Back to Journals » OncoTargets and Therapy » Volume 13
A Novel Imidazopyridine Derivative Exhibits Anticancer Activity in Breast Cancer by Inhibiting Wnt/β‑catenin Signaling
Authors He LJ, Yang DL ![]() , Chen HY, Huang JH, Zhang YJ, Qin HX, Wang JL, Tang DY
, Chen HY, Huang JH, Zhang YJ, Qin HX, Wang JL, Tang DY ![]() , Chen ZZ
, Chen ZZ
Received 26 June 2020
Accepted for publication 1 September 2020
Published 9 October 2020 Volume 2020:13 Pages 10111—10121
DOI https://doi.org/10.2147/OTT.S266752
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Leo Jen-Liang Su
Liu-Jun He,1 Dong-Lin Yang,1 He-Ying Chen,1– 3 Jiu-Hong Huang,1 Ya-Jun Zhang,1 Hong-Xia Qin,1 Juan-Li Wang,1 Dian-Yong Tang,1 Zhong-Zhu Chen1
1National & Local Joint Engineering Research Center of Targeted and Innovative Therapeutics, Chongqing Key Laboratory of Kinase Modulators as Innovative Medicine, Chongqing Engineering Laboratory of Targeted and Innovative Therapeutics, Chongqing Collaborative Innovation Center of Targeted and Innovative Therapeutics, College of Pharmacy & International Academy of Targeted Therapeutics and Innovation, Chongqing University of Arts and Sciences, Chongqing 402160, People’s Republic of China; 2Division of Molecular Nephrology and the Creative Training Center for Undergraduates, The Ministry of Education Key Laboratory of Laboratory Medical Diagnostics, The College of Laboratory Medicine, Chongqing Medical University, Chongqing 400016, People’s Republic of China; 3The Undergraduates Class of 2016 Entry the College of Laboratory Medicine, Chongqing Medical University, Chongqing 400016, People’s Republic of China
Correspondence: Dong-Lin Yang; Zhong-Zhu Chen
National & Local Joint Engineering Research Center of Targeted and Innovative Therapeutics, Chongqing Key Laboratory of Kinase Modulators as Innovative Medicine, Chongqing Engineering Laboratory of Targeted and Innovative Therapeutics, Chongqing Collaborative Innovation Center of Targeted and Innovative Therapeutics, College of Pharmacy & International Academy of Targeted Therapeutics and Innovation, Chongqing University of Arts and Sciences, Chongqing 402160, People’s Republic of China
Tel/Fax +86-23-61162836
Email [email protected]; [email protected]
Background: Breast cancer exhibits poor prognosis and high relapse rates following chemotherapy therapeutics. Thus, this study aims to develop effective novel agents regulating the core molecular pathway of breast cancer such as Wnt/β-catenin signaling.
Methods: The present study screened a novel inhibitor, called “C188”, using MTT assay. The molecular formula of C188 is C21H15FN4O3 and the molecular weight is 390. Flow cytometry and Western blotting were employed to assess cell cycle arrest after treatment with C188. Wound-healing and transwell assays were applied to measure the cell migration and invasion viability. The regulatory effects of C188 on Wnt/β‑catenin signaling and localization of β‑catenin in the nucleus were investigated by Western blotting and immunofluorescence.
Results: We found that C188 significantly suppressed proliferation and growth in a dose- and time-dependent manner in breast cancer cells, but not in normal breast cells. The inhibitory effect was caused by cell cycle arrest at the G1-phase which is induced by C188 treatment. Additionally, C188 dramatically inhibited cell migration of breast cancer cells in a dose-dependent manner. The migration inhibition was attributed to the suppression of Wnt/β‑catenin signaling and localization of β‑catenin in the nucleus mediated by regulating phosphorylation of β‑catenin and its subsequent stability. Furthermore, the target genes, including Axin 2, c-JUN, and c-Myc, were downregulated due to the decrease of β‑catenin in the nucleus after exposure to C188.
Conclusion: C188 treatment resulted in the downregulation of cyclin D which led to cell cycle arrest at the G1 phase, and the inhibition of cell migration, indicating that C188 may be an effective novel therapeutic candidate as a potential treatment for human breast cancer.
Keywords: C188, breast cancer, proliferation, cell cycle, cell migration, Wnt/β-catenin
Introduction
Breast cancer is the leading cause of cancer morbidity and mortality among women worldwide. Breast cancer is a heterogeneous disease and currently the major therapy involves mastectomy with chemo- and radio-therapy.1 However, breast cancer exhibits high tolerance and relapse rates to traditional treatments.2 Hence, it is urgent to develop effective therapeutic agents which have the anticancer ability to breast cancer on the one hand and minimize the side-effects on the other.
The Wnt/β-catenin signaling has significant implications in both cellular homeostasis and embryonic development, and abnormal activation has been involved in the carcinogenesis such as breast cancers.3–6 The mutations of β-catenin are rare, while its expression and nuclear accumulation is frequently abnormal in breast cancer, demonstrating a constitutive activation in Wnt/β-catenin signaling. β-catenin is a vital protein in this signaling and is regulated by the destruction complex consisting of Axin, adenomatous polyposis coli (APC), glycogen synthase kinase 3 (GSK3), casein kinase 1 (CK1), and β-transducing repeat-containing protein (β-TrCP).7 The destruction complex induces β-catenin phosphorylation at several sites containing Ser45/Thr41/Ser37/Ser33, resulting in its subsequent ubiquitination and proteasomal degradation. However, β-catenin accumulates in the cytoplasm and enters into the nucleus causing aberrant Wnt signaling activation. Subsequently, β-catenin transfers into the nucleus from cytoplasm and forms a transcription complex with T-cell-specific transcription factor (TCF)/LEF-1, contributing to the transcription of downstream target genes responsible for the progression and metastasis of cancers.8 Previous studies showed that primary cells from breast tumors as well as breast cancer cell lines expressed several Wnt ligands and Frizzled (Fzd) receptors.9,10 Notably, expression of low-density-lipoprotein-related protein 6 (LRP6) and disheveled (DVL), and phosphorylated DVL proteins are upregulated in human breast cancer cells. Fzd combines with LRP6 to form a receptor complex which can be bound by Wnt proteins. Then, the combination of cytoplasmic part of Fzd with DVL can offer a platform for the interaction between the LRP tail and Axin, which finally leads to the activation of Wnt/β-catenin signaling.11,12 Therefore, agents targeting Wnt/β-catenin signaling are potentially anticancer medicine.
Imidazopyridine derivatives, consisting of aromatic aldehydes and a pyridine group, have been reported to be effective inhibitors against tumor growth and advancement in several carcinomas, including pancreatic, lung, breast, and prostate cancers.13–16 Additionally, a recent study showed that a novel Glycogen synthase kinase 3α/β (GSK3α/β) inhibitor, CG0009, which is an Imidazopyridine derivative, can result in the activation of pro-survival signal of β-catenin and slight upregulation of the β-catenin target genes, c-Jun and c-Myc, but not cyclin D1. CG0009-mediated cyclin D1 depletion overwhelms the pro-survival signal of β-catenin, leading to cell death in breast cancer cell lines.17 However, the report toward the effect of imidazopyridine derivatives on breast cancer by suppressing Wnt/β-catenin signaling and its downstream targets is less. Therefore, we found C188, an imidazopyridine derivative, is an effective and potent anticancer inhibitor targeting Wnt/β-catenin signaling. Our data, to the best of our knowledge, is the first to demonstrate the anticancer effect of this novel imidazopyridine derivative on breast cancer cells and shows C188 may be a promising lead therapeutic agent for future studies.
Materials and Methods
Reagents and Antibodies
The C188 compound was purchased from TagerMol (C188-0056, Shanghai, China). The molecular formula of C188 is C21H15FN4O3 and the molecular weight is 390.38 (Supplemental Figure S1). 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) was purchased form Sangon Biotech (A600799-0005, Shanghai, China). MG132 (S2619) was purchased from Selleck. Propidium iodide (PI, ST512) and Crystal Violet Staining Solution (C0121) were obtained from Beyotime Biotechnology (Shanghai, China). The CDK4(12790S), CDK6(3136S), Cyclin D1(55506S), p21(2947S), p27(3686S), β-Actin(3700S), Axin1(2087S), LRP6(2560S), Dvl3(3218S), β-Catenin(8480S), Phospho-β-Catenin (Ser33/37)(2009S), GSK-3β(12456S), Phospho-GSK-3β (Ser9)(5558S), Axin2 (2151S), c-JUN (9165S), and c-Myc (18583S) antibodies were purchased from Cell Signaling Technology (Danvers, USA).
Cell Lines and Culture
The human breast cancer cell lines MCF-7, T47-D, normal human breast cell (Hs578-Bst), and normal human rectal mucosal cell (FHC) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Normal human bronchial epithelial cell of lung (HBE) was purchased from Mingzhou Biology Company (Ningbo, China). MCF-7, T47-D, and HBE cell lines were cultured with high-glucose DMEM (Hyclone, SH30022.01, USA) medium supplemented with 10% fetal bovine serum (FBS, Gibco, 10,100,147, Australia origin). Hs578-Bst was cultured in ATCC Hybri-Care Medium (Catalog No. 46-X) supplemented with 1.5 g/L sodium bicarbonate, 30 ng/mL mouse epidermal growth factor (EGF), and 10% FBS. FHC was cultured in DMEM:F12 (Gibco, 11,330,032, USA) medium with 10% FBS, 10 ng/mL cholera toxin, 0.005 mg/mL insulin, 0.005 mg/mL transferrin, and 100 ng/mL hydrocortisone. Cells were cultivated in an incubator with a humidified atmosphere of 5% CO2 at 37°C. Both MCF7 and T47-D cell lines are estrogen receptor (ER), progesterone receptor (PR) positive, and human epidermal growth factor receptor-2 (HER2) negative.
Cell Viability Assay
The effect of C188 on MCF-7, T47-D, Hs578-Bst, FHC, and HBE was determined by MTT assay. Briefly, cells were harvested with a density of 80% confluence, counted and seeded into 96-well plates (4×103 cells per well) containing 100 μL complete medium. After incubating for 24 hours, cells were supplemented with another 100 μL complete medium containing various concentrations of C188 (0, 5, 10, 20, 40, 80 μM) and incubated for 0, 12, 24, 36, and 48 hours, respectively. At each time point, all of the wells were added with 20 μL MTT solution (5 mg/mL) and incubated for another 4 hours. Next, the medium was removed, and 200 μL DMSO (Dimethylsulfoxide) was added to each well. The absorbance was measured at 570 nm by a microplate reader (Bio-Tek, Winooski, VT, USA) after shaking the plate on the table shaker for 10 minutes. All experiments were repeated three times, the cell viability curve of MCF-7 and T47-D was analyzed by GraphPad Prism 6.0.
Colony Forming Assay
Human breast cancer cell lines (MCF-7 and T47-D) were harvested at the density of 80% confluence, and then seeded into the 6-well plate with the density of 600 cells per well supplemented with 2 mL completed medium. After incubating for 24 hours, the cells were treated with C188 at the concentrations of 0, 10, 20, and 40 μM for 10 days. The medium was removed and the wells were washed with PBS. Then, cells were fixed with 4% paraformaldehyde for 20 minutes at room temperature. Paraformaldehyde was discarded and cells were stained with crystal violet staining solution at room temperature for 30 minutes. The colonies were washed with PBS three times and the plate was scanned.
Immunoblotting
Both MCF-7 and T47-D cells were treated with C188 for 48 hours at the concentrations of 0, 10, 20, and 40 μM, and then lysed in RIPA buffer (150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 25 mM Tris-HCl (pH 7.4), and 1 mM EDTA (pH 8.0)) supplemented with protease and phosphatases inhibitors (Roche, Mannheim, Germany). The concentration of protein was measured by the BCA (Bicinchoninic acid) Kit (Beyotime, P0010, Shanghai, China), and then the samples were loaded with 30 μg total protein into sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using corresponding gel concentration. The proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore Corporation, MA, USA), and the membranes were blocked with Quickblock (Beyotime, P0235, Shanghai, China) at room temperature for 1 hour. Then, the membranes were incubated with corresponding primary antibody at 4°C for 12 hours, and washed with TBST for 5 times×5 minutes. Subsequently, the membranes were incubated with IRDye 800CW goat anti-mouse IgG (H+L) or IRDye 680LT donkey anti-rabbit IgG (H+L) (Licor, USA) at room temperature for 1 hour and detected by an Odyssey two-color infrared fluorescence imaging system (Licor, USA).
Flow Cytometry Analysis
The flow cytometry (Becton Dickinson, USA) was used to measure the effect of C188 on the cell cycle distribution of MCF-7 and T47-D cell lines. Cells were treated with C188 at concentrations of 0, 10, 20, and 40 μM, respectively, and then cells were harvested and washed with cold PBS twice. Next, the harvested cells were fixed using 70% ethanol at 4°C for 12 hours. Cells were further washed with cold PBS twice and stained with 50 μg/mL propidium iodide (PI, Beyotime, ST512, Shanghai, China) and RNase A (Beyotime, ST578, Shanghai, China) for 30 minutes at 37°C. Finally, cells were protected from light and analyzed using flow cytometry.
Immunofluorescence
MCF-7 and T47-D cells were counted and seeded onto the CellCarrierTM-96-well plate (PerkinElmer) at a density of 6×103 cells per well. Cells were treated with C188 at the concentrations of 0, 10, 20, and 40 μM for 48 hours, and washed with PBS for 3 times×5 minutes. Then, cells were fixed with 4% paraformaldehyde at room temperature for 30 minutes and blocked with QuickBlock™ Blocking Buffer (Beyotime, P0260, Shanghai, China) containing 0.1% Triton X100 at 37°C for 30 minutes. Next, cells were washed with PBS and further incubated with anti-β-catenin at 4°C for 12 hours. Cells were incubated with Alexa Fluor 488-conjugated antibody for 1 hour (Thermo, A32731, Waltham, USA) at room temperature, and then incubated with DAPI for 15 minutes at room temperature. The subcellular localization and expression of β-Catenin were analyzed by the High Content analysis system (PerkinElmer, Waltham, MA, USA).
Wound-Healing Assay
To evaluate the effect of C188 on the migration of MCF-7 and T47-D cells, the wound-healing assay was used. Briefly, both cell lines were seeded in the 6-well plate at a density of 1.5×106 cells per well and grown for 24 hours to reach the confluence. The cells were scraped by a 200-µL pipette tip to create a wound. Each well was washed with PBS to remove the floating cells, and then treated with C188 at the concentrations of 0, 10, 20, and 40 μM. After another 48 hours, the wound gap was captured and analyzed by the microscope (Olympus, Tokyo, Japan). Finally, the wound healing area was measured using Image J software and calculated with “(Free area (0 h)-Free area (48 h))/Free area (0 h) * 100%” formula.
Transwell Assay
For the transwell assay, a 24-well transwell chamber with 8 μm pore size (Corning, NY, USA) was used to evaluate the cell migration. Cells were resuspended in 200 µL medium without serum containing 0, 10, 20, and 40 μM of C188, respectively, and seeded onto the upper chamber. Only 500 µL complete medium was placed in the lower chambers of a 24-well plate. After being incubated for 48 hours, cells were fixed with 4% paraformaldehyde followed by staining with 0.2% crystal violet staining solution, and cells which had migrated into the lower chamber were counted using a microscope.
Statistical Analysis
All data were conducted on more than three independent experiments. Statistical analysis was performed using GraphPad Prism 6. t-test analysis was applied to calculate the significance between two groups. The data were displayed as the mean±SD, and significance was set at P<0.05.
Results
Dose-Dependent Effect of C188 on Cell Viability and Tumorigenicity in Breast Cancer Cells
Given the anticancer effect of imidazopyridine scaffold compounds, we explored whether C188, which is shown in Figure 1A and Supplemental Figure S1, can suppress proliferation and viability of breast cancer cells. After exposure to 20 compounds including C188, only C188 can significantly decrease cell proliferation in a dose- and time-dependent manner rather than other 19 compounds in breast cancer cell lines MCF7 and T47-D (Supplemental Figure S2 and Figure 1B). The IC50 values of C188 to MCF7 and T47-D were 24.4 and 23 μM, respectively (Figure 1B). Moreover, C188 had little effect on normal human breast cell (Hs578-Bst), human rectal mucosal cell (FHC), and human bronchial epithelial cell of lung (HBE) at the same concentrations (Figure 1C), indicating the low toxicity to normal cells. To further evaluate the effect of C188 on the tumorigenicity in vitro, colony formation assay was performed to investigate its influence on long-term survival of breast cancer cells. Consistently, in comparison with the DMSO treating group, the number and size of colonies were efficiently reduced after exposed to 10 and 20 μM C188, and rare colony was formed when treated with 40 μM (Figure 1D and E), implying that it may be a potential inhibitor for breast cancer therapy.
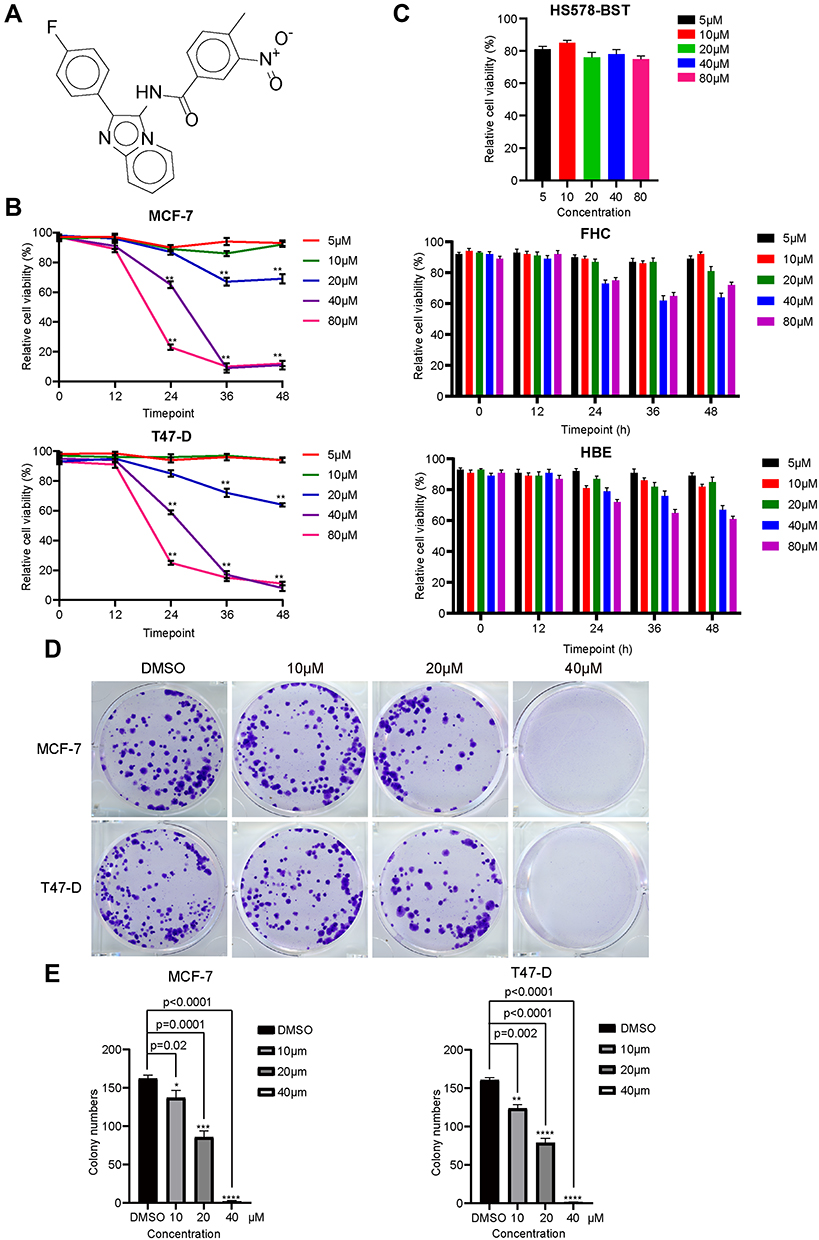 |
Figure 1 C188 suppresses proliferation and growth in a dose-dependent manner in breast cancer cells. (A) Chemical structural formula of C188. (B) MCF7 and T47-D cells were treated with the indicated concentrations of C188 for 12, 24, 36, and 48 hours. Relative cell viability was measured with MTT assay. (C) The cytotoxicity and specific effect of C188 on Hs578-Bst, FHC, and HBE cells. The normal human breast cell (Hs578-Bst), normal human rectal mucosal cell (FHC), and normal human bronchial epithelial cell of lung (HBE) were treated with 5, 10, 20, 40, and 80 μM C188, respectively. Hs578-Bst was treated for 24 hours, while FHC and HBE cells were treated for 12, 24, 36, and 48 hours. Relative cell viability was measured with MTT assay. (D) Colony formation assay was conducted to investigate growth in vitro after treatment with DMSO, 10, 20, and 40 μM C188 for 10 days. The colonies were visualized with the images. (E) The corresponding histogram shows the colony numbers of C188 treatment and DMSO control groups. Values represent the mean±SD (n=3). (*P<0.05; **P<0.01; ***P<0.005; ****P<0.0001). |
C188 Blocks Cell Cycle Procession by Arresting Breast Cancer Cells into G1-Phase
To deeply assess the mechanism responsible to C188-induced cell proliferation inhibition, cell cycle distribution was analyzed using flow cytometry in breast cancer cells treated with or without the inhibitor. Control and treated cells were harvested and stained with PI and then detected by flow cytometry. As shown in Figure 2A and B, the results showed that C188 influenced the cell cycle progression in a concentration-dependent manner in MCF7 and T47-D cell lines. Representative histograms further indicated that the percentage of G1-phase cells dramatically increased after exposure to various concentrations of C188 (from 30.2 to 50.2% for MCF7 cells; from 37.7 to 48.2% for T47-D cells), suggesting that this compound induced cell cycle arrest at G1-phase. To further confirm the results, immunoblotting was explored to assess the G1-phase related protein levels. C188 triggered a significant decrease in expression of CDK4/6 and cyclin D, as well as an increase in p21 and p27 (Figure 2C and D). Taken together, our data suggests that C188 promotes cell cycle arrest at G1 phase by regulating the expression or stability of its related proteins.
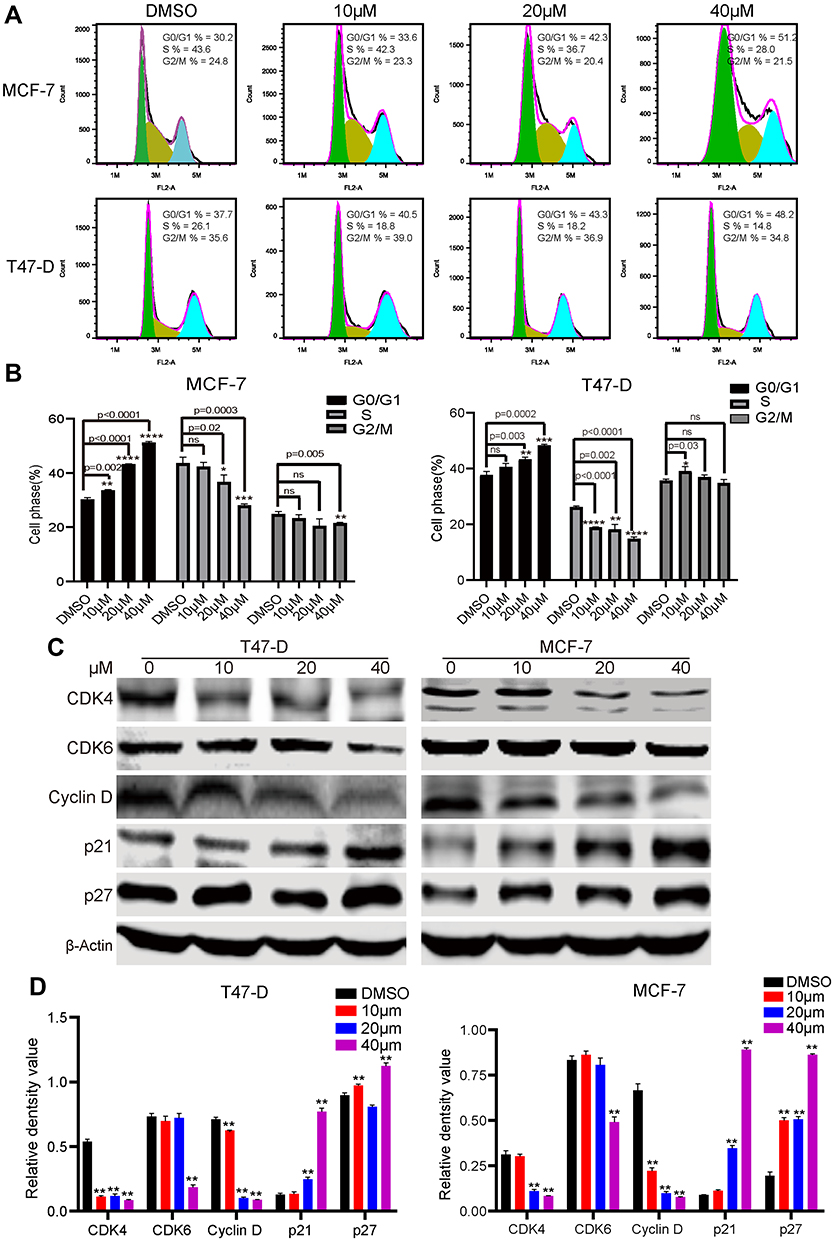 |
Figure 2 C188 induces cell cycle arrest at G1-phase by regulating the associated proteins. (A) Cell cycle of MCF7 and T47-D was analyzed by flow cytometry following treatment with C188 for 48 hours. DMSO as the control group. Graph is representative of the percentage mean of three independent experiments. FL2-A: the fluorescent area. (B) Histograms represent the percentages of cell distribution in G0/G1-, S- and G2/M-phase. The data are shown as the means±SD (n=3). (C) Effect of C188 on expression level of G1-phase related proteins. Cyclin D, CDK4, CDK6, p21, and p27 in MCF7 and T47-D cells were detected by immunoblotting. β-actin was used as a loading control. (D) The statistical analysis of relative density value of western bands presented in (C). Values represent the mean±SD (n=3). (*P<0.05; **P<0.01; ***P<0.005; ****P<0.0001; ns represents no significant difference). |
C188 Inhibits Cell Migration of Breast Cancer Cells in a Dose-Dependent Manner
Cell migration is one of the critical malignant processes exhibited by many types of cancer tumor, thus we further evaluated the effect of this compound on migratory activity of breast cancer cells. Interestingly, the wound healing assay indicated that the wound-closure of C188 treating cells versus control cells is dramatically reduced in both MCF7 and T47-D cell lines (Figure 3A). Consistently, as shown in Figure 3B, our in vitro transwell migration assay showed that treatment with C188 considerably decreased the number of cells reaching the bottom wells compared with the vehicle control, suggesting the inhibition effect of C188 on cell migration. Thus, these data demonstrate that C188 may negatively regulate cell migratory ability of breast cancer in vitro.
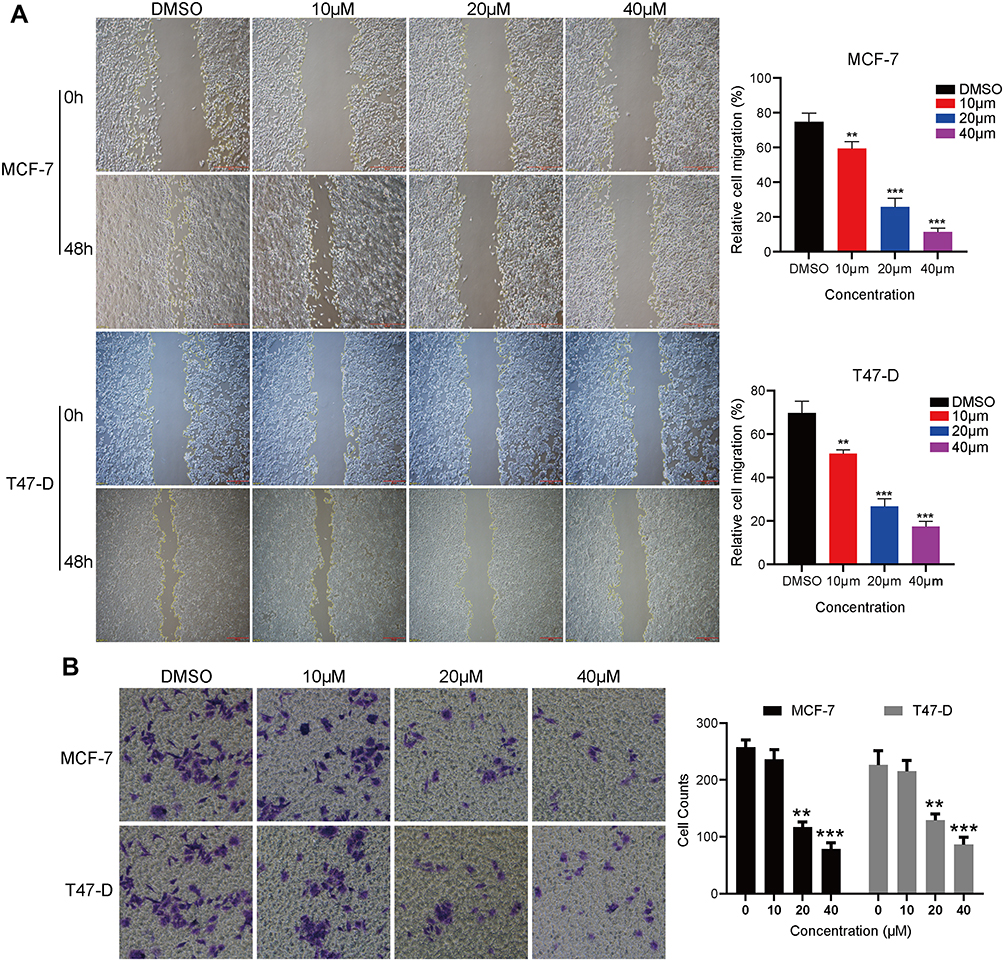 |
Figure 3 C188 reduces cell migration of breast cancer cells. (A) MCF7 and T47-D cells were plated in a 6-well culture plate until 90% confluence. The wound was scratched on monolayer culture using a 200 μL pipette tip, and the cells were then treated with indicated concentrations of C188 for 48 hours. DMSO as the control group. The inhibitory effect of C188 was determined by comparing the wound closure. (B) MCF7 and T47-D cells were seeded in the insert of 24-well plates in media containing 0, 10, 20, and 40 μM C188 in the upper chamber. After 24 hours incubation, the migrated cells were stained with crystal violet and the cells migrating into the lower chamber were counted. Representative images are indicated at 40x magnification. Data were collected from three independent experiments (**P<0.01; ***P<0.005). |
C188 Inhibits Wnt/β-Catenin Signaling in Breast Cancer Cells
It has been reported that Wnt/β-catenin signaling correlates with cell migration and invasion in a variety of malignancies.18–20 Additionally, the nuclear accumulation of β-catenin resulting from mutational inactivation of APC (Adenomatous Polyposis Coli) contributes to tumorigenesis by promoting transcription of its downstream targets such as c-Myc and cyclin D1, which are important proteins for the control of cell cycle progression and proliferation.21,22 Therefore, to investigate whether the Wnt/β-catenin signaling is involved in the mode of C188-induced proliferation suppression and migration inhibition, we detected the protein levels relating to Wnt/β-catenin pathway using immunoblotting in MCF7 and T47-D cell lines. As shown in Figure 4A, the scaffold protein Axin1 was upregulated while Dishevelled3 (Dvl3) and LRP6 that contribute to the stability of β-catenin were significantly downregulated after exposure to C188. Moreover, in comparison with vehicle group, the phosphorylation level at GSK3β (Glycogen synthase kinase-3) serine 9 after treatment with C188 was dramatically reduced in a dose dependent manner. Next, we further investigated the influence of this compound on the phosphorylation and stability of β-catenin. The result showed that the phosphorylation site at β-catenin serine 33/37 was considerably increased in comparison with control group, while the protein level of total β-catenin in immunoblotting and immunofluorescence was decreased in a dose-dependent manner after exposure to C188 (Figure 4A and B), implying that the Wnt/β-catenin signaling was inhibited by regulating the phosphorylation and protein stability of these components involving this pathway. We next evaluated the mechanism responsible for C188 induced β-catenin degradation. To illustrate this issue, we investigated whether the combination of C188 with MG132 could rescue the β-catenin expression level reduced by C188 treatment. As indicated in Figure 4C, β-catenin protein level was effectively rescued by MG132 addition in both T47-D and MCF7 cells, suggesting that C188 promotes a β-catenin decrease by the ubiquitin proteasome system. To further investigate the effect of C188 on the nuclear localization of β-catenin, immunofluorescence was performed after treatment with indicated concentrations of this compound. As shown in Figure 4D, the green immunofluorescent signal which co-localized with the nucleus represented with DAPI staining was considerably reduced in a dose-dependent manner after treatment with C188, demonstrating that treatment with this compound not only decreases the expression level of β-catenin, but also inhibits its entrance into the nucleus. To further employ whether the targeted genes were influenced due to the decrease of β-catenin locating at the nucleus, we detected the expression of β-catenin targeting genes such as Axin 2, c-Jun, and c-Myc. As indicated in Figures 2C and 4E, C188 can significantly down-regulate the expression of cyclin D1, Axin 2, c-Jun, and c-Myc in a dose-dependent manner, implying the transcription inhibition of β-catenin targeting genes because of the decrease of nuclear β-catenin. Taken together, these observations demonstrated that β-catenin decrease due to accumulation of ser33/37 phosphorylation induced by C188 results in inhibition of cell migration. The decrease of β-catenin further represses transcription of cyclin D1, Axin 2, c-Jun, and c-Myc, leading to the cell cycle arrest and proliferation suppression in breast cancer cells.
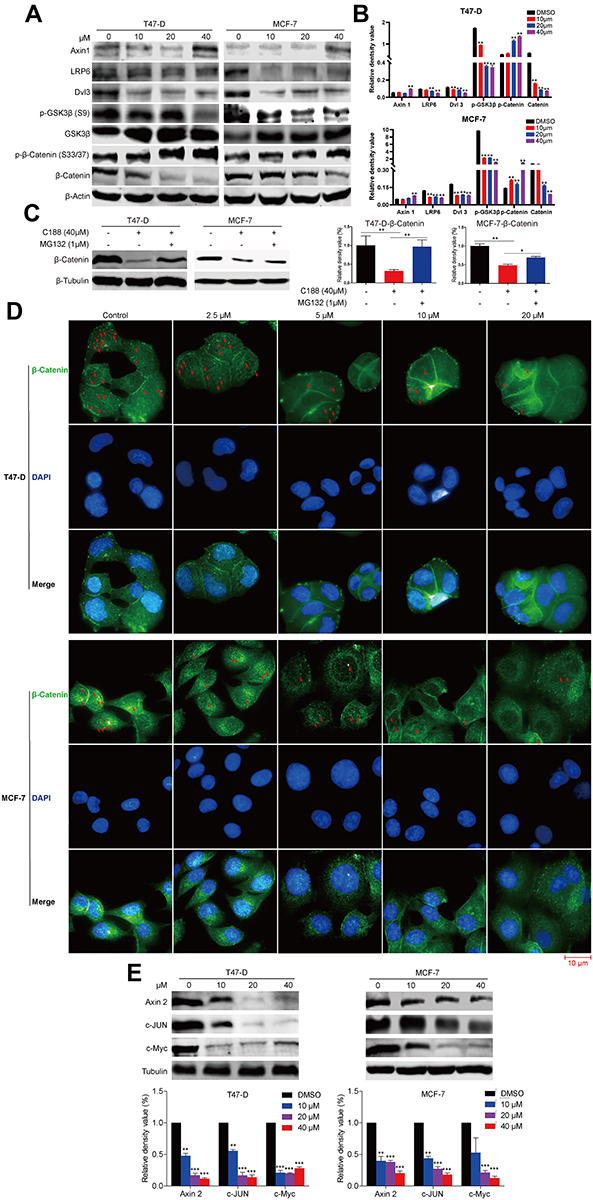 |
Figure 4 C188 inhibits Wnt/β-catenin signaling by regulating the stability of β-catenin in breast cancer cells. (A and B) MCF7 and T47-D cells were treated with the indicated concentrations of C188 for 48 hours. DMSO was used as the control group. The phosphorylation and expression levels of Wnt pathway-related proteins were investigated by immunoblotting. β‑actin was used as a loading control. The results were representative of three independent experiment (**P<0.01). (C) MCF7 and T47-D cells were exposed to 40 μM C188 and 1 μM MG132 for 12 hours to evaluate whether MG132 could rescue the protein expression of β-catenin. β‑tubulin was loaded as a control (*P<0.05; **P<0.01). (D) Both MCF7 and T47-D cells were exposed to different concentrations of C188 for 48 hours, and then β-catenin localizing in the nucleus was detected with immunofluorescence. The nucleus was confirmed with DAPI (4,6-diamidino-2-phenylindole) staining. The red arrow represented the signal of β-catenin localizing in the nucleus. Scale bar: 10 μm. (E) MCF7 and T47-D cells were exposed to the indicated concentrations of C188 for 48 hours. DMSO was used as the control. The expression levels of Axin 2, c-Jun, and c-Myc proteins were evaluated by immunoblotting. β‑tubulin was used as a loading control. The results were representative of three independent experiment (**P<0.01; ***P<0.005). |
Discussion
Increasing reports conducted in the world declared the utterly poor prognoses and high recurrence rate of breast cancer, suggesting that current therapies need to be improved for the effective cure of certain cancer cells.23 At present, surgery combined with chemo- and radiotherapy is a general therapeutic approach. However, surgery causes some disadvantages such as bleeding, damage to healthy tissues and infections. Moreover, chemo- and radio-therapy also result in a series of side-effects, including an impaired immune system and hair loss.2,23 Thus, it is necessary to develop novel agents that contribute to the inhibition of proliferation and growth of breast cancer cells. Previous reports have confirmed that compounds including imidazopyridine core have attracted considerable attention because of their broad bioactivities. In this study, we screened a compound, C188, exhibiting high activity to both MCF7 and T47-D cell lines which are classified into luminal A subtype that is ER- and PR-positive and HER2-negative.24,25 It is inferred that the effect of C188 may depend on ER and PR, suggesting that this inhibitor has the potential in curing luminal A subtype breast tumor. Furthermore, we evaluate its effect on antiproliferation activity, cell cycle, cell migration, and possible underlying mechanism.
Cell cycle arrest is an important pathway for the inhibition of tumor cell procession. Here, we indicated that C188 dramatically reduced breast cancer cells proliferation and growth in vitro in a dose- and time-dependent manner, demonstrating its anticancer effect on breast cancer. Furthermore, the flow cytometric analysis of cell cycle data revealed that a high percentage of MCF7 and T47-D cells was arrested in the G1 phase because of exposure to C188. Consistently, immunoblotting showed that G1/S phase-related proteins, such as CDK4/6 and cyclin D, were downregulated, while p21 and p27 were upregulated. Cell migration, which is another vital characteristic for tumor survival, is critical for endothelial cells to form blood vessels during angiogenesis, and is necessary for cancer cell growth and metastasis.13 In our current study, the results showed that the significant migration decrease was induced in a dose-dependent manner following treatment with C188. These data demonstrated that C188 had the potential application for breast cancer therapy of luminal A subtype after studying the toxicity, pharmacology, and pharmacodynamics.
Aberrant existence of the Wnt/β-catenin signaling has been associated with the procession and metastasis of breast cancer,6 and primary cell originated from breast tumors or cancer cell lines were demonstrated to express several Wnt ligands and Fzd receptors.9,10 Meanwhile, LRP6, DVL, and phosphorylated DVL proteins have been indicated in advanced breast cancer cells.11,12,25 Drugs targeting this pathway potentially decrease the tumor proliferation and growth.26–28 Moreover, Wnt/β-catenin signaling has been implicated in cell migration and invasion in a variety of malignancies.18–20 Previous studies also demonstrated that β-catenin accumulation in nucleus increased transcription of its downstream targets including c-Myc and cyclin D1, which are important proteins contributing to cell cycle progression and proliferation.21,22 In this present study, it revealed that C188 induced the increase of scaffold protein Axin 1 as well as the decrease of Dvl3 and LRP6 protein levels, which maintained the stability of β-catenin. Furthermore, the phosphorylation level at ser9 of GSK3β, which is phosphorylated by upstream element PI3K/Akt cell survival pathway leading to the activity inhibition of GSK3β, could be dramatically inhibited by C188. Subsequently, active GSK3β phosphorylated β-catenin at Ser33, Ser37, and Thr41 in response to C188 treatment, which results in β-catenin degradation. Interestingly, MG132 can partially rescue the level of β-catenin which is reduced after treatment with C188, implying that the ubiquitin-proteasome pathway is involved in the C188 induced degradation of β-catenin. Cell migration of tumor cells and cyclin D1 expression level have been reported to be implicated with β-catenin. Consistently, our result indicated that cell migration activity and cyclin D1 level were inhibited by C188-induced blockage of Wnt/β-catenin signaling and decrease of β-catenin localized in nucleus. Therefore, we speculate that C188 may be a novel inhibitor which inhibits the Wnt/β-catenin pathway and entrance of β-catenin into the nucleus, and then suppresses the proliferation and cell migration as well as cell cycle by regulating transcription of Axin 2, c-Jun, c-Myc, and cyclin D1. More recently, one study has found that imidazopyridine attenuate the survival of hepatocellular carcinoma by targeting protein tyrosine phosphatase SHP-1.29 Additionally, imidazopyridine analogs may be the inhibitors of AKT1 demonstrated by computational investigation.30 Thus, one of our future studies with C188 is the identification of cellular targets that confer its promising anti-proliferative activity. Furthermore, the in vivo effect of C188 is not well known. Therefore, our future subjects will focus on evaluating the in vivo antitumor viability, toxicity, pharmacology, and pharmacodynamics, which will further help us design rational analogs for improving anti-cancer activity and target specificity.
In conclusion, the findings presented in this study provide a novel inhibitor which induces cell proliferation, migration, and cell cycle arrest in the G1 phase in both MCF7 and T47-D cell lines. Interestingly, C188 also leads to the inhibition of Wnt/β-catenin signaling by promoting the decrease of β-catenin expression and nuclear localization which is mediated through the suppression of p-GSK-3β. Our findings favorably imply C188 as a potential therapeutic candidate for breast cancer because of its capability to inhibit Wnt/β-catenin signaling.
Data Sharing Statement
All data generated and/or analyzed during this study are included in this published article.
Acknowledgments
The authors would like to thank the Basic Research and Frontier Program of Chongqing Science and Technology Bureau, grant number cstc2018jcyjAX0219; Science and Technology Research Program of Chongqing Municipal Education Commission, grant number KJQN201801304, KJQN201901331; Scientific Research Foundation of the Chongqing University of Arts and Sciences, grant number 2017RBX11, 2017ZBX05 and 2017ZBX07.
Disclosure
The authors declare that they have no competing interests for this work.
References
1. O’Sullivan CC, Loprinzi CL, Haddad TC. Updates in the evaluation and management of breast cancer. Mayo Clin Proc. 2018;93:794–807. doi:10.1016/j.mayocp.2018.03.025
2. Liang Y, Zhou Y, Deng S, Chen T. Microwave‐assisted syntheses of benzimidazole‐containing selenadiazole derivatives that induce cell‐cycle arrest and apoptosis in human breast cancer cells by activation of the ROS/AKT pathway. Chemmedchem. 2016;11(20):2339–2346. doi:10.1002/cmdc.201600261
3. Boras-Granic K, Hamel PA. Wnt-signalling in the embryonic mammary gland. J Mammary Gland Biol Neoplasia. 2013;18(2):155–163. doi:10.1007/s10911-013-9280-x
4. Clevers H. Wnt/β-catenin signaling in development and disease. Cell. 2006;127(3):469–480. doi:10.1016/j.cell.2006.10.018
5. Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and β-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5(9):691. doi:10.1038/nrg1427
6. Pohl S-G, Brook N, Agostino M, Arfuso F, Kumar AP, Dharmarajan A. Wnt signaling in triple-negative breast cancer. Oncogenesis. 2017;6(4):e310.
7. Li VS, Ng SS, Boersema PJ, et al. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012;149(6):1245–1256. doi:10.1016/j.cell.2012.05.002
8. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. doi:10.1016/j.cell.2017.05.016
9. Milovanovic T, Planutis K, Nguyen A, et al. Expression of Wnt genes and frizzled 1 and 2 receptors in normal breast epithelium and infiltrating breast carcinoma. Int J Oncol. 2004;25(5):1337–1342.
10. Benhaj K, Akcali KC, Ozturk M. Redundant expression of canonical Wnt ligands in human breast cancer cell lines. Oncol Rep. 2006;15(3):701–707.
11. Nagahata T, Shimada T, Harada A, et al. Amplification, up‐regulation and over‐expression of DVL‐1, the human counterpart of the Drosophila disheveled gene, in primary breast cancers. Cancer Sci. 2003;94(6):515–518. doi:10.1111/j.1349-7006.2003.tb01475.x
12. Schlange T, Matsuda Y, Lienhard S, Huber A, Hynes NE. Autocrine WNT signaling contributes to breast cancer cell proliferation via the canonical WNT pathway and EGFR transactivation. Breast Cancer Res. 2007;9(5):R63. doi:10.1186/bcr1769
13. Li G-Y, Jung KH, Lee H, et al. A novel imidazopyridine derivative, HS-106, induces apoptosis of breast cancer cells and represses angiogenesis by targeting the PI3K/mTOR pathway. Cancer Lett. 2013;329(1):59–67. doi:10.1016/j.canlet.2012.10.013
14. Yun S-M, Jung KH, Lee H, et al. Synergistic anticancer activity of HS-173, a novel PI3K inhibitor in combination with Sorafenib against pancreatic cancer cells. Cancer Lett. 2013;331(2):250–261. doi:10.1016/j.canlet.2013.01.007
15. Lee H, Kim SJ, Jung KH, et al. A novel imidazopyridine PI3K inhibitor with anticancer activity in non-small cell lung cancer cells. Oncol Rep. 2013;30(2):863–869. doi:10.3892/or.2013.2499
16. Muniyan S, Chou Y-W, Ingersoll MA, et al. Antiproliferative activity of novel imidazopyridine derivatives on castration-resistant human prostate cancer cells. Cancer Lett. 2014;353(1):59–67. doi:10.1016/j.canlet.2014.07.002
17. Kim HM, Kim C-S, Lee J-H, et al. CG0009, a novel glycogen synthase kinase 3 inhibitor, induces cell death through cyclin D1 depletion in breast cancer cells. PLoS One. 2013;8(4):e60383. doi:10.1371/journal.pone.0060383
18. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103(2):311–320. doi:10.1016/S0092-8674(00)00122-7
19. Birchmeier W, Hulsken J, Behrens J. E-cadherin as an invasion suppressor. Ciba Found Symp. 1995;189:124–141.
20. Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14(15):1837–1851.
21. Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc Natl Acad Sci. 1999;96(10):5522–5527. doi:10.1073/pnas.96.10.5522
22. Tetsu O, McCormick F. β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422. doi:10.1038/18884
23. Thakur KK, Bordoloi D, Kunnumakkara AB. Alarming burden of triple-negative breast cancer in India. Clin Breast Cancer. 2018;18(3):e393–e399. doi:10.1016/j.clbc.2017.07.013
24. Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13(4):215. doi:10.1186/bcr2889
25. King TD, Suto MJ, Li Y. The wnt/β‐catenin signaling pathway: a potential therapeutic target in the treatment of triple negative breast cancer. J Cell Biochem. 2012;113(1):13–18. doi:10.1002/jcb.23350
26. Egashira I, Takahashi‐Yanaga F, Nishida R, et al. Celecoxib and 2, 5-dimethylcelecoxib inhibit intestinal cancer growth by suppressing the Wnt/β-catenin signaling pathway. Cancer Sci. 2017;108(1):108–115. doi:10.1111/cas.13106
27. Reddivari L, Charepalli V, Radhakrishnan S, et al. Grape compounds suppress colon cancer stem cells in vitro and in a rodent model of colon carcinogenesis. BMC Complement Altern Med. 2016;16(1):278. doi:10.1186/s12906-016-1254-2
28. Manigandan K, Manimaran D, Jayaraj RL, Elangovan N, Dhivya V, Kaphle A. Taxifolin curbs NF-κB-mediated Wnt/β-catenin signaling via up-regulating Nrf2 pathway in experimental colon carcinogenesis. Biochimie. 2015;119:103–112. doi:10.1016/j.biochi.2015.10.014
29. Su J-C, Chang C-H, Wu S-H, Shiau C-W. Novel imidazopyridine suppresses STAT3 activation by targeting SHP-1. J Enzyme Inhib Med Chem. 2018;33(1):1248–1255. doi:10.1080/14756366.2018.1497019
30. Gu X, Wang Y, Wang M, Wang J, Li N. Computational investigation of imidazopyridine analogs as Protein kinase B (Akt1) allosteric inhibitors by using 3D-QSAR, molecular docking and molecular dynamics simulations. J Biomol Struct Dyn. 2019;1–16. doi:10.1080/07391102.2019.1705185
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.