphates (dNTPs), and DNA-purification kit were purchased from Tiangen Biotech (Beijing, PRC). A DNA ligation Kit, DNA markers, and restriction endonucleases were obtained from Takara Biotech (Dalian, PRC). Our laboratory prepared the rabbit antialpaca IgG antibody. Isopropyl-β-d-thiogalactoside (IPTG), horseradish peroxidase (HRP)-conjugated goat antirat IgG, and anti-His monoclonal antibody were provided by Beijing Biosynthesis Biotechnology (Beijing, PRC). Quartz crystal microbalance (QCM) measurements were performed using a Q-Sense D300 instrument with 5 MHz quartz crystals deposited with gold electrodes (14 mm diameter) on a single side of the surface (Q-Sense, Gothenburg, Sweden). All other reagents were at least analytical grade for this study, and were purchased from Sigma-Aldrich (St Louis, MO, USA).
Animals
BALB/c-nu/nu female mice, approximately 4 weeks old and 18–20 g, were housed in specific pathogen-free conditions. Female Sprague Dawley rats, approximately 4–5 weeks old and 100±10 g, were purchased from Shanghai SLAC Laboratory Animal Co Ltd (laboratory animal production license SCXK [Hu]). The Institutional Animal Ethics Committee for Experimentation on Animals of Tongji University approved all animal experiments.
Preparation of rSPA antigen
A 245 bp fragment of the rSPA gene (verified via the National Center for Biotechnology Information website) was optimized based on E. coli codon usage, synthesized by GenScript, and subcloned into the pET-44a expression vector. Then, the recombinant plasmid pET-44a-rSPA was transformed into BL21(DE3) E. coli bacteria. The transformants were inoculated in Lysogeny broth medium containing 70 μg/mL ampicillin and incubated overnight at 37°C/250 rpm. Expression was induced with different concentrations of IPTG (0.5, 0.8, and 1 mM) in various induction times and temperatures (2, 4, and 8 hours and 25°C, 30°C, 37°C). The expressed protein was examined by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis gel and then purified by 1.0 mL nickel nitrilotriacetic acid resin (Qiagen, Valencia, CA, USA). Immunoblotting and enzyme-linked immunosorbent assay (ELISA) using anti-rSPA-poly-ant evaluated the purified protein, and GST protein was used as a control.
Immunization and construction of anti-rSPA-VHH libraries
Immunization and construction of anti-rSPA VHH libraries have been described in detail elsewhere.27–29 Briefly, a 2-year-old male alpaca was immunized weekly subcutaneously with at least 2 mg rSPA antigen for a period of 5 weeks. Nodular masses at the subcutaneous injection sites were observed after each immunization to ensure that the immunization procedure was performed appropriately. Blood was collected from the jugular vein prior to each injection, and the sera were used to monitor the immunization process, determined by indirect ELISA. The serum antibody titers were determined by the presence of a positive result at the highest possible dilution. Fourteen days after the last immunization, peripheral blood (200 mL) was collected and lymphocytes were isolated. Then, ribonucleic acid (RNA) was extracted using a Pure RNA Isolation Kit (Roche, Nutley, NJ, USA), and complementary DNA (cDNA) was synthesized via a Revert Aid First-Strand cDNA Synthesis Kit (Fermentas, Vilnius, Lithuania). Nested polymerase chain reaction was performed to amplify the VHH fragments by using primers specific for HcAbs, which were designed according to the constant region (CH2) of alpaca HcAbs.30 The amplified VHH fragments were ligated into the phagemid vector pCANTAB5E after being digested with the SfiI restriction enzyme. Next, the ligated vector was subsequently transformed into TG1 E. coli cells. The VHH repertoire was expressed in phage particles after a rescue procedure with the helper phage M13K07.
Panning and screening of anti-rSPA-specific nanobodies
The panning protocol was utilized as described elsewhere.27–29 Three consecutive rounds of panning using immunotubes that were coated with rSPA antigen were used to enrich the anti-rSPA-VHH libraries. Individual colonies that were specific for rSPA after the third round of panning were tested in a phage ELISA. Briefly, clones were grown in 2× YT medium with 100 μg·mL−1 ampicillin and 0.1% glucose medium. They were then infected with M13K07 helper phage and incubated at 37°C for 30 minutes. Then, 50 μg·mL−1 kanamycin was added and the clones amplified overnight at 37°C with shaking. The cultured clones were next centrifuged, and 100 μL of supernatant containing the recombinant phage particles was then added to rSPA precoated microtiter plate wells. After 2 hours’ incubation at 37°C, microtiter plate wells were washed three times with phosphate-buffered saline (PBS)-Tween 20 followed by the addition of anti-M13 HRP conjugate (1/5,000). VHH phages that were bound to rSPA were detected by adding 100 μL of HRP substrate (KPL, Gaithersburg, MD, USA). After 15 minutes of incubation, the reaction was stopped using 1 M H3PO4, and absorption at 405 nm was measured. The unique VHH genes of the clones that scored positive in phage ELISA were selected for sequencing analysis.
Expression and purification of nanobodies
After sequence confirmation, the clones with higher affinity were selected for prokaryotic expression. The production and purification of the selected clones was described earlier. Then, Western immunoblotting and ELISA confirmed the biological activity of the purified protein. As the expression vector contains His-tag, anti-His antibody was used as a secondary antibody. At the same time, the purified protein concentrations were measured by bicinchoninic acid assay.
Biophysical analyses of Nb17
First, Nb17 was adjusted to a concentration of 10 μg/mL in PBS. A goat anti-rSPA-poly-ant was used as control. Both were incubated at various temperatures (0°C, 10°C, 30°C, 50°C, 70°C, and 100°C) and at various pH values (3.5, 6.5, 7.0, 7.3, 7.5, 8.0, and 8.5 for 2 hours). After incubation, Nb17 and rSPA-poly-ant were subsequently incubated for 30 minutes at room temperature. To test antibody binding, all samples were detected using the standard procedure as described in the following. Nb17 and rSPA-poly-ant were transferred to rSPA-coated microplates. After incubation for 1 hour, microplates were blocked to reduce a specific binding with PBS containing 1% bovine serum albumin and 0.05% Tween 80. Then, Nb17 and rSPA-poly-ant was used as the second antibody. As the third step, goat antimouse conjugated with HRP was used.
Immunohistochemistry
In order to determine the binding activity of the purified anti-rSPA nanobodies to SPA, we performed immunohistochemistry using the frozen sections of the lung, heart, liver, spleen, kidney, and muscle tissue of rats. A goat anti-rSPA-poly-ant was used as a positive control for the primary antibody. The anti-rSPA nanobodies (Nb6 and Nb17) were used for the experimental groups, while an unrelated nanobody was used as the negative-control group. The secondary antibody used in both the experimental and the negative-control groups was mouse anti-His monoclonal antibody, and in the positive-control group, it was rabbit antigoat IgG.
Analysis of binding ability by QCM
The binding ability of Nb17 to rSPA antigen was further evaluated by QCM. The frequency change (Δf) is used to measure the ability of protein–protein binding. The formula of Sauerbrey − Δf =−2.6×105 f0ΔM/A − proved that for nanobodies with gold substrate antigen binding, the better the quality of the greater amount of change (ΔM), the greater the frequency, and the stronger the effect of protein binding. In this formula, f0 means quartz-oscillator base frequency (MHz), Δf is frequency quartz-oscillator frequency-shift value of the variable (Hz), ΔM is electrode material deposited on the mass of the variable (g), A is the area of the working electrode (cm2), and a negative number represents quality. The procedures included 1) 0.1% mercaptopropionic acid at electrode surfaces to produce a carboxylic acid-terminated monolayer, 2) activation by 5 mg/mL ethylene dichloride and 10 mg/mL N-hydroxysuccinimide to generate a stable acyl amino ester intermediate for 1 hour, and 3) adding 1 mL Nb17 and control nanobody to gold substrates. At 37°C 1-hour incubation under continuous monitoring, working ranges from 102 to 105 cells/mL with reproducibility <10% relative standard deviation were obtained. The frequency change (Δf) indicated the ability of protein–protein binding.
Analysis of targeting efficiency in vivo
Since there was a high degree of homology (95%) in the amino acid sequence of SPA between the Sprague Dawley rats and BALB/c-nu/nu female mice and it is much easier to image mice, we next used nude mice (six for each group) for in vivo imaging. The nude mice were randomly divided to an experimental group (receiving Nb17) and a control group (receiving a goat anti-rSPA-poly-ant), which were labeled with fluorescein isothiocyanate (FITC). The free FITC group were also assigned as controls. The experimental group was intravenously injected with 10 μL FITC-Nb17 (1 mg/kg), and the control group with the same amount of anti-rSPA-poly-ant, followed by imaging at multiple time points (0 and 15 minutes, and 1, 2, 3, and 6 hours) using a Xenogen small-animal in vivo imaging system (PerkinElmer, Waltham, MA, USA). For the imaging experiments, all the mice were first anesthetized with urethane.
Acute and chronic toxicity tests
Twenty Sprague Dawley female rats were randomly divided into four groups: 1) the acute toxicity group, 2) the acute toxicity control group, 3) the chronic toxicity group, and 4) the chronic toxicity control group. The acute toxicity group was intravenously injected with 10 mg/kg Nb17 and monitored for 1 week. The acute-negative control group was treated with the same volume of sterile normal saline. The chronic toxicity group was intravenously injected with Nb17 at a dose of 0.1 mg/kg Nb17 once daily for 3 months. The chronic-negative control group was treated with the same volume of saline daily for 3 months. Liver and kidney functions were then analyzed. Tissue injury of rat lung, liver, spleen, and kidney tissues stained with hematoxylin and eosin was investigated at the indicated time points.
Statistical analysis
All experimental data were statistically analyzed using SPSS 16.0 software. Continuous variables are expressed as mean ± standard deviation. Experimental data were analyzed by one-way analysis of variance, followed by least significant difference t-tests, when significant differences existed among groups. P-values of less than 0.05 were considered statistically significant.
Results
Preparation of antigen
The recombinant vector pET-44a-rSPA containing a 245-amino acid fragment of SPA was created as described in the Materials and methods section and transformed into BL21 E. coli. The optimum expression condition of the recombinant protein was found to be 0.8 mM IPTG at 37°C for 6 hours. The molecular weight of the expressed protein was estimated to be approximately 27 kDa, suggesting a correct size of typical rSPA protein. Immunoblotting analysis demonstrated that the expressed protein specifically bound to anti-rSPA polyclonal antibody at 27 kDa, whereas no band was seen in a control GST protein (Figure 1A). ELISA also showed robust binding activity between rSPA and goat anti-rSPA-poly-ant compared to the control protein (Figure 1B). Overall data suggest that we successfully constructed, produced, and purified rSPA.
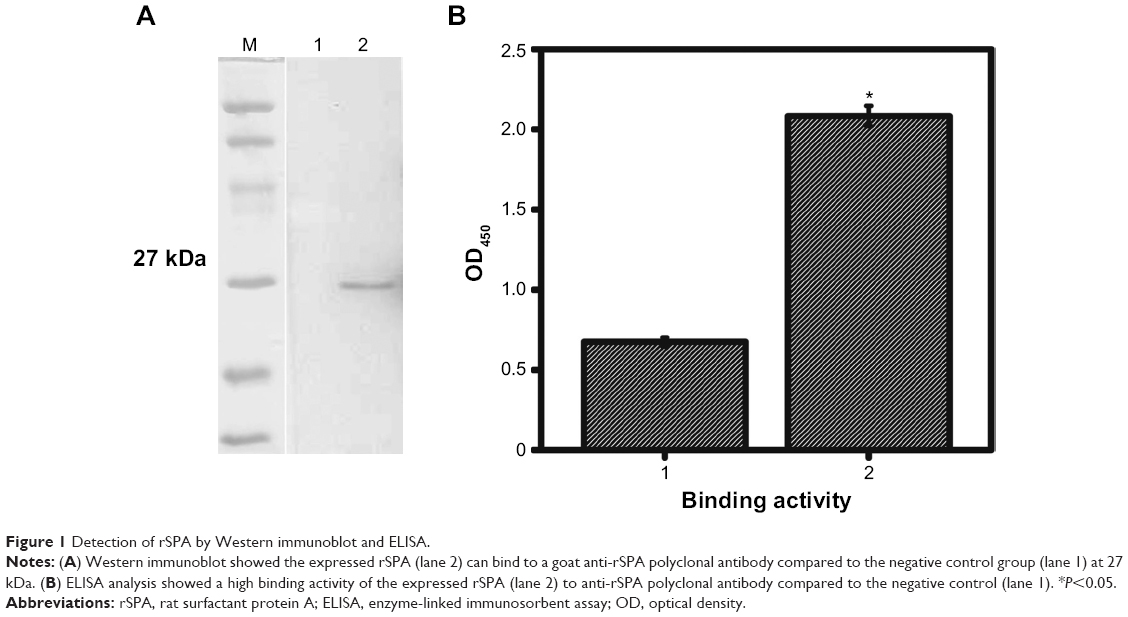 | Figure 1 Detection of rSPA by Western immunoblot and ELISA. |
Alpaca immunization, generation, and screening of anti-rSPA nanobody library
An alpaca was immunized with the recombinant rSPA, and blood serum was separated before each immunization. Rabbit anti-alpaca IgG labeled with HRP was prepared as a second antibody. Our results showed that the antibody titer gradually increased with increasing immunization times, and reached an effective concentration of 1:12,000 after the fourth immunization (Table 1).
 | Table 1 Alpaca immune antibody titer |
Lymphocytes were purified from 200 mL peripheral blood of the rSPA-immunized alpaca and total messenger RNA from 2.9×109 lymphocytes was extracted using Trizol reagent to obtain cDNA. The VHH fragments with a size of 500 bp were generated by polymerase chain-reaction amplification. A VHH library with 5.93×105 fragments was obtained. After three consecutive rounds of in vitro panning using the rSPA antigen, phage particles, which bound specifically to rSPA, were successfully enriched (Table 2). A total of 31 random clones with high affinity for rSPA were identified by phage ELISA. DNA-sequencing analysis revealed that six clones among them were IgG2a subtype, and 25 clones were IgG3 subtype (Figure 2A and B). Cluster analysis showed that 31 individual clones contained 16 different sequences, among which Nb17, 2, and 4 contained significant repeats (identical sequences), and Nb12, 18, 8, and 20 and Nb6, 16, 25, and 7 contained another two types of repeats, which indicated significant enrichment of these three isotypes (Figure 2C). After initial testing of binding specificity for the antigen, we designated two of them (Nb17, Nb6) and performed further characterization.
 | Table 2 Enrichment after three rounds of panning |
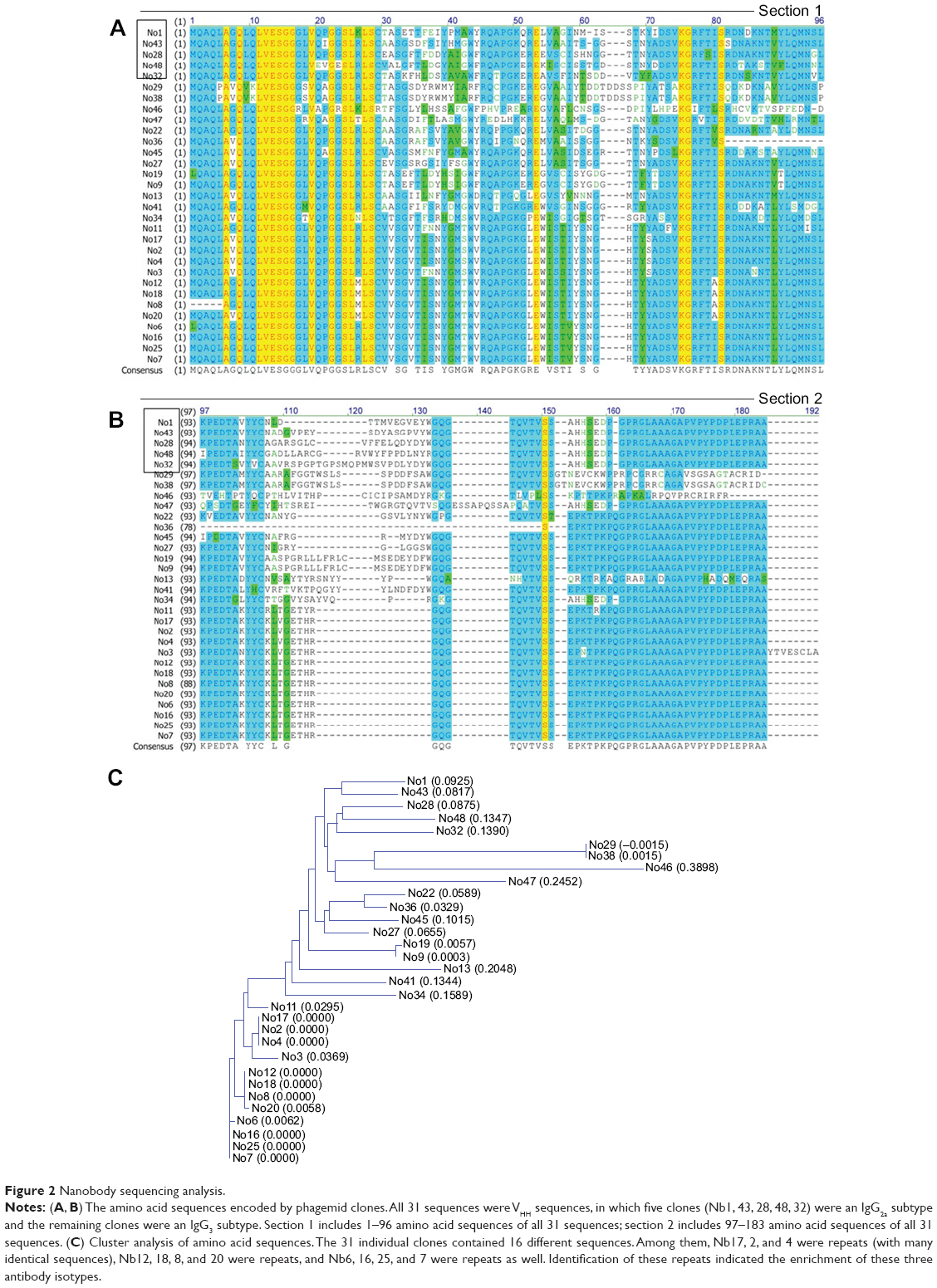 | Figure 2 Nanobody sequencing analysis. |
Production and binding specificity of anti-rSPA nanobodies
Two clones, Nb6 and Nb17, were chosen for further characterization. Expression conditions of both sequences were analyzed using various temperatures and IPTG concentrations. Optimal expression levels were attained at 25°C for 4 hours with 0.8 mM IPTG. The molecular weights of these proteins were approximately 19 kDa and were soluble. The concentration of the purified proteins was approximately 25 mg/mL. With immunoblotting, rSPA reacted well with purified Nb6 and Nb17 with a single band of 19 kDa (Figure 3A). ELISA also showed that the purified Nb6 and Nb17 had positive reactivity to rSPA (Figure 3B).
 | Figure 3 Analysis of the binding specificity of Nb6 and Nb17 to rSPA. |
To investigate further the binding ability of Nb6 and Nb17 to naturally occurring rSPA, immunohistochemistry was performed on cryosections of rat lung, heart, liver, spleen, kidney, and muscle. Our results showed that both Nb6 and Nb17 preferentially bound to rat lung tissue (Figure 4, brown staining), but did not show binding to the heart, liver, spleen, kidney, or muscle, and the same-degree reaction with the lung tissues by anti-SPA-poly-ant (positive control) was also detected. However, the binding ability of Nb6 was weaker than that of Nb17 and anti-SPA-poly-ant (Figure 4). No significant binding reaction was observed in the negative-control group.
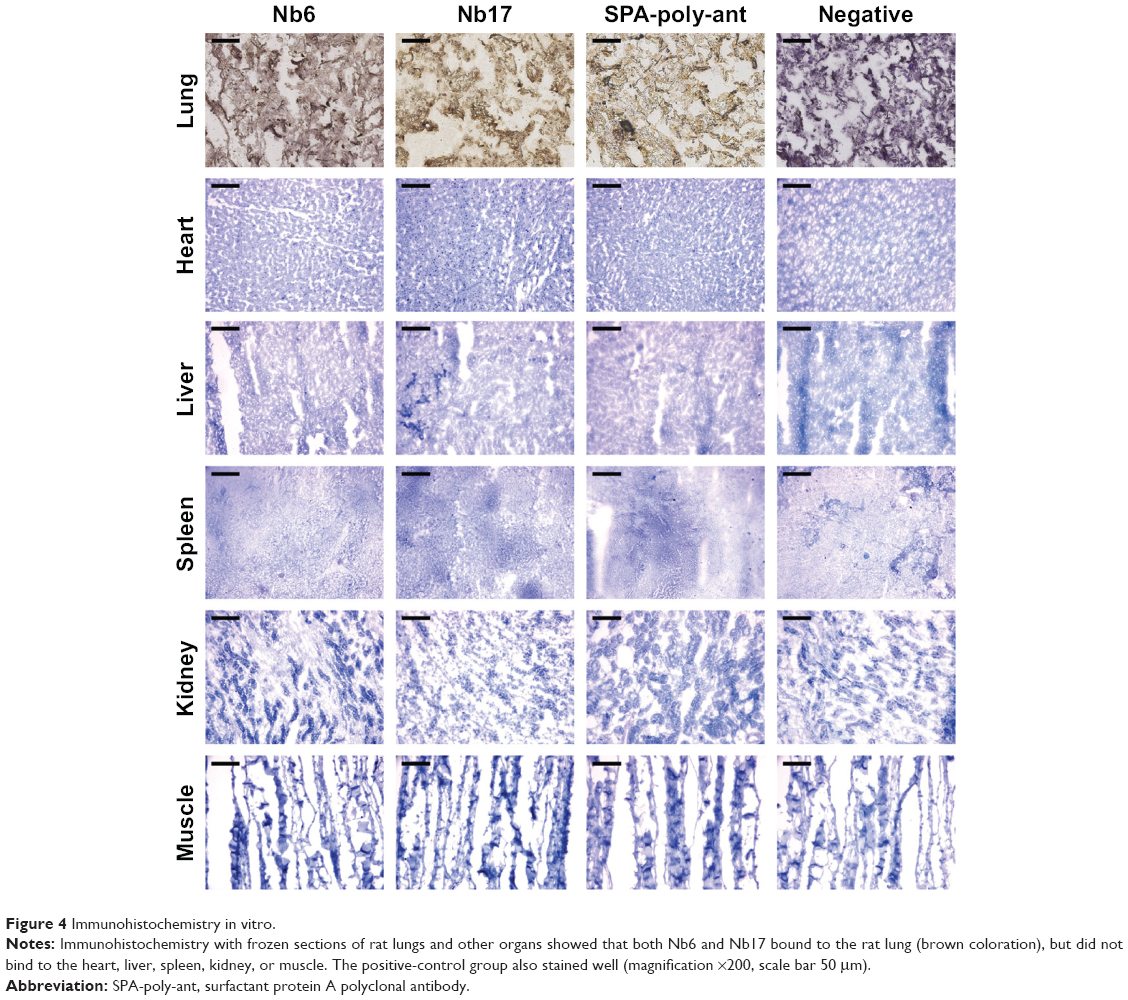 | Figure 4 Immunohistochemistry in vitro. |
Since Nb17 showed a higher binding ability to rSPA in immunohistochemistry than Nb6, we chose Nb17 to evaluate its targeting ability further. In order to investigate the biophysical feature of Nb17, a standard antigen-specific ELISA was performed to test whether binding could be observed at various temperatures and various pH values. As shown in Figure 5, contrary to conventional rSPA-poly-ant, Nb17 was found to be far more stable. In more detail, for Nb17, it is clearly shown that it specifically bound to rSPA even at temperatures of 90°C and pH values of 3.5 or at 8.0. However, rSPA-poly-ant showed almost no antigen binding when the temperature was above 70°C and acid or alkaline conditions were extreme.
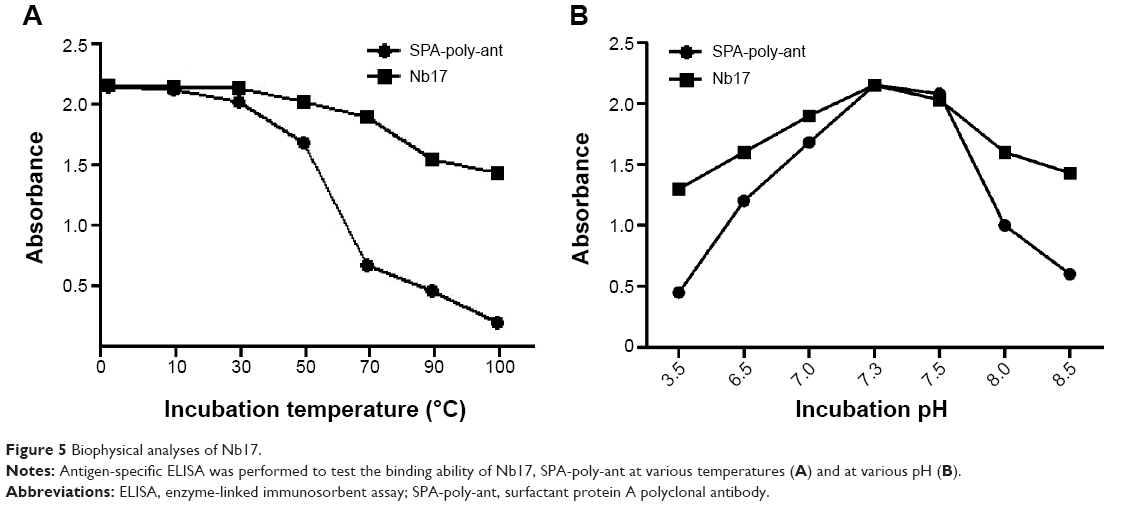 | Figure 5 Biophysical analyses of Nb17. |
We next performed QCM based on the frequency change of quartz crystal resonance, which has been widely used to characterize protein features, including protein–protein interaction. To confirm the specific binding of Nb17 to rSPA, an irrelevant nanobody that did not recognize rSPA was generated and used as a negative control. As Figure 6A shows, the frequency change of Nb17 (Δf17) was significantly higher than that of the control nanobody (ΔfCTR) by the passage of time, indicating the high binding ability between Nb17 and rSPA. These results suggest that our Nb17 possesses high specificity for rSPA.
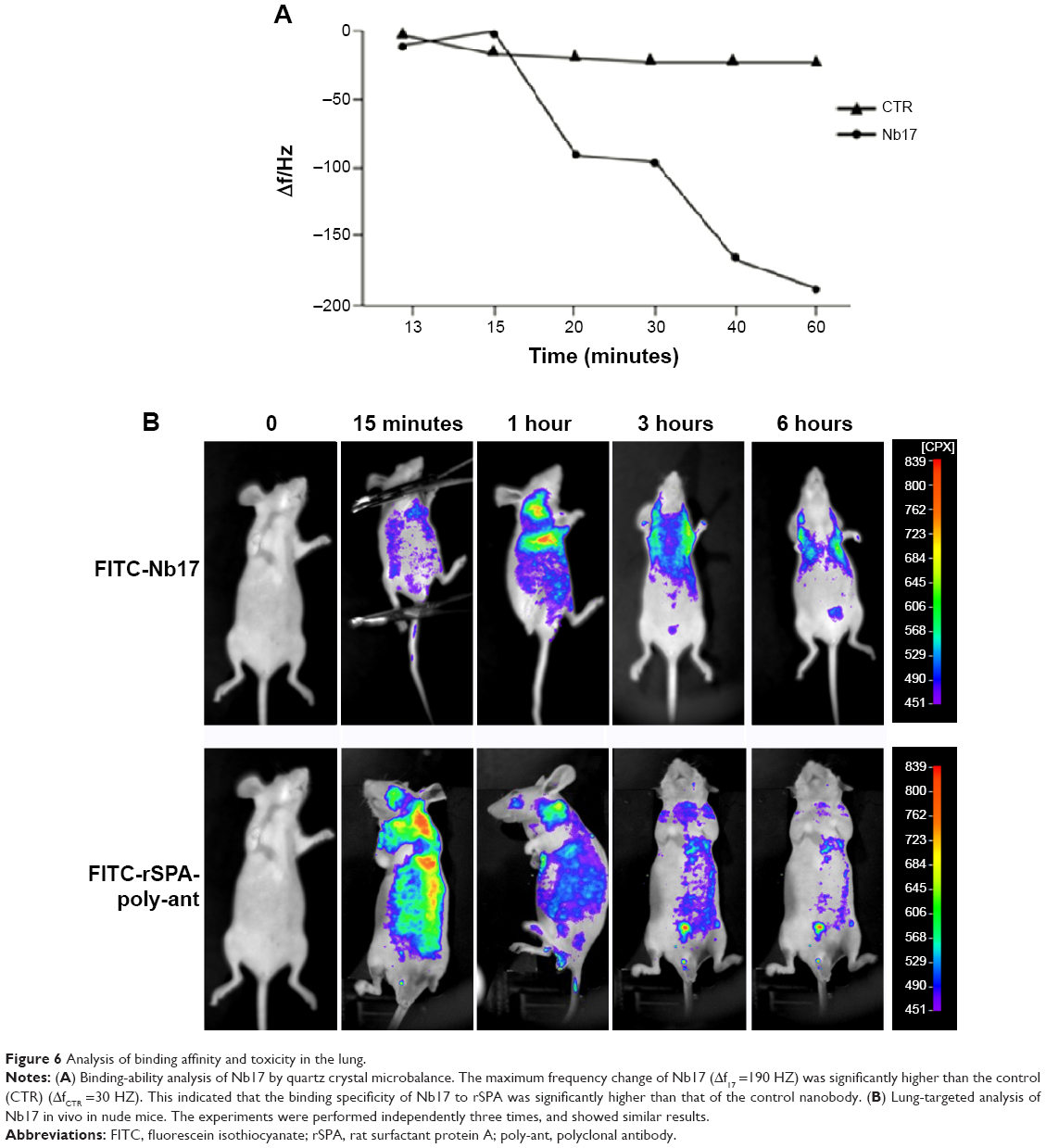 | Figure 6 Analysis of binding affinity and toxicity in the lung. |
Analysis of the targeting ability of Nb17 in mice
We chose a mouse model for in vivo imaging to detect the lung-targeting ability of Nb17, as rats have a high homology (95%) to the mouse SPA amino acid sequence. As shown in Figure 6B, we injected FITC-labeled Nb17 (FITC-Nb17) or FITC-SPA-poly-ant (control). FITC-Nb17 began to accumulate in mouse lungs at 15 minutes after tail-vein injection, and was almost completely cleared from the bladder at 6 hours after injection. However, no concentrated FITC was detected in any other organs. SPA-poly-ant also began to enter mouse lungs at 15 minutes after intravenous injection, but its binding to rat lungs was much lower than that of the Nb17-FITC. We labeled the protein according to the reaction between the R-amino group on lysine of protein and carbon–sulfur bond amine on fluorescence to form a FITC-protein conjugate (fluorescence antibody). A whole IgG antibody has 86 lysine residues, and 15–20 of them can be used for the reaction. As a nanobody, Nb17’s size is only 10% of an IgG antibody, so the overall fluorescence that can bind is much less than that of a whole IgG antibody. FITC-SPA-poly-ant were also seen in other organs at high concentration besides lungs, and almost completely removed from the lungs and bladder of rats at 6 hours after injection. The free FITC group failed to show organ-specific accumulation, and was completely cleared from the kidney and the bladder of rats (data not shown).
Analysis of acute and chronic toxicity
Serum alanine transaminase, aspartate transaminase, blood urea nitrogen, and serum creatinine were determined to assess the acute (1-week) and chronic (3-month) toxicity in rats as detailed earlier. No statistically significant differences were found between any of the four groups by multiple-comparison analysis (P>0.05, Table 3). Therefore, overall data indicated that Nb17 had not induced significant toxicity. We further analyzed the histological alterations caused by Nb17 using cryosections of the lung, liver, spleen, and kidney in acute toxicity, chronic toxicity, and control groups. Our results showed that there were no apparent histological alterations by morphological assessment in tissues of the lung, liver, spleen, or kidney of rats between the Nb17 groups and controls using hematoxylin and eosin staining (Figure 7).
 | Table 3 Liver- and kidney-function test |
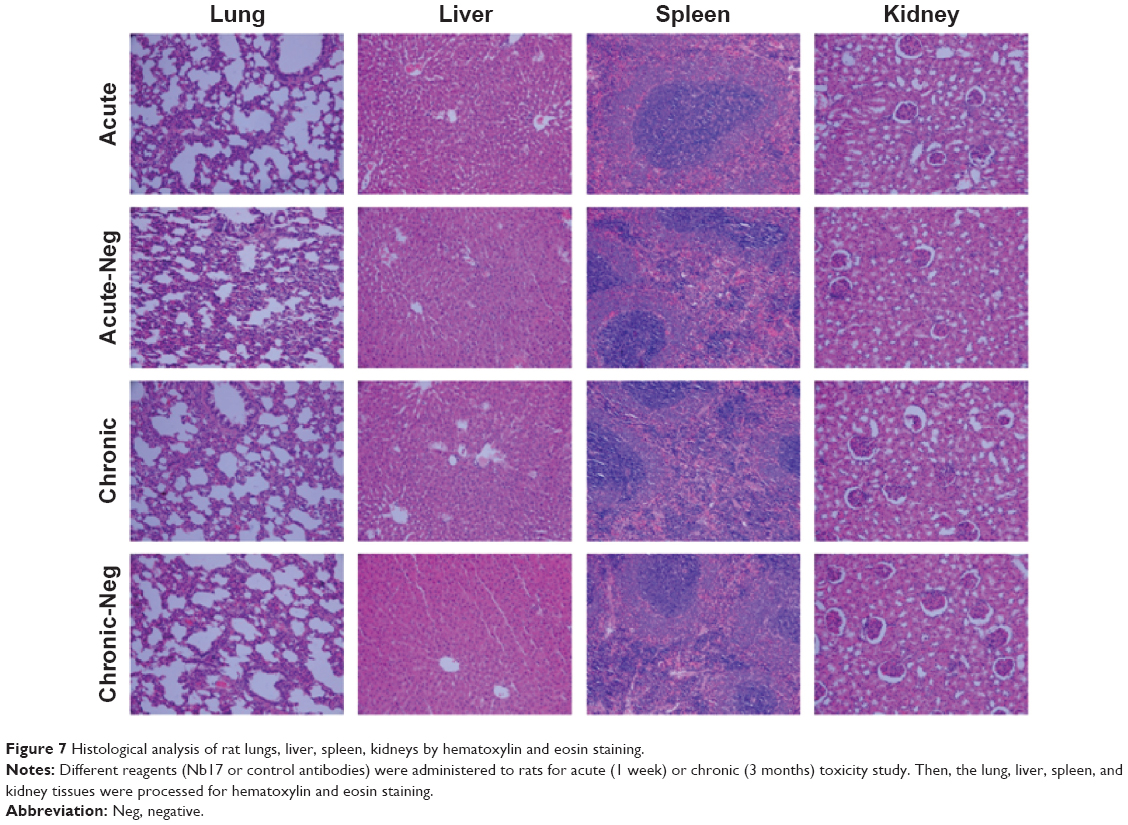 | Figure 7 Histological analysis of rat lungs, liver, spleen, kidneys by hematoxylin and eosin staining. |
Discussion
Active targeting drugs are believed to have favorable properties, such as concentrated drugs at the site of the disease, enhanced therapeutic effects, reduced systemic side effects, and lowered costs. However, development of active lung-targeting drugs has been impeded by the difficulty in identifying specific lung targets and their appropriate ligands. Thus far, studies on lung targeting have been largely dependent on the mechanical retention of pulmonary capillary and airway inhalation, in which both have intrinsic limitations. To explore a new way for lung targeting, peptide targeting to the pulmonary vasculature was developed by screening phage-peptide libraries in vivo after intravenous injection into the lungs of mice, due to the differences in vascular endothelial cells from different organs.31–33 However, this method has met limited success in clinical application, in part because of the increased risk of thrombosis. Therefore, it is necessary to explore a more effective lung-targeting strategy.
rSPA is one of the most abundant pulmonary surfactant proteins (SPs), and is expressed on alveolar epithelial type II cells and Clara cells. In addition, rSPA messenger RNA can also be detected in the epithelial cells of the middle ear, jejunum, colon, and serosa mesothelium, but in much lower protein levels.14 In our previous work, we also showed that rSPA can serve as a lung-targeting molecule. However, natural SPA cannot meet experimental requirements, since it is primarily extracted from bronchoalveolar lavage fluid, and this procedure is associated with very complicated steps and a low yield. In this study, we generated recombinant rSPA by a prokaryotic expression system, and successfully used the rSPA as an immunogen to produce lung-targeting nanobodies.
Ligands for ideal lung targeting require not only a high affinity to the target but also unique characteristics, including low molecular weight, low immunogenicity, and high tissue penetration. As the binding between antigen and antibody has high affinity and stability, anti-SPA antibody may be suitable for lung-targeting ligands. However, conventional antibodies (such as a polyclonal and monoclonal antibodies) consisting of two heavy and two light chains suffer from a number of drawbacks, such as high molecular weight (150 kDa), high immunogenicity, low tissue penetration, and high costs in production. Recent technical advances have allowed the transformation of those conventional antibody molecules into small antibody molecules (eg, Fab and single-chain variable fragments) through genetic engineering technology, but these molecules are not satisfactory for specific ligands because of their decreased stability and affinity to their antigens. Interestingly, these drawbacks of conventional antibodies may be solved by using single-chain antibody fragments (VHHs or nanobodies). A nanobody is the smallest antibody molecule, with intact antigen-binding capability, strong stability, and tissue penetration. Nanobodies have been applied in multiple fields of medicine as a novel antibody drug.34–36 However, there have been no reports of any anti-rSPA nanobodies for lung-targeting ligands.
The ability to isolate a specific nanobody from immunized phage libraries has been used by several groups.27–29 In the present study, we successfully immunized an alpaca using our recombinant rSPA, and constructed a size of 5.93×105 cfu immunized library. Fortunately, 31 clones with high affinity for rSPA were successfully screened using a phage ELISA. After DNA sequencing, 16 different VHH sequences were obtained. Nanobodies are highly expressed in a variety of microorganisms, since they are encoded by a single gene, small molecular weight, and simple structure.37–40 The methods for preparation of nanobodies are straightforward compared to the procedures for genetic engineering antibody preparation. The two nanobodies (Nb6 and Nb17) in our current study also can be routinely prepared under normal culture conditions, establishing a simple and efficient approach for nanobody preparation.
The binding ability of the nanobodies (Nb6 and Nb17) with rSPA was assessed and validated by immunoblotting and ELISA. Our data indicated that both Nb6 and Nb17 can specifically bind to naturally occurring rSPA. Using immunohistochemistry, the binding ability of Nb17 was as strong as the positive-control group (a commercial anti-SPA-poly-ant), while Nb6 was relatively weak. One possible explanation for this phenomenon is that in spite of the high degree of similarity between the sequences of Nb17 and Nb6, a tiny difference in variable region contributes enormously to binding strengths.
When antibodies were tested for functional binding at various temperatures and at various pH conditions to their antigen, major differences were observed between the Nb17 and rSPA-poly-ant. It is possible that the extreme conditions resulted in dissociation of the polyantibody structure, thereby exposing the hydrophobic interfaces on both heavy and light chains. The exposed hydrophobic interfaces subsequently aggregated and precipitated, resulting in nonfunctional molecules. However, contrary to conventional antibodies, because of the efficient refolding after chemical or thermal denaturation of nanobodies and an additional disulfide bond between complementary determining regions CDR1 and CDR3, improved protein stability can be obtained.41,42
To determine further the lung-targeting ability of the recombinant nanobody, we performed in vivo imaging by labeling Nb17 with FITC (FITC-Nb17). FITC-Nb17 began to accumulate in the lung at 15 minutes after intravenous injection, but was completely metabolized by the kidney and bladder at 6 hours after injection. Compared to SPA-poly-ant, FITC-Nb17 had much higher accumulation and longer retention in the lung. Importantly, no obvious accumulation of free FITC (a negative control) was detected in any organs. This finding strongly suggests not only strong lung-targeting capability for Nb17 but also limited retention in the liver and spleen. Although it is impossible to have no cross-reactivity, background retention in those organs appeared to be minimal.
In addition to analyzing in vivo biodistribution, we also carried out acute and chronic toxicity tests with the anti-rSPA nanobodies. Our results confirmed the safety of the nanobodies in rats, which is consistent with previous reports.43 In fact, the nanobodies had small molecular weights with only one domain and without an Fc domain of a traditional antibody, thus avoiding complement activation caused by the Fc domain. Additionally, the nanobodies bore a high degree of homology with the human VH gene-family sequence, which may contribute to their low immunogenicity and high safety profile in the context of using nanobodies in future clinical therapeutics.
Acknowledgments
This study was funded by grants from the National Science Foundation of China (91442103, 81170011, and 81200045); the Science and Technology Commission of Shanghai Municipality (12DJ1400103, 124119a9000, 12DZ1942500, and 12411950105) and the Health Bureau Program of Shanghai Municipality (SHDC12014120, 2013SY047). The authors would like to thank Kirby Huber (Department of Basic Sciences, School of Medicine and Health Sciences, University of North Dakota), a native English speaker, for proofreading of this manuscript.
Author contributions
All authors contributed toward data analysis, drafting, and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Raissy HH, Kelly HW, Harkins M, Szefler SJ. Inhaled corticosteroids in lung diseases. Am J Respir Crit Care Med. 2013;187(8):798–803. | ||
Zervas E, Samitas K, Gaga M, Beghe B, Fabbri LM. Inhaled corticosteroids in COPD: pros and cons. Curr Drug Targets. 2013;14(2):192–224. | ||
Karra N, Nassar T, Laenger F, Benita S, Borlak J. Safety and proof-of-concept efficacy of inhaled drug loaded nano- and immunonanoparticles in a c-Raf transgenic lung cancer model. Curr Cancer Drug Targets. 2013;13(1):11–29. | ||
Dandekar P, Venkataraman C, Mehra A. Pulmonary targeting of nanoparticle drug matrices. J Aerosol Med Pulm Drug Deliv. 2010;23(6):343–353. | ||
Zhang X, Sun P, Bi R, Wang J, Zhang N, Huang G. Targeted delivery of levofloxacin-liposomes for the treatment of pulmonary inflammation. J Drug Target. 2009;17(5):399–407. | ||
Wei Y, Zhao L. Passive lung-targeted drug delivery systems via intravenous administration. Pharm Dev Technol. 2014;19(2):129–136. | ||
Irwin RS, Richardson ND. Side effects with inhaled corticosteroids: the physician’s perception. Chest. 2006;130(1 Suppl):41S–53S. | ||
Maya S, Kumar LG, Sarmento B, et al. Cetuximab conjugated O-carboxymethyl chitosan nanoparticles for targeting EGFR overexpressing cancer cells. Carbohydr Polym. 2013;93(2):661–669. | ||
Kouchakzadeh H, Shojaosadati SA, Tahmasebi F, Shokri F. Optimization of an anti-HER2 monoclonal antibody targeted delivery system using PEGylated human serum albumin nanoparticles. Int J Pharm. 2013;447(1–2):62–69. | ||
Banerjee R, Tyagi P, Li S, Huang L. Anisamide-targeted stealth liposomes: a potent carrier for targeting doxorubicin to human prostate cancer cells. Int J Cancer. 2004;112(4):693–700. | ||
Omidfar K, Amjad Zanjani FS, Hagh AG, Azizi MD, Rasouli SJ, Kashanian S. Efficient growth inhibition of EGFR over-expressing tumor cells by an anti-EGFR nanobody. Mol Biol Rep. 2013;40(12):6737–6745. | ||
Heukers R, Altintas I, Raghoenath S, et al. Targeting hepatocyte growth factor receptor (Met) positive tumor cells using internalizing nanobody-decorated albumin nanoparticles. Biomaterials. 2014;35(1):601–610. | ||
Li L, Wartchow CA, Danthi SN, et al. A novel antiangiogenesis therapy using an integrin antagonist or anti-Flk-1 antibody coated 90Y-labeled nanoparticles. Int J Radiat Oncol Biol Phys. 2004;58(4):1215–1227. | ||
Akiyama J, Hoffman A, Brown C, et al. Tissue distribution of surfactant proteins A and D in the mouse. J Histochem Cytochem. 2002;50(7):993–996. | ||
Kingma PS, Whitsett JA. In defense of the lung: surfactant protein A and surfactant protein D. Curr Opin Pharmacol. 2006;6(3):277–283. | ||
Barreira ER, Precioso AR, Bousso A. Pulmonary surfactant in respiratory syncytial virus bronchiolitis: the role in pathogenesis and clinical implications. Pediatr Pulmonol. 2011;46(5):415–420. | ||
Chen XY, Wang SM, Li N, et al. Creation of lung-targeted dexamethasone immunoliposome and its therapeutic effect on bleomycin-induced lung injury in rats. PloS One. 2013;8(3):e58275. | ||
Chapman AP. PEGylated antibodies and antibody fragments for improved therapy: a review. Adv Drug Deliv Rev. 2002;54(4):531–545. | ||
Hamers-Casterman C, Atarhouch T, Muyldermans S, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363(6428):446–448. | ||
Rahbarizadeh F, Ahmadvand D, Sharifzadeh Z. Nanobody; an old concept and new vehicle for immunotargeting. Immunol Invest. 2011;40(3):299–338. | ||
Muyldermans S, Baral TN, Retamozzo VC, et al. Camelid immunoglobulins and nanobody technology. Vet Immunol Immunopathol. 2009;128(1–3):178–183. | ||
Ebrahimizadeh W, Mousavi Gargari S, Rajabibazl M, Safaee Ardekani L, Zare H, Bakherad H. Isolation and characterization of protective anti-LPS nanobody against V. cholerae O1 recognizing Inaba and Ogawa serotypes. Appl Microbiol Biotechnol. 2013;97(10):4457–4466. | ||
van der Linden RH, Frenken LG, de Geus B, et al. Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim Biophys Acta. 1999;1431(1):37–46. | ||
Huang L, Muyldermans S, Saerens D. Nanobodies: proficient tools in diagnostics. Expert Rev Mol Diagn. 2010;10(6):777–785. | ||
Van Bockstaele F, Holz JB, Revets H. The development of nanobodies for therapeutic applications. Curr Opin Investig Drugs. 2009;10(11):1212–1224. | ||
De Groeve K, Deschacht N, De Koninck C, et al. Nanobodies as tools for in vivo imaging of specific immune cell types. J Nucl Med. 2010;51(5):782–789. | ||
Alvarez-Rueda N, Ladjemi MZ, Behar G, et al. A llama single domain anti-idiotypic antibody mimicking HER2 as a vaccine: immunogenicity and efficacy. Vaccine. 2009;27(35):4826–4833. | ||
Ardekani LS, Gargari SL, Rasooli I, et al. A novel nanobody against urease activity of Helicobacter pylori. Int J Infect Dis. 2013;17(9):e723–e728. | ||
Roovers RC, Laeremans T, Huang L, et al. Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR nanobodies. Cancer Immunol Immunother. 2007;56(3):303–317. | ||
Doyle PJ, Arbabi-Ghahroudi M, Gaudette N, et al. Cloning, expression, and characterization of a single-domain antibody fragment with affinity for 15-acetyl-deoxynivalenol. Mol Immunol. 2008;45(14):3703–3713. | ||
Giordano RJ, Edwards JK, Tuder RM, Arap W, Pasqualini R. Combinatorial ligand-directed lung targeting. Proc Am Thorac Soc. 2009;6(5):411–415. | ||
Wu M, Pasula R, Smith PA, Martin WJ 2nd. Mapping alveolar binding sites in vivo using phage peptide libraries. Gene Ther. 2003;10(17):1429–1436. | ||
Rajotte D, Ruoslahti E. Membrane dipeptidase is the receptor for a lung-targeting peptide identified by in vivo phage display. J Biol Chem. 1999;274(17):11593–11598. | ||
Heukers R, van Bergen En, Henegouwen PM, Oliveira S. Nanobody-photosensitizer conjugates for targeted photodynamic therapy. Nanomedicine. 2014;10(7):1441–1451. | ||
Bakherad H, Mousavi Gargari SL, Rasooli I, et al. In vivo neutralization of botulinum neurotoxins serotype E with heavy-chain camelid antibodies (VHH). Mol Biotechnol. 2013;55(2):159–167. | ||
Siller-Matula JM, Krumphuber J, Jilma B. Pharmacokinetic, pharmacodynamic and clinical profile of novel antiplatelet drugs targeting vascular diseases. Br J Pharmacol. 2010;159(3):502–517. | ||
Ryckaert S, Pardon E, Steyaert J, Callewaert N. Isolation of antigen-binding camelid heavy chain antibody fragments (nanobodies) from an immune library displayed on the surface of Pichia pastoris. J Biotechnol. 2010;145(2):93–98. | ||
Teh YH, Kavanagh TA. High-level expression of camelid nanobodies in Nicotiana benthamiana. Transgenic Res. 2010;19(4):575–586. | ||
Han Z, Zhang B, Wang YE, Zuo YY, Su WW. Self-assembled amyloid-like oligomeric-cohesin scaffoldin for augmented protein display on the Saccharomyces cerevisiae cell surface. Appl Environ Microbiol. 2012;78(9):3249–3255. | ||
Lentz EM, Garaicoechea L, Alfano EF, Parreno V, Wigdorovitz A, Bravo-Almonacid FF. Translational fusion and redirection to thylakoid lumen as strategies to improve the accumulation of a camelid antibody fragment in transplastomic tobacco. Planta. 2012;236(2):703–714. | ||
Monegal A, Olichon A, Bery N, Filleron T, Favre G, de Marco A. Single domain antibodies with VH hallmarks are positively selected during panning of llama (Lama glama) naïve libraries. Dev Comp Immunol. 2012;36(1):150–156. | ||
Harmsen MM, De Haard HJ. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol. 2007;77(1):13–22. | ||
Vu KB, Ghahroudi MA, Wyns L, Muyldermans S. Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol Immunol. 1997;34(16–17):1121–1131. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.