Back to Journals » The Application of Clinical Genetics » Volume 14
A Novel Frameshift Mutation, Deletion of HBB:c.199_202delAAAG [Codon 66/67 (-AAAG)] in β-Thalassemia Major Patients from the Western Region of Uttar Pradesh, India
Authors Chauhan W ![]() , Afzal M, Zaka-ur-Rab Z, Noorani MS
, Afzal M, Zaka-ur-Rab Z, Noorani MS
Received 8 December 2020
Accepted for publication 22 February 2021
Published 1 March 2021 Volume 2021:14 Pages 77—85
DOI https://doi.org/10.2147/TACG.S294891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Waseem Chauhan,1 Mohammad Afzal,1 Zeeba Zaka-ur-Rab,2 Md Salik Noorani3
1Department of Zoology, Aligarh Muslim University, Aligarh, India; 2Department of Pediatrics, Jawahar Lal Nehru Medical College, Aligarh Muslim University, Aligarh, India; 3Department of Botany, Jamia Hamdard, New Delhi, India
Correspondence: Mohammad Afzal
Human Genetics and Toxicology Laboratory, Department of Zoology, Aligarh Muslim University, Aligarh, India
Tel +919897839601
Email [email protected]
Purpose: Beta thalassemia is one of the most common inherited disorders in India with heterogenous clinical phenotypes from silent carrier to clinically severe ones. Our study aimed to characterize the mutation spectrum in thalassemia patients who are coming to the hospital for follow-up from the western region of Uttar Pradesh India.
Patients and Methods: For the study, a case series of the retrospective bi-centre study was conducted. The patients from two thalassemia centers in the major hospitals (LLRMC Meerut, and JNMC, Aligarh administered by the Ministry of Health and Family Welfare (MoHFW)) in the Western Uttar Pradesh, India were considered for the study. A total of 77 blood samples were obtained from individuals (both related and unrelated) diagnosed with β-thalassemia after their consent. After DNA extraction, HBB gene amplification, mutation-specific polymerase chain reaction and gene sequencing were carried out to analyze the mutations.
Results: In this study, seven different types of mutations were reported for the first time in Western Uttar Pradesh, India. A novel frameshift mutation, deletion of 4 nucleotides Codon 66/67 (-AAAG) in exon 2 region, is reported for the first time. IVS 1– 5 (G>C) and Codon 41/42 (-CTTT) are the most frequently reported mutations. The molecular spectrum for these two cases consists of 44 and 42 alleles out of 108 alleles, respectively.
Conclusion: A total of 108 β-thalassemia alleles were studied from 46 homozygous and 31 compound heterozygous patients. All the individuals were from 20 districts of the Western Uttar Pradesh, India.
Keywords: codon 41/42-CTTT, IVS I-5G>C, beta globin, mutation, beta-thalassemia major
Introduction
β-thalassemia is a monogenic and heterogeneous group of an inherited disorder of haemoglobin synthesis where a mutated beta-globin gene hinders/stops its protein production. These β-thalassemia phenotypes are related to the large number of mutations that affect the HBB gene on chromosome 11p15.5 (OMIM number +141900). Till now, more than 300 mutations have been reported (Database Ithanet and HbVar). They are the most common in the Mediterranean region, the Indian subcontinent, South East Asia, and the Middle East,1 which have populations with high-frequency of disease carriers; however, a few yet most unique mutations pertaining to the area it belongs have been reported. Some of these mutations are also rare to be found with other sets of populations. This autosomal recessive condition afflicts thousands of people of many ethnicities worldwide.2 The β-thalassemia carrier frequency in the African continent varies from 1% to 13%, in comparison to Europe, where the carrier frequency ranges from 0.12% in Finland to 15% in Cyprus. Similarly, in California (United States), one identified new-born with β-thalassemia is found for every 55,000 birth.1,3,4
In India, the carrier frequency of β-thalassemia spans from 0.3% to 15%, depending on the ethnic subgroup.1 The first case after 1938 was reported from India by Mukherji, (1938).5 According to WHO report on guidelines and management of Hb disorders, the carrier frequency of all types of β-thalassemia traits in India is approximately 3–4%.6 The severe forms of β-thalassemia include 30–40 million carriers and almost 8000 to 10,000 births each year. Therefore, it is estimated that 10% of the total born thalassemia homozygotes or compound heterozygotes globally adds to the national health burden at significant rates.7
Mutational analysis exhibits the sequence variants ranging from point mutations to large deletions. Such mutations affect HBB gene transcription, messenger RNA processing, translation, or alterations in the gene product structure. Out of all, 21 mutations account for more than 80% of all β-thalassemia determinants.8 Few mutational studies have been reported from western Uttar Pradesh populations for β -thalassemia.9 As a result, our hospital-based study finds its relevance as it focuses on transfusion-dependent patients’ mutational status. The present study is also focused on reporting the types of mutations and variants in the HBB gene among the β-thalassemia patients belonging to 20 districts out of 29 in Western Uttar Pradesh. The study was conducted using allele-specific PCR and direct sequencing for the identification of mutation.
Patients and Methods
Study Design and Patients
For this study, a case series of the retrospective bi-centre study was selected. The patients from two thalassemia centres in the major hospitals (LLRMC Meerut and JNMC, Aligarh, administered by the Ministry of Health and Family Welfare (MoHFW)) in Western Uttar Pradesh, India, were taken for the study. Samples were collected during the period from March 2018 to October 2019. The inclusion criteria for thalassemia were: (i) diagnosis of thalassemia major (ii) diagnosis and initiation of transfusion before the age of 2 years, (iii) blood transfusion frequency in every month, or even for a lesser period, and (iv) with consent for their inclusion. Based on these criteria, the authors of this study reviewed the medical files of all the thalassemia patients and identified 77 potential thalassemia major patients from more than 150 thalassemia cases registered at the centres included in this study; 73 out of 77 subjects were from unrelated families and rest 4 were twin siblings.
Genomic DNA Extraction
A total of 77 blood samples were obtained from individuals (both related and unrelated) and diagnosed with β-thalassemia after their consent. About 5 mL of the patient’s blood was collected in red-topped tubes coated with ethylenediaminetetraacetic acid (EDTA) as an anticoagulant. The DNA was extracted using gDNA Blood Mini Kit (Chromous Biotech, Bengaluru, India). The kit was used according to the manufacturer’s instructions, and samples were stored at -20 °C until further use.
Designing of Allele-Specific Primers
Before designing allele-specific primers, we amplified and then sequenced the whole HBB gene from 10 randomly selected samples to get an idea about prevalent mutations among the patients. Forward primer (P2F) 5ʹ-GCCAAGGACAGGTACGGCTGTCATC- 3ʹ, reverse primer (P2R) 5ʹ-CAGTTTAGTAGTTGGACTTAGG-3ʹ, and a primer for walking (HBBR) 5ʹ-CTCTTTCTTTCAGGGCAATAATG-3ʹ were used for the amplification and sequencing of the whole HBB gene of 1678 bp in length, respectively.10 Based on the sequence analysis of these ten randomly selected samples, the allele-specific primers for most prevalent mutations (IVS I-5 (G>C), Codon 3 (T>C) Codon 41/42 (-CTTT), and Codon 66/67 (-AAAG) (Table 1)) were designed using online tool http://biotools.nubic.northwestern.edu/OligoCalc.html.11
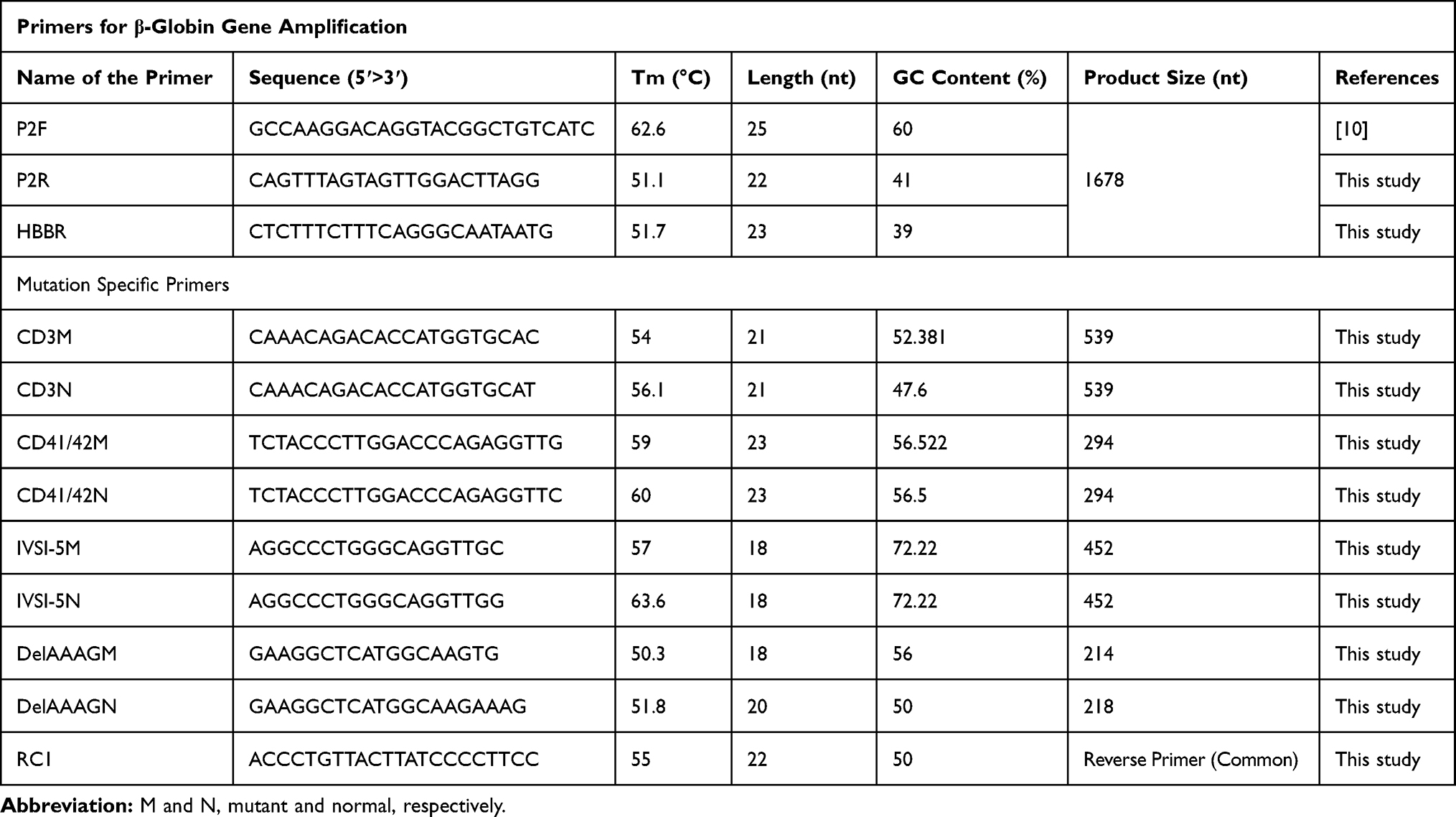 |
Table 1 List of Primers with Their Tm, GC Content, and Product Size Used for HBB Gene and Prevalent Mutations |
HBB Gene Amplification
For the amplification of the HBB gene (1678 bp), a polymerase chain reaction was set up with 100 ng/mL of genomic DNA, 2 mM of dNTPs, 10X nPfu-Forte Buffer, 2.5 U/μL nPfu-Forte DNA polymerase (NEB), 5 pmol/μL of each primer (P2F, P2R and HBBR in Table 1), and MilliQ water in a final volume of 20 μL. Amplification was done for 35 cycles of denaturation (94°C for 60 s), annealing (55°C for 60 s), and extension (72°C for 120 s) on a T100 thermal cycler (Bio-Rad, USA).
Common Mutation Detection
For the detection of the three most common mutations of the HBB gene viz. IVS I-5 (G>C), Codon 41/42 (-CTTT), Codon 3 (T>C), and the confirmation of Codon 66/67 (-AAAG) (novel mutation), an allele-specific polymerase chain reaction (Ugozzoli and Wallace, 1991) was set up by using primers given in Table 1. A negative and positive control (Normal primer with wild-type DNA from the healthy individual) was used to ensure the absence of contamination and primer specificity, respectively. For this, a reaction mixture of 10 μL having 100 ng/mL of genomic DNA, 1 U Taq DNA polymerase (GeNei, Bengaluru, India), 10 mM of dNTPs, 10X Taq buffer, 10 pmol/μL of each primer, and MilliQ water were made in 200 μL PCR tube. Amplification was done for 35 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 45 s, and extension at 72°C for 60 s using a T100 thermal cycler (Bio-Rad, USA). The amplified product was electrophoresed in 2.0% agarose gel for 40 minutes in 1X TAE Buffer. Gel pictures were recorded on a Gel Documentation system (AlphaImager, USA).
HBB Sequencing and Analysis
The HBB genes that were not amplified through the common mutation detection primers were also sequenced. Purification of PCR products was done by using HiYieldTM Gel/PCR DNA Extraction kit (RBC, Taiwan). PCR products were directly sequenced by using the Sanger sequencing method through a service providing company 1st Base, Malaysia. Sequencing results were submitted in Genbank (NCBI) with accession no. MT941910, MT941911, MT941912, and MT941913. The patients’ sequences were compared with National Centre for Biotechnology Information (NCBI) Reference sequences (>NG_000007.3, Homo sapiens chromosome 11, GRCh38.p13 Primary Assembly) by using an alignment tool Multalin.12 Mutation frequency was determined by direct counting. Ithanet and HbVar databases were used for the identification of the mutations reported in other populations.13
Bioinformatic Tools
To calculate the probability of deleterious effects of novel mutation, different bioinformatic tools available online were used, viz EXPASY Translate tool (https://web.expasy.org/translate/) to translate the DNA sequence to protein sequence. Mview tool (https://www.ebi.ac.uk/Tools/services/web/toolresult.ebi?jobId=mview) was used for BLAST, and Swiss-Model was used for modeling and function prediction.
Ethical Statement
The work conducted received approval from the Institutional ethics committee, Jawaharlal Nehru Medical College & Hospital, Aligarh Muslim University, Aligarh, India. All experiments were examined and approved before the experimental setup. Moreover, all the patients included in this study gave their written informed consent to participate and analyze their medical records.
Results
A total of 108 β-thalassemia alleles were studied from 46 homozygous and 31 compound heterozygous patients. All the individuals were from 20 districts of Western Uttar Pradesh, India. Sequence analysis of the HBB gene in the populations revealed that seven different HBB gene mutations were observed among some common mutations in 108 alleles, two deletions, and five point-mutations. One novel mutation was found in exon-2 [Codon 66/67 (-AAAG)] (Tables 2 and 3). Out of seven mutations, three of them (Codon 3 (T>C), Codon 41/42 (-CTTT), and Codon 66/67 (-AAAG)) were present in the exon region, and the rest four mutations were in the intron region. Among the genotypes of β-thalassemia patients, the most prevalent mutations were Codon 41/42 (-CTTT) and IVS I-5 (G>C). The second most common mutation was found in Codon 3 (T>C) (Table 2, Figure 1, and Supplementary Figures 1–4).
 |
Figure 1 Chromatograms of DNA sequences depicting the HBB Mutations at different positions. (A) Codon 3 (T>C) and (B) Codon 41/42 (-CTTT), mutations are present in exons, while (C–F) are the presentation of mutations that are present in intronic region of β-globin gene. Red arrows are used for marking the exact position of mutations. Sequence analysis was done using Finch TV 1.4.0. (FinchTV chromatogram viewer is a popular desktop application, developed by Geospiza, Inc.). |
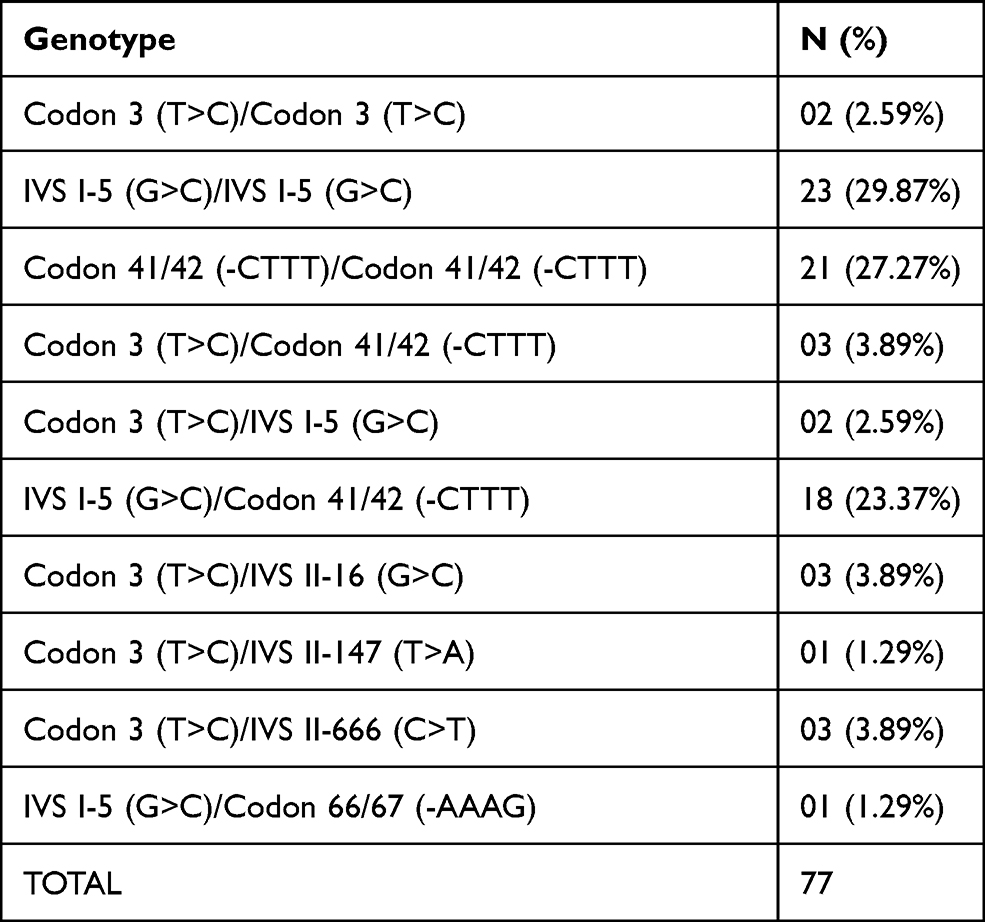 |
Table 2 Genotype of 77 Clinically Manifested β-Thalassemia Major Patients (Homozygous and Compound Heterozygous) |
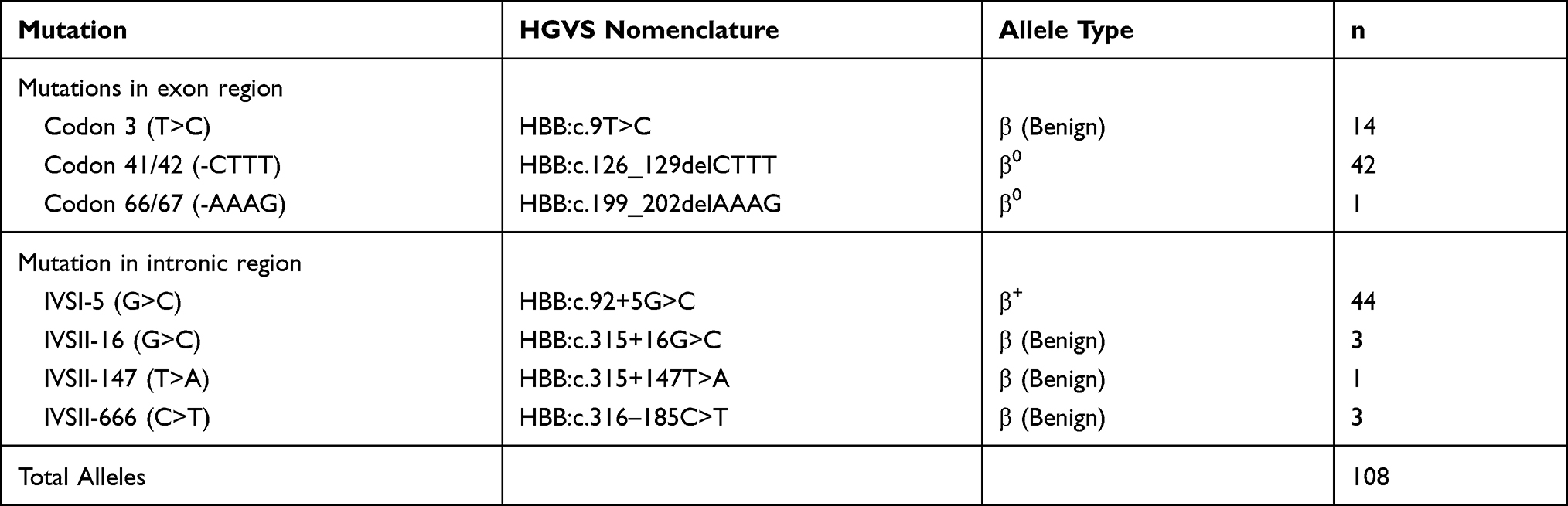 |
Table 3 Genotypic Frequencies of Common and Rare Mutations Found in the Population of Western Region of Uttar Pradesh, India |
Discussion
Mutations in Exon Region
Codon 3 (T>C)
Codon 3 (T>C) was found in 14 (18.18%) individuals. In which only two individuals were homozygous, and the rest were compound heterozygous with IVS I-5 (G>C), Codon 41/42 (-CTTT), IVS II-16, IVS II-147, and IVS II-666 (Table 2). In this mutation, the third codon of the HBB gene is changed from CAT to CAC (rs713040). Both CAT and CAC code for the same amino acid (histidine). This silent mutation has no apparent effect in thalassemia as previously observed in patients of Bangladeshi origin.14 The presence of this variant in this population can be related to human migration. Since this mutation is silent in nature, the severity of the diseases in individuals having this mutation might be possible due to the presence of another deleterious mutation (s).
Codon 41/42 (-CTTT)
The codon 41/42 mutation was due to the deletion of four nucleotides (-CTTT) in the exon-2 of the HBB gene and has been reported in 42 (54.54%) patients. Homozygous individuals for this mutation were 21 (27.27%). Other 21 β-thalassemia patients were found in the heterozygous (compound) state with Codon 3 (T>C) and IVS I-5 (G>C), 3 (3.89%), and 18 (23.37%), respectively. Codon 41/42 (-CTTT) is a severe mutation and quite common in this subcontinent (1.3% in the Maldives to up to 37.3% in Malaysia, Thailand, Singapore, and China, in the world population as well).15 A frameshift mutation, deletion of 4 nucleotides, codons 41/42, TTCTTT to TT, was first reported in an Asian Indian with β0-thalassemia by Kazazian et al.16
A Novel Codon 66/67 (-AAAG)
A five-year-old male child was found in the compound heterozygous state (β+/β0) with point mutation IVS I-5 (G>C) and Codon 66/67 (-AAAG). This novel deletion of AAAG (HBB: c.199_202delAAAG) located at chr11:5226691-5226694, and presents in exon-2 of the HBB gene (Figure 2A). Health records, HPLC, and CBC data of this patient recorded from the hospital and patient’s handbook showed critically reduced BMI (15.9 kg/m2), reflecting severe thinness, a prolonged blood transfusion history (Since the age of 8 months), and the elevated HbA2 (4.7%) and HbF (37%) than that of the normal range. Moreover, the evaluation of markedly reduced CBC data (MCV = 73.4fl and MCH = 22.5pg) shows the existence of ineffective erythropoiesis. The examination of peripheral blood smear shows morphologically changed RBCs as anisopoikilocytosis; dacryocytes, and acanthocytes (Figure 2B). The translation of this mutated gene sequence to protein sequence by using Expasy translate tool (https://web.expasy.org/translate/) followed by BLAST with normal beta-globin protein (Mview Tool) predicts the production of a truncated β-globin chain due to frameshifting (Figure 2C). Using bioinformatics tool (Swiss-Model), modeling of the normal structure of the beta-globin protein exhibits binding affinity towards heme molecules (Figure 2D (i) (ii) (ii)). In contrast, the functional and structural deleterious effects of this frameshift mutation were prognosticated that the truncated globin chain (Figure 2D (iv), (v)) shows the absent affinity towards heme molecules.17,18 The family’s molecular study could not be done because the parents of the child refused to give consent.
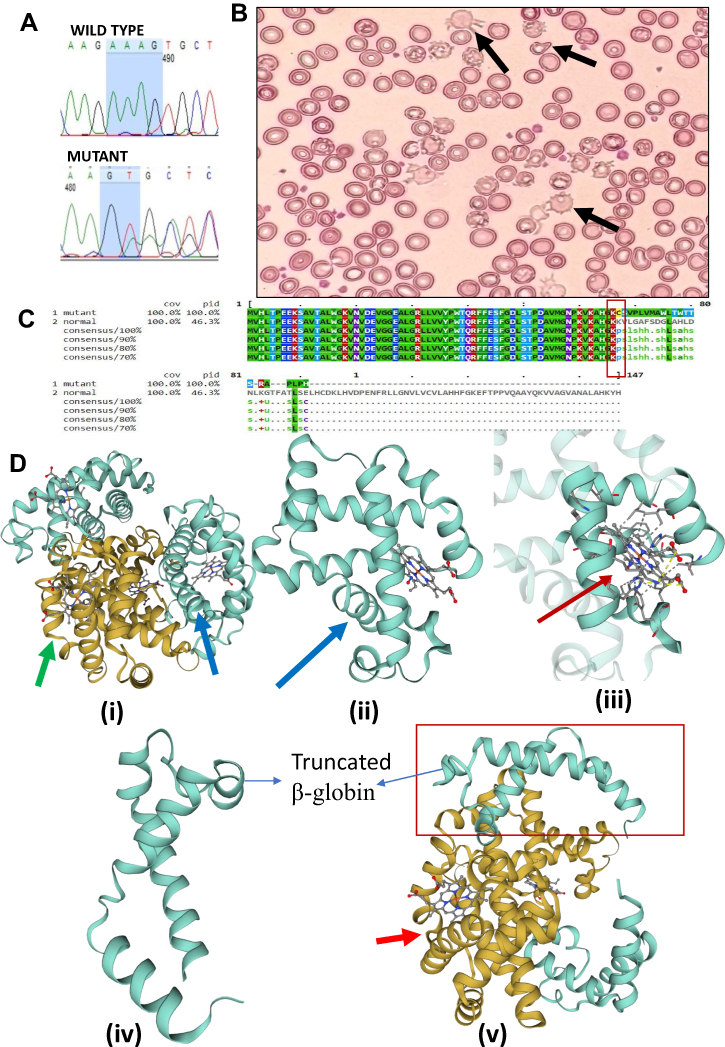 |
Figure 2 (A) Sanger sequencing chromatogram confirms the deletion of four nucleotides (AAAG) from beta-globin gene (HBB). (B) The peripheral blood smear stained with Leishman’s staining showing Anisopoikilocytosis; Acanthocytes (Irregularly spaced blunted projections from the margin of the cells) and Dacryocytes or Teardrop erythrocytes can be seen (marked by black arrows). (C) The Mview of Multiple Sequence Alignment (EMBL-EBI) of two beta-globin protein sequences; first one is of mutant with deletion of AAAG and second one is a normal protein sequence (control). This deletion is responsible for the frameshifting which results into stop codon and ultimately cause truncated β-globin chain (Rectangular region showing point of frameshifting). (D) The modelling using online software Swiss-Model shows the truncated protein of 87 aa has lost its binding capacity towards heme molecule as compared to normal beta-globin protein (147 aa). (i), (ii) and (iii) depicting model for normal beta-globin structure and its binding with heme molecule, whereas the figure (iv and v) shows truncated protein of 87 amino acids, which is devoid of all amino acids that are involved in binding with heme molecules. Notes: Green colored arrow was used to mark α-globin protein, the blue colored arrow showing normal beta-globin protein, while red-colored arrow depicts the binding of normal beta-globin (SMTL ID: 6kye.1) with the heme molecule, Swiss-Model used “SMTL ID: 5sw7.1.B” as template for modelling. |
Mutations in Intron Region
IVS I-5 (G>C)
It is the most common mutation in Asian countries (Table 2) like Pakistan, India, Sri Lanka, the Maldives, and among the Malays from Malaysia and Singapore. In the present study IVS I-5 (G>C) mutation was found in 44 (57.14%) individuals of the thalassemia cases, as previously reported as 73.5% in the eastern region, 49.36% in the western region, 54.35% in the northern region and 57.41% in the southern region of India.19 This mutation lies in the splicing region that causes an alteration in the splicing process.
IVS II-16 (G>C), IVS II-147 (T>A), and IVS II-666 (C>T)
IVSII-16 (G>C) is in the alternative splicing region of mRNA, which might lead to differential expression. This polymorphism is quite common in Indian populations. Fifty-five populations studied all over India have revealed this C allele frequency ranging from 0.21 in Indo-Europeans to 0.66 in Dravidian linguistic groups.20 On the other hand, IVS II-147 (T>A) and IVS II-666 (C>T) are studied less. Similar to the IVS II-16 (G>C) these polymorphisms are also located in the intron-2, therefore clinically benign or silent.
Genotype and Phenotype
All the beta-thalassemia major affected individuals exhibited heterogeneity in the disease’s phenotypes; therefore, our study also shows the inconsistently increased values of HbA2, HbF, and Ferritin as reported in the various reports. All the patients were having a highly elevated level of serum ferritin (Between 1500–3000 ng/mL) suggested iron overload and chances of Liver Injury due to increased liver iron concentration.21 For patients whose mutations were non-deleterious, the severity of the diseases was also stated by Codon 3 (T>C), IVS II-666 (C>T), and IVS II-16 (G>C), which is only possible with the existence of some other deleterious mutations. Individuals with IVS I-5 (G>C) and Codon 41/42 (-CTTT) mutations were having highly increased HbA2 levels (>4.0%) with the reduced amount of MCV and MCH. A Patient was having a compound heterozygous state of mutations IVS I-5 (G>C) and Codon 66/67 (-AAAG) was showing similar phenotypes as found in the case of Codon 41/42 (-CTTT) except the level of HbF (Table 4).
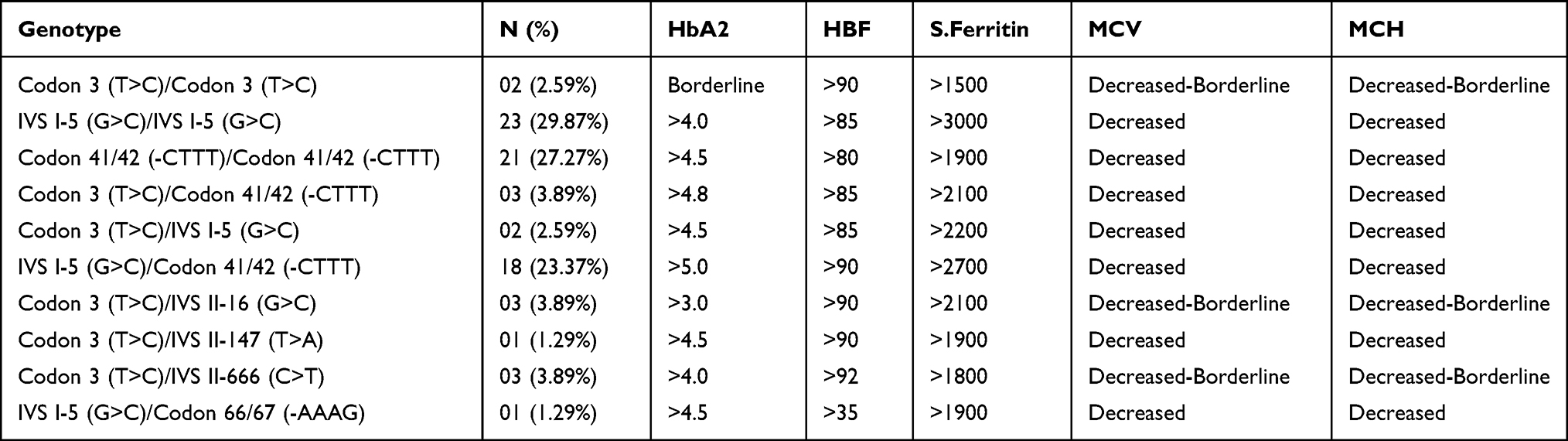 |
Table 4 Genotype-Phenotype Correlation: All the Patients Were Having Markedly Increased Level of S. Ferritin, HbA2, and HbF Level, and Decreased Values of MCV and MCH. Phenotypes of Patients with Silent Mutations Viz. Codon 3 (T>C)/IVS II-16 (G>C) or IVS II-147 (T>A) or IVS II-666 (C>T) Shows Possibilities of the Presence of Other Deleterious Mutations |
Conclusion
This study reports seven different types of mutations for the first time in Western Uttar Pradesh, India, including a novel frameshift mutation (deletion of 4 nucleotides (Codon 66/67 (-AAAG)) reported in the exon-2 region. Bioinformatic tools prognosticated the effects of this novel deletion on the beta-globin chain’s functions and structure as truncated and non-functional protein due to lack of those amino acids involved in binding with heme molecules. IVS 1–5 (G>C) and Codon 41/42 (-CTTT) are the most frequently reported mutations. The molecular spectrum for these two cases consists of 44 and 42 alleles out of 108 alleles, respectively. The allele of Codon 3 (T>C) is a Bangladeshi variant found in 14 individuals. This study also reports other intronic mutations (Polymorphisms) {IVS II-16 (G>C), IVS II-147 (T>A), and IVS II-666 (C>T)}.
Acknowledgments
The authors are grateful to the Chairman, Department of Zoology, AMU, and Head, Department of Botany, Jamia Hamdard, for providing necessary laboratory facilities. Indian Council of Medical Research (ICMR), India, is highly acknowledged for providing financial support during the study. Grant Number [3/1/3-JRF-2016/HRD-32013].
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–712.
2. Angastiniotis M, Modell B. Global epidemiology of hemoglobin disorders. Ann N Y Acad Sci. 1998;850:251–269. doi:10.1111/j.1749-6632.1998.tb10482.x
3. Modell B, Darlison M, Birgens H, et al. Epidemiology of haemoglobin disorders in Europe: an overview. Scand J Clin Lab Invest. 2007;67(1):39–69. doi:10.1080/00365510601046557
4. Michlitsch J, Azimi M, Hoppe C, et al. Newborn screening for hemoglobinopathies in California. Pediatr Blood Cancer. 2009;52(4):486–490. doi:10.1002/pbc.21883
5. Mukherji M. Cooley’s anaemia (erythroblastic or Mediterranean anaemia). Indian J Pediatr. 1938;5(1):1–7. doi:10.1007/BF02957712
6. WHO. Joint WHO-TIF meeting on management of hemoglobin disorders (2nd: 2008: Nicosia, Cyprus). Geneva: World Health Organization; 2008. Available from: https://apps.who.int/iris/handle/10665/43969.
7. Mohanty D, Colah RB, Gorakshakar AC, et al. Prevalence of β-thalassemia and other haemoglobinopathies in six cities in India: a multicentre study. J Community Genet. 2013;4(1):33–42. doi:10.1007/s12687-012-0114-0
8. Huisman THJ, Carver MF, Baysal EA. Syllabus of Thalassemia Mutations. Augusta, GA: The Sickle Cell Anemia Foundation; 1997.
9. Christopher AF, Kumari A, Chaudhary S, Hora S, Ali Z, Agrawal SC. Unique pattern of mutations in β-thalassemia patients in Western Uttar Pradesh. Indian J Hum Genet. 2013;19(2):207–212. doi:10.4103/0971-6866.116119
10. Pirastru M, Manca L, Trova S, Mereu P. Biochemical and molecular analysis of the Hb Lepore Boston Washington in a Syrian homozygous child. Biomed Res Int. 2017;2017:1261972. doi:10.1155/2017/1261972
11. Kibbe WA. OligoCalc: an online oligonucleotide properties calculator’. Nucleic Acids Res. 2007;35(webserver issue):W43–W46. doi:10.1093/nar/gkm234
12. Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16(22):10881–10890. doi:10.1093/nar/16.22.10881
13. George E, Li HJ, Fei YJ, et al. Types of thalassemia among patients attending a large university clinic in Kuala Lumpur, Malaysia. Hemoglobin. 1992;16(1–2):51–66. doi:10.3109/03630269209005676
14. Ibn Ayub M, Moosa MM, Sarwardi G, Khan W, Khan H, Yeasmin S. Mutation analysis of the HBB gene in selected Bangladeshi beta-thalassemic individuals: presence of rare mutations. Genet Test Mol Biomarkers. 2010;14(3):299–302. doi:10.1089/gtmb.2009.0160
15. Colah R, Gorakshakar A, Nadkarni A. Global burden, distribution and prevention of β-thalassemias and hemoglobin E disorders. Expert Rev Hematol. 2010;3(1):103–117. doi:10.1586/ehm.09.74
16. Kazazian HH
17. Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi:10.1093/nar/gky427
18. Bienert S, Waterhouse A, de Beer TA, et al. The swiss-model repository-new features and functionality. Nucleic Acids Res. 2017;45(D1):D313–D319. doi:10.1093/nar/gkw1132
19. Edison ES, Shaji RV, Devi SG, et al. Analysis of beta globin mutations in the Indian population: presence of rare and novel mutations and region-wise heterogeneity. Clin Genet. 2008;73(4):331–337. doi:10.1111/j.1399-0004.2008.00973.x
20. Kulkarni GD, Kulkarni SS, Kadakol GS, et al. Molecular basis of β-thalassemia in Karnataka, India. Genet Test Mol Biomarkers. 2012;16(2):138–141. doi:10.1089/gtmb.2011.0035
21. Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV. Serum ferritin: past, present and future. Biochim Biophys Acta. 2010;1800(8):760–769. doi:10.1016/j.bbagen.2010.03.011
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.