Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
A Novel Cerebroprotein Hydrolysate, CH1, Ameliorates Chronic Focal Cerebral Ischemia Injury by Promoting White Matter Integrity via the Shh/Ptch-1/Gli-1 Signaling Pathway
Authors Cao W, Zhang C, Chen R, Wu Q, Xu R, Zhang L, Zhang X
Received 2 November 2020
Accepted for publication 14 December 2020
Published 23 December 2020 Volume 2020:16 Pages 3209—3224
DOI https://doi.org/10.2147/NDT.S289990
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
Wen Cao,1 Cong Zhang,1 Rong Chen,2,3 Qianqian Wu,1 Renhao Xu,2,3 Lan Zhang,1 Xiangjian Zhang1– 3
1Department of Neurology, Second Hospital of Hebei Medical University, Shijiazhuang, Hebei 050000, People’s Republic of China; 2Hebei Collaborative Innovation Center for Cardio-Cerebrovascular Disease, Shijiazhuang, Hebei 050000, People’s Republic of China; 3Hebei Vascular Homeostasis Key Laboratory for Neurology, Shijiazhuang, Hebei 050000, People’s Republic of China
Correspondence: Xiangjian Zhang
Department of Neurology, Second Hospital of Hebei Medical University, 215 Hepingxi Road, Shijiazhuang, Hebei 050000, People’s Republic of China
Tel +86 15803210578
Fax +86 31166002822
Email [email protected]
Purpose: Strokes are devastating as there are no current therapies to prevent long-term neurological deficits. Previous studies reported that cerebroprotein hydrolysate (CH) plays a role in neuronal protection in acute phase after ischemic stroke, while the long-term effects of CH upon brain plasticity and neurological outcomes after stroke are still uncertain. To address these gaps, we assessed the effect of a new cerebroprotein hydrolysate, CH1, on long-term gray and white matter integrity as well as axonal plasticity in the late phase after ischemic stroke and the potential mechanisms.
Methods: Adult male mice were subjected to permanent distal middle cerebral artery occlusion (dMCAO), followed by daily intraperitoneal injection of CH1 for 14 days. Motor function was measured weekly through behavioral neurological evaluations. Gray matter intensity and white matter intensity were examined by immunofluorescence staining. The sonic hedgehog (Shh) inhibitor cyclopamine (CYC) was injected to determine the involvement of the Shh pathway in the therapeutic effects of CH1.
Results: We found that intraperitoneal delivery of CH1, compared to vehicle administration, significantly improved long-term neurological outcomes at various times and promoted neuronal viability at 14 days but not at 28 days after stroke. Importantly, CH1 mitigated stroke-induced white matter injury and facilitated axonal plasticity in the late stage after stroke.
Conclusion: These results unveil a previously unappreciated role for CH in the repair of white matter and brain plasticity after stroke.
Keywords: ischemic stroke, cerebroprotein hydrolysate, axonal plasticity, white matter integrity, Shh pathway
Introduction
Ischemic stroke is the leading cause of death and disability in adults worldwide.1 To date, the only FDA-approved medication for ischemic stroke is recombinant tissue plasminogen activator (rtPA), but its use is confined to a narrow time window of 4.5 h, and it can cause intracranial hemorrhage as well as inflammatory response in brain capillaries and subsequently neuronal cell damage after stroke as deleterious side effects.2–4 Most candidate neuroprotective drugs have found mismatches between preclinical studies and clinical trials, partly due to a failure to activate long-term brain repair processes.5 Therefore, effective strategies to promote long-term functional assessments after stroke are critical beyond the hyperacute phase of ischemia.
Stroke causes neuronal damage within gray matter and axonal injury within white matter tracts.6,7 Upon cerebral ischemia, the brain initiates self-repair mechanisms and undergoes a post-stroke neurorestorative process, mainly including angiogenesis, neurogenesis and axonal plasticity.8 Previous studies in the stroke field have mainly focused on neuronal cell death mechanisms.9,10 However, assessments of gray matter injury do not always coincide with behavioral function impairments; therefore, an increasing amount of research has addressed the assessment of axonal plasticity within the white matter, which has been demonstrated to be strongly correlated with ischemia.10 Extensive rewiring of remaining neuronal connections in the white matter during the axonal plasticity process leads to post-stroke cortical map rearrangement and functional improvement.11 Therefore, strategies targeting axonal repair and the formation of new connections are needed for stroke recovery.
Sonic hedgehog (Shh), a secreted glycoprotein, is an endogenous activator of the Hedgehog pathway. Shh initiates signaling by binding to its cell surface receptor, Patched-1 (Ptch-1). Then, Ptch-1 is inactivated, releasing its suppression of smoothened (Smo). The transcription factor Gli-1 is activated and translocated into the nucleus, inducing the transcription of Gli-1 target genes. Our recent study has demonstrated that the endogenous Hedgehog pathway is activated after ischemic stroke, promoting neurogenesis in the subventricular zone in vivo and promoting neurite outgrowth in vitro.12 Meanwhile, activation of the Shh signaling pathway promotes axonal regeneration following stroke.13 Therefore, Shh and its downstream signaling molecules can serve as potential therapeutic targets to induce axonal plasticity after stroke.
Cerebroprotein hydrolysate (CH) is a mixture of low-molecular-weight neuropeptides derived from purified porcine brain tissue. The ability to penetrate biological membranes and pass through the blood-brain barrier makes CH a strong candidate for clinical application. In vivo, CH has been demonstrated to improve long-term functional outcomes, promote neurogenesis,14,15 attenuate neuroinflammation16 and inhibit free radical formation.15 Importantly, several randomized clinical trials in patients also revealed the therapeutic value of CH in the field of stroke research.17–21 However, the effect of CH on axonal plasticity within the white matter following ischemic stroke is not known in detail.
In this study, we sought to explore the potential application of a new cerebroprotein hydrolysate, known as cerebroprotein hydrolysate (I) (CH1). We aimed to evaluate the neuroprotective effect of CH1 against long-term neurological deficits and axonal plasticity in the late phase in a mouse model of distal middle cerebral artery occlusion (dMCAO) and determine whether this benefit was associated with activation of the Shh pathway.
Materials and Methods
Animals and Experiments
A total of 221 adult male C57BL/6 mice (23 to 25 g; 8 to 12 weeks) were obtained from Vital River Laboratory Animal Technology Co. 3, Ltd. (Beijing, China). All animal procedures were approved by the Institutional Animal Care and Management Committee of the Second Hospital of Hebei Medical University (Permit No. HMUSHC-130,318) and performed in accordance with the Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize animal suffering and the number of animals used. Mice were group-housed in a specific-pathogen-free (SPF)-grade, temperature- and humidity-controlled animal facility with free access to food and water under a 12/12-hour light/dark cycle. All mice were the same age at the beginning of the experiments. All surgeries and outcome measurements were performed by investigators blinded to the experimental group assignments.
dMCAO Model
Focal cerebral ischemia was induced in adult male mice by permanent occlusion of the right common carotid artery (CCA) and right distal middle cerebral artery (MCA) in a sterile operating room, as described previously.22
Briefly, mice were anesthetized with 3% (vol/vol) isoflurane in 67%:30% (vol/vol) N2O/O2; anesthesia was maintained with 1.5% (vol/vol) isoflurane. After a skin incision was made at the midline of the neck, the right CCA was dissected and permanently ligated. Then, another incision was made between the right ear and eye, and a craniotomy was performed to expose the right MCA. The distal MCA was permanently coagulated by low-intensity bipolar electrocautery without damaging the brain surface. The rectal temperature and the right temporalis muscle temperature were maintained at 37.0±0.5°C during surgery by using a heating pad and a heat lamp, respectively.
Regional cortical cerebral blood flow (CBF) was measured using two-dimensional laser speckle techniques to evaluate whether the dMCAO models were conducted successfully according to the manufacturer’s instructions (Supplementary Methods section in the Supplementary materials). Mice were excluded if they died or developed subarachnoid hemorrhage during operation or if their regional cortical CBF was not reduced to less than 30% of the baseline level. A total of 8 mice were excluded because of death or failing to meet the criteria. The attrition rate is about 3.6%. Throughout the text, we refer to this ischemia modeling procedure as “dMCAO”.
Drug and Reagent Administration
CH1 was purchased from Hebei ZhiTong Biopharma Pharmaceutical Co., Ltd. with a Chinese FDA ratification code of GuoYaoZhunZi-H20051737/H20051738. CH1 was administered daily by intraperitoneal (i.p.) injection for 14 days after dMCAO. All the mice were randomly divided into 6 groups using a random number table generated by the software program SPSS 22.0 (SPSS Inc., Chicago, IL, USA). These 6 groups were as follows: Sham group (sham) – sham-operated mice underwent the same surgical procedures as the dMCAO mice except for the occlusion of the CCA and MCA; vehicle group (vehicle) – mice were subjected to dMCAO and then received i.p. injections of normal saline in the same volume as the experimental CH1 injections; CH1 groups – mice received i.p. injections of CH1 at 5 mg·kg−1 (CH1-L), 10 mg·kg−1 (CH1-M), or 20 mg·kg−1 (CH1-H) once daily for 14 days after dMCAO; CH1+cyclopamine (CYC) groups – mice were treated with CH1 combined with CYC (1 mg·mL−1, 10 mg·kg−1); this combination was administered daily by i.p. injection for 14 days after stroke.
Behavioral Tests
Rotarod
In order to assess sensorimotor balance and coordination after stroke, mice were placed on an accelerating rotating drum as described previously.22 In brief, mice were forced to run on a rotarod accelerating from 4 to 40 rpm within a 300-s duration. The latency of each mouse to fall off the machine was recorded by an investigator who was blinded to the treatments. All mice were trained for 3 days before stroke, and the average latency on the last day before dMCAO was used as the prestroke (baseline) level. The test was repeated at 3 days, 7 days, 14 days, and 28 days after dMCAO.
mNSS
The modified neurological severity score (mNSS) is a composite evaluation standard of motor, sensory, reflex, and balance tests, graded on a scale ranging from 0 to 18 (normal score, 0; maximal deficit score, 18).23 A higher score indicates worse neurological function. All neurological function evaluations were recorded by an investigator blinded to the treatment groups.
Adhesive Removal Test
An adhesive removal test was performed to assess sensorimotor function and postural asymmetry in ischemic mice.24,25 Briefly, two pieces of adhesive tape (0.2 × 0.3 cm2) were attached to the lesioned forepaws of the mice with equal pressure. The mice were placed in a Perspex box; contact time (the latency of the mouse to show a tactile response to the presence of the adhesive strips) and removal time (the latency of the mouse to remove the adhesive strips) were collected, with a maximum period of 120 s.
TTC
The infarct volume was evaluated by 2,3,5-triphenyltetrazolium chloride (TTC) staining at day 14 and day 28 after dMCAO.26 Briefly, under deep anesthesia, the fresh brain of each mouse was rapidly removed and sliced into 6 coronal sections. Then, sections were immersed in 2% TTC at 37°C for 15 min. Infarct volume was calculated with NIH ImageJ software (National Institutes of Health, RRID SCR_003070) by an observer blinded to the experimental groups. Infarct volumes were determined using the following equation,27 which corrects for edema: Infarct volumes (%)=[Total Lesion Volume - (Right Hemisphere Volume - Left Hemisphere Volume)]/Left Hemisphere Volume × 100.
Assessment of Cerebral Cortical Expansion
Under deep anesthesia with isoflurane at 14 days and 28 days after stroke, the brains of mice were fixed by paraformaldehyde perfusion and then removed. Images of the whole brains were captured by a microscopic digital camera (AxioCam, Zeiss) and then analyzed by the ImageJ system. The injury contralateral width (W.contra) from the lateral edge to the midline and the injury ipsilateral width (W.ipsi) from the edge of cortical cavitation to the midline were measured. Measurements were performed at the midpoint of the forebrain. Cortical width index (CWI) = W.ipsi × 100%/W.contra.28
Nissl Staining
We conducted Nissl staining to evaluate cortical neuronal density. The paraffifinized brain sections (5mm) were stained with 0.1% cresyl violet (Sigma, USA). Then, the cell morphology in cerebral cortex was observed under an ×200 optical microscope (Zeiss, Germany). Numerous Nissl bodies were observed, indicating that the neurons had a high ability to synthesize proteins. When the neurons were damaged, the number of Nissl corpuscles decreased significantly. The number of stained cells was counted randomly from four high-power fields in the ischemic penumbra and calculated using Image J.
Western Blotting
Proteins were isolated from brain tissue and analyzed using the standard SDS-PAGE method. Immunoreactivity was determined by semiquantitative gel density scanning. The following antibodies were used: rabbit anti-Shh (1:1000, Boster), rabbit anti-Ptch-1 (1:500, Abcam), mouse anti-Smo (1:300, Proteintech), rabbit anti-Gli-1 (1:500, Bioworld), rabbit anti-GAP43 (1:5000, NOVUS), and rabbit anti-GAPDH (1:10,000, Bioworld). Membranes were scanned with an Odyssey infrared scanner (LICOR Bioscience, Lincoln, NE, USA) and analyzed with ImageJ software. The relative densitometric values were quantified with respect to GAPDH.
Tract-Tracer Injections and Quantification of Axonal Sprouting
Poststroke axonal remodeling was assessed using anterograde tracing as described previously.28 At 14 days after stroke, mice were anesthetized and fixed in a stereotaxic frame with the skull exposed. The tract tracer biotinylated dextran amine (BDA, 10,000 MW; Molecular Probes, Eugene, OR) was dissolved in saline at a concentration of 10% (wt/vol) and injected at two sites in the contralesional (left) primary motor cortex (M1; 2 µL per injection; stereotaxic coordinates relative to bregma: 1. anteroposterior (AP) 0.6 mm, mediolateral (ML) 1.2 mm, dorsoventral (DV) 1.5 mm; 2. AP 0.0 mm, ML 1.8 mm, DV 1.70 mm)29 2 weeks after dMCAO or the sham operation. The micropipette was left in place for 10 min after the injection. BDA was allowed to be anterogradely transported for an additional 2 weeks to trace the organization of the pyramidal tracts. Two weeks later, the mice were perfused with 0.9% (wt/vol) NaCl, followed by 4% (wt/vol) paraformaldehyde. The entire brain was processed and immersed in 4% paraformaldehyde overnight. Tissue sections (35 µm) were incubated in streptavidin-FITC to analyze the regeneration of BDA+ fibers after injury. The number of midline-crossing BDA-positive fibers (from the intact side to the lesioned side) was quantified manually by an investigator blinded to the experimental groups.
Immunofluorescence Staining
Briefly, coronal brain sections fixed in 4% PFA were permeabilized in 0.5% Triton X-100 for 20 min. After being washed once in PBS and blocked with 10% donkey serum for 1 h at 37°C, tissues were incubated with primary antibodies at 4°C overnight. The following primary antibodies were used: rat anti-myelin basic protein (MBP; 1:400, Abcam); mouse anti-non-phosphorylated neurofilament H (SMI-32; 1:100, Biolegend), mouse anti-NeuN (1:500, BD Biosciences), and rabbit anti-Gli-1 (1:50, Bioworld). On the second day, after three washes in PBS, sections were incubated with appropriate secondary antibodies at room temperature for 1 h. After three washes in PBS, brain sections were then coverslipped with Fluoromount-G containing 4′,6-diamidine-2-phenylindole (DAPI; Southern Biotech, Birmingham, AL) to stain the cell nuclei.
Statistical Analysis
Data are presented as the mean ± SD. GraphPad Prism software (GraphPad Software Inc., CA, USA) was used for statistical analysis. The data were first tested for normality and Levene’s test was used to assess the uniformity of the variance. Nonparametric one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was performed for comparisons among more than two groups. A two-tailed Student’s t-test was performed for two-group comparisons. Nonparametric analyses were performed with the Kruskal–Wallis H-test for mNSS. ImageJ software was used to quantify the band intensity in Western blot images. Quantification was performed from at least three independent experiments. The criterion for statistical significance was set to P < 0.05.
Results
Poststroke CH1 Administration Alleviates Long-Term Neurological Deficits in dMCAO Mice
The dMCAO model was successfully conducted, which were examined by cerebral blood flow measurements (Fig. S1A-C). We first examined the effects of various CH1 doses (5, 10, and 20 mg/kg) on long-term neurological deficits at various times after dMCAO (Figure 1A).
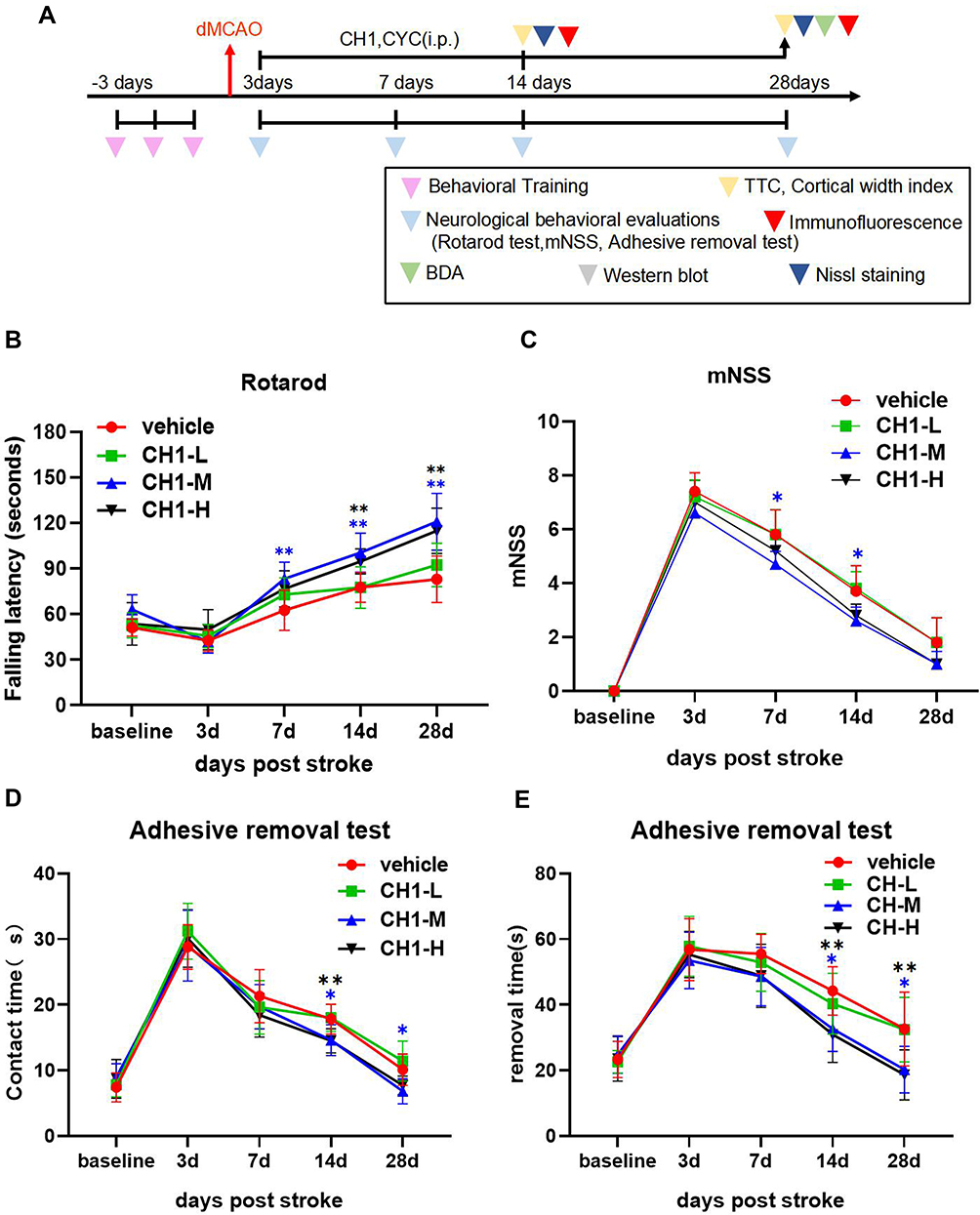 |
Figure 1 Treatment with CH1 promotes long-term functional recovery after stroke. (A) Illustration of timeline for animal experiments. (B–E) Mice were subjected to dMCAO and i.p. injected with different concentrations (5 mg/kg, 10 mg/kg, 20 mg/kg) CH1 treatments. Sensorimotor functions were assessed before (baseline) and up to 28 days after dMCAO by the Rotarod (B), mNSS evaluation (C) and adhesive removal test (D and E). *P< 0.05, **P< 0.01 by one-way ANOVA or Kruskal–Wallis H-test, n = 10 mice per group. |
As illustrated in all experiments, postischemic neurobehavioral performance was considerably worse than baseline at day 3 after dMCAO and gradually returned to baseline 7 days later. According to the rotarod results, 5 mg/kg CH1 (CH1-L) was not associated with a statistically significant improvement in neurological function at any time point. Both 10 mg/kg (CH1-M) and 20 mg/kg (CH1-H) CH1, compared with vehicle, significantly improved neurological function at 7, 14 and 28 days after stroke (Figure 1B).
The results of the adhesive removal test were consistent with the rotarod data: both 10 mg/kg and 20 mg/kg CH1 mice exhibited better contact time and removal time than the vehicle group 14–28 days after dMCAO (Figure 1D and E). Both parameters showed nearly complete recovery at 28 days after dMCAO.
The mNSS scores indicated severe and persistent neurological deficits after dMCAO compared with baseline. Treatment with 10 mg/kg CH1 significantly alleviated neurological deficits in late-stage poststroke mice (Figure 1C).
Taken together, our findings indicate that both 10 mg/kg and 20 mg/kg CH1 potentiated sensorimotor function in the late stage of stroke. Among the tested doses, 10 mg/kg was the lowest dose that significantly improved poststroke performance in all tests. The higher dose (20 mg/kg) produced no further improvement in behavioral performance. Therefore, 10 mg/kg CH1 was selected as the optimal dose for the following studies.
Poststroke CH1 Administration Reduced Infarct Volume and Increased the Cortical Width Index at 14 Days but Not 28 Days After Stroke
Next, we studied the effects of CH1 on neuropathological outcomes correlated with improvements in functional recovery at 14 days and 28 days after stroke. As expected, the infarct volume was dramatically reduced in CH1 (10 mg/kg)-treated mice compared with the vehicle group at 14 days after stroke (Figure 2A and B). Interestingly, at 28 days after ischemia, the infarct volumes were comparable between CH1- and vehicle-treated mice. Meanwhile, the CH1 group exhibited a significantly higher cortical width index than the vehicle-treated group at 14 days after stroke (Figure 2C and D), but the cortical width index of the CH1 group was comparable to that of the vehicle group at 28 days after stroke.
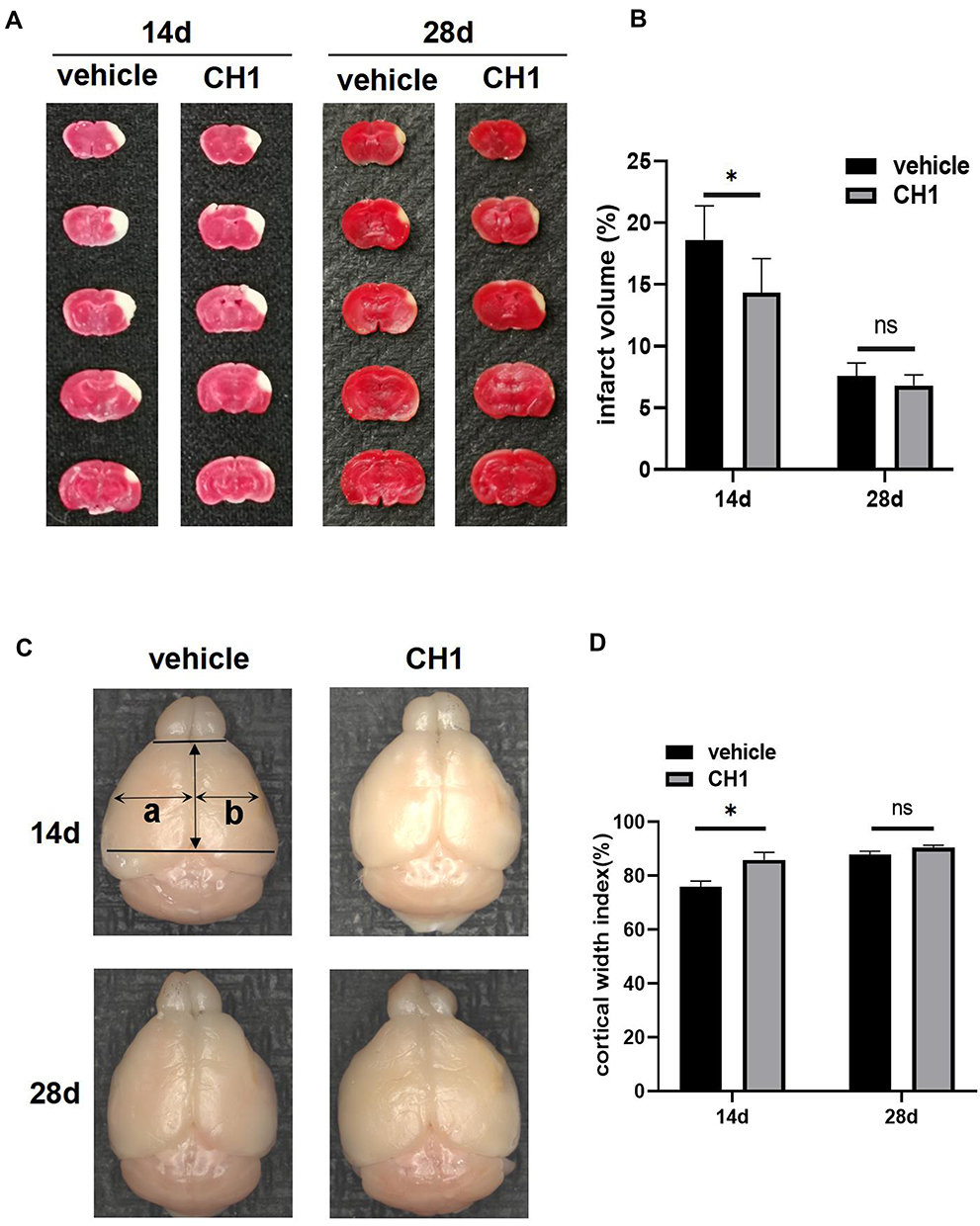 |
Figure 2 Treatment with CH1 reduced infarct volume and increased cortical width index at 14d post-stroke but not at 28d. (A and B) Representative images (A) and statistical graph (B) of infarct volume stained with TTC 14d and 28d after dMCAO in vehicle and CH1 group. Absence of red stain represents infarct area. (C and D) Ex vivo images (C) and statistical graph (D) of cortical width index 14d and 28d after dMCAO, a and b mark the maximum width from the midline to the edge of the noninfarcted and infarcted hemispheres, respectively. *P< 0.05, ns: no significance by t-test, n = 6 mice per group. |
These findings suggested that CH1 can reduce infarct volume and increase the cortical width index at 14 days after stroke, but the neuropathological outcomes did not coincide with behavioral function impairments between the CH1 and vehicle groups at 28 days after stroke.
Poststroke CH1 Treatments Promoted Neuron Survival at 14 Days After dMCAO but Not at 28 Days
Next, to determine the reason why no significant difference was observed in neuropathological outcomes between the CH1 and vehicle groups at 28 days after stroke, we explored microstructural changes within the gray matter. We assessed the total number of viable neurons (NeuN+ cells) in the ischemic penumbra at 14 days and 28 days after dMCAO.30 Consistent with the neuropathological outcomes discussed above, CH1 significantly increased the number of viable neuronal somata in the ischemic penumbra at 14 days after ischemic stroke (Figure 3A and C). However, at 28 days after ischemia, the numbers of neuronal somata were comparable between CH1- and vehicle-treated mice. Therefore, the viable neurons within the gray matter cannot account for the improved neurological outcomes by CH1 at 28 days poststroke. Meanwhile, we conducted Nissl staining of the coronal brain sections from the sham, vehicle and the CH1 treated mice at 14 days and 28 days after ischemic stroke. As shown in Figure 3B, after stroke, the cells were disorderly, the number of Nissl bodies significantly reduced, the cell gap increased and many vacuoles formed. Neurons with abundant Nissl bodies were significantly decreased after stroke compared with the sham group, which was restored by CH administration at 14d post-stroke. However, at 28d post-stroke, there is still no significance between dMCAO group and CH1 group (Figure 3D).
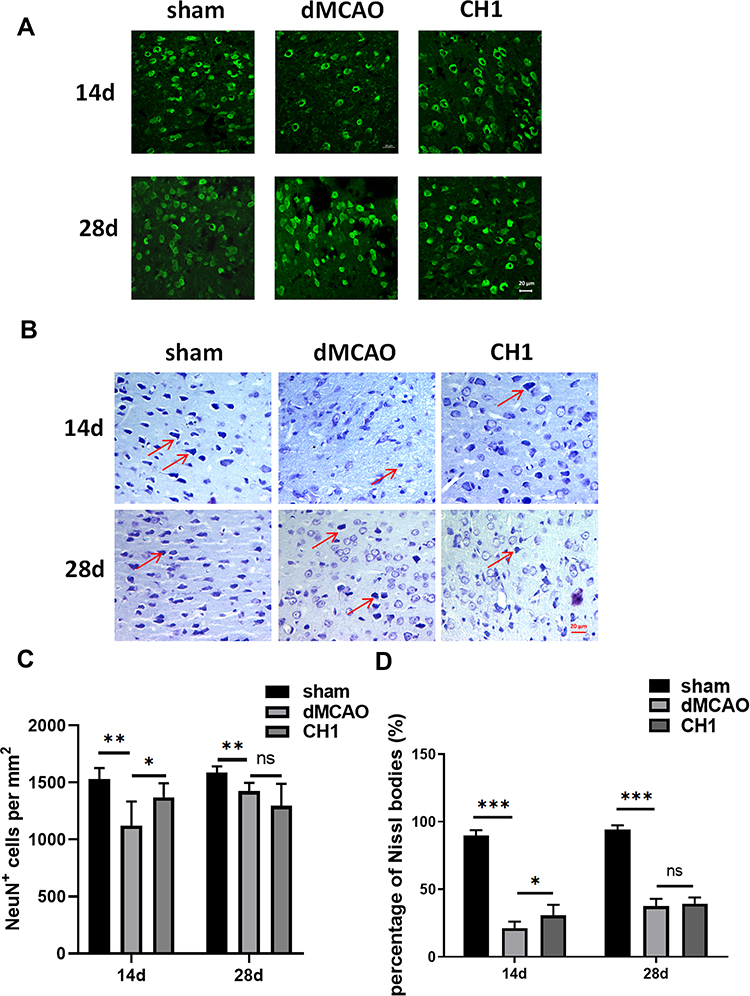 |
Figure 3 Post-stroke CH1 treatments promoted neuron survival at 14 day after dMCAO but not at 28day. (A) Representative images of NeuN positive cells in the ischemic penumbra after stroke at 14 and 28 days in different groups. Scale bar = 20 μm. (B) Nissl-stained images of brain cortical neurons in the ischemic penumbra after stroke at 14 and 28 days in different groups. Scale bar = 20 μm. (C) Quantification of NeuN positive cells numbers per mm2 in the ischemic penumbra after stroke at 14 and 28 days in different groups. (D) Quantification of percentage of Nissl bodies in the ischemic penumbra after stroke at 14 and 28 days in different groups. *P< 0.05, **P< 0.01, ***P<0.001, ns: no significance by one-way ANOVA, n = 6 mice per group. |
This makes us suspect that in the late stage of stroke, the recovery of neurological outcomes with CH1 treatment is no longer due to the protection of neurons but the recovery of white matter.
Poststroke CH1 Treatments Improved White Matter Intensity at 28 Days After Stroke
Emerging evidence has revealed that white matter injury is significantly correlated with neurological deficits after stroke.
Therefore, we investigated the impact of CH1 on the microstructural changes in white matter intensity (WMI) 28 days after dMCAO by double-label immunostaining for MBP (a marker of mature myelin oligodendrocytes, indicating myelin) and SMI-32 (a marker of non-phosphorylated neurofilament H, indicating demyelination) in the peri-infarct cortex (CTX) and striatum (STR).31
In sham-operated animals, abundant staining with MBP was visible in both CTX and STR, whereas SMI-32 staining was barely detectable in these regions. dMCAO caused an increase in SMI-32 immunofluorescence and a concomitant reduction in MBP in CTX and STR. These pathological changes resulted in an increased ratio of SMI-32 to MBP fluorescence intensity, which has been established as an indicator of white matter injury, in the dMCAO group compared with the sham group (Figure 4A–C).
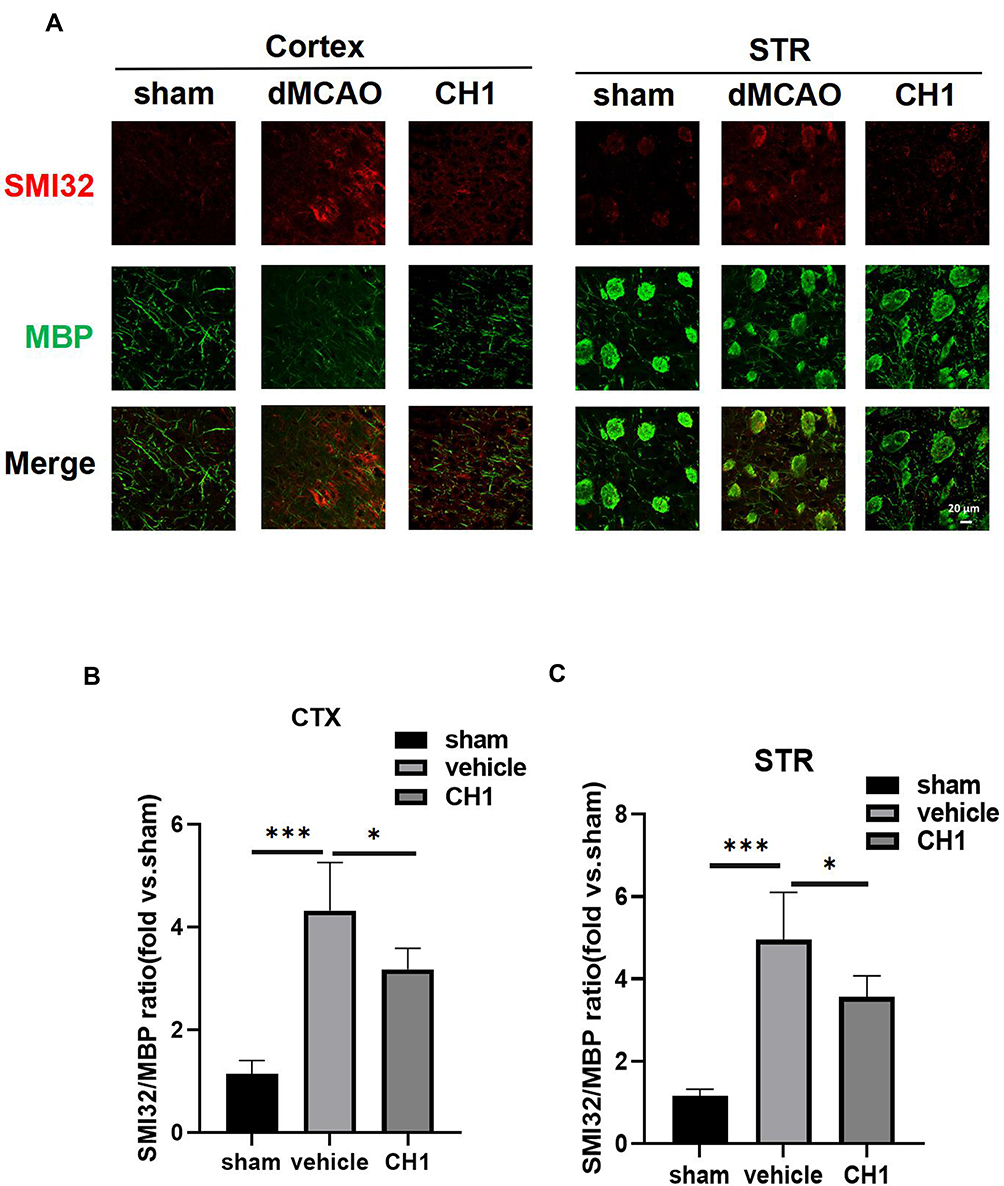 |
Figure 4 Post-stroke CH1 treatment mitigates white matter injury. (A) Representative images of MBP (green) and SMI-32 (red) in peri-infarct cortex (CTX) and striatum (STR) or the corresponding areas in sham-operated mice at 28 days after MCAO. Scale bar = 20μm. (B and C) Quantification of the ratio of SMI-32 to MBP fluorescence intensity in the cortex (B), and striatum (C) of the ischemic hemisphere. *P< 0.05, ***P<0.001, by one-way ANOVA, n = 5 mice per group. |
Strikingly, poststroke intraperitoneal CH1 injections, compared to vehicle alone, substantially reduced the ratio of SMI32 to MBP in both CTX and STR, suggesting that CH1 permanently improved WMI (Figure 4A–C).
These results indicated that poststroke CH1 administration significantly facilitated the restoration of white matter structural integrity after ischemia.
Intraperitoneal Delivery of CH1 Enhanced Axonal Plasticity in Poststroke Mice
Postischemic white matter injury takes the forms of demyelination in the early phase and secondary axonal degeneration as well as impaired nerve conductivity in the late phase.32 Therefore, we suspected that CH1 potently ameliorated WMI and improved neurological outcomes by promoting axonal plasticity after ischemic stroke. We examined axonal connections and sprouting in the corpus callosum (CC) and peri-infarct CTX in each group.
Stroke induces axonal sprouting within motor, premotor, and somatosensory cortical areas, and these new connections are correlated with functional motor recovery. As shown in Figure 5A, axons projecting from neurons within the BDA-injected region cross the CC (the green line), reaching the denervated contralateral neurons in the peri-infarct areas, and branch extensively to form synaptic connections 28 days after stroke. Consistent with previous studies, ischemic stroke attack significantly increased the BDA-positive area in the peri-infarct regions and the number of midline-crossing axonal fibers (Figure 5B–D). CH1 treatment, compared with vehicle treatment, further improved the BDA-positive area in the peri-infarct regions and the number of midline-crossing tracts compared with the vehicle group (Figure 5B–D).
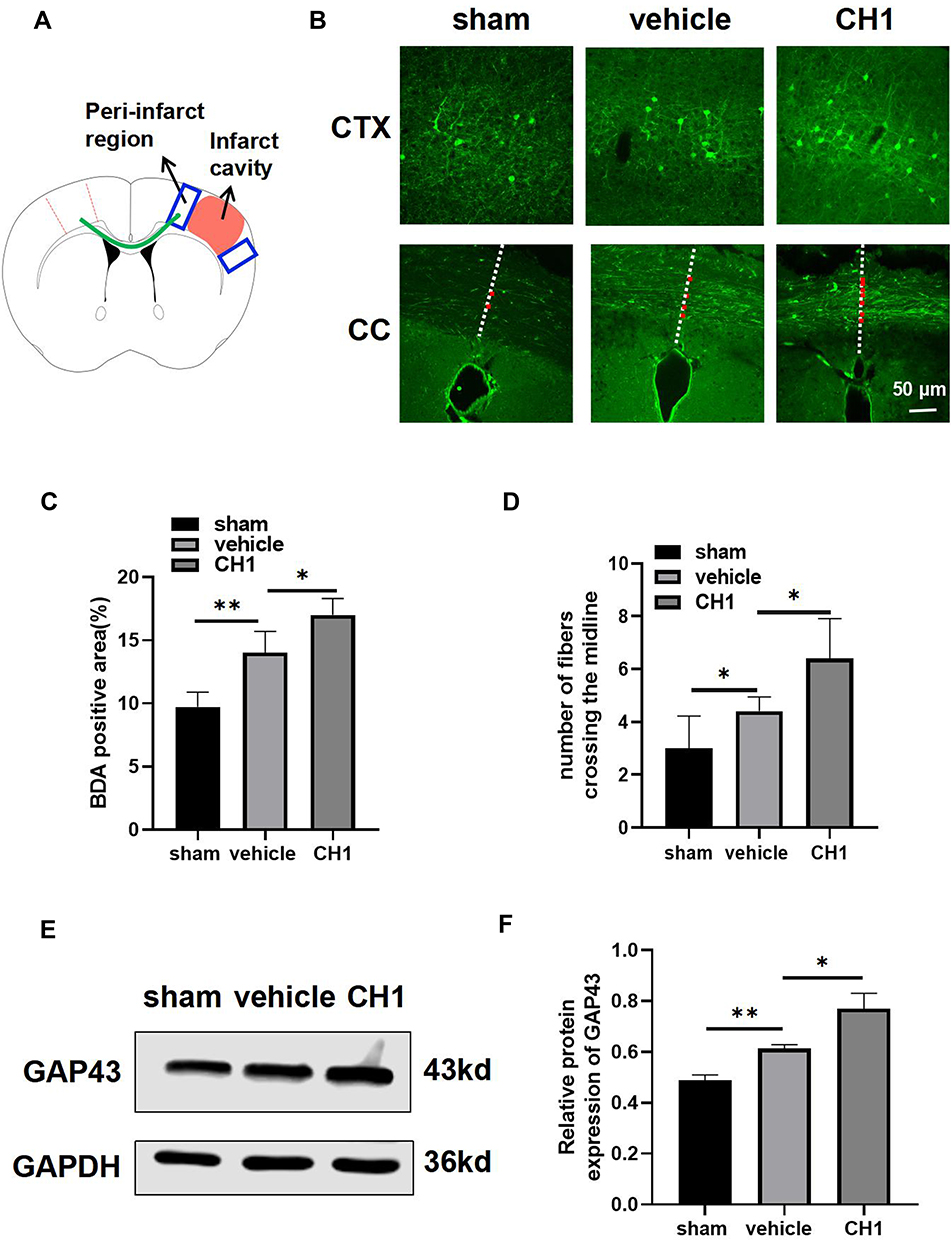 |
Figure 5 CH1 boosts poststroke axonal plasticity in dMCAO mice. (A) Schematic diagram of tract-tracing experiments. Mice received stereotaxic injections of BDA in the contralesional (left) motor cortex at 14 d after dMCAO. BDA fluorescent signal was examined at in the peri-infarct cortex (CTX) (depicted in blue box) and corpus callosum (CC) (depicted in green curve) at 28 d after dMCAO. (B) Representative photomicrographs images of streptavidin-FITC BDA+ label (green) area in the peri-infarct cortex and projections from the left hemisphere into the right hemisphere. The midline is depicted in white, dashed lines. Red dots indicate midline-crossing axons at the level CC. Scale bar = 50μm. (C) Quantification of BDA fluorescence intensity in the peri-infarct cortex. (D) Quantification of midline-crossing axons numbers per section in the CC. (E and F) Representative Western blot images (E) and quantitative data (F) of GAP43. *P< 0.05, **P< 0.01 by one-way ANOVA, n =4–5 mice per group. |
Our data also revealed that CH1 upregulated the protein expression of GAP43 at day 28 after surgery (Figure 5E and F), demonstrating that CH1 enhanced axonal regeneration at day 28 after cerebral ischemia.
These results revealed the effect of CH1 on axonal plasticity during the recovery period after dMCAO.
CH1 Promoted Axonal Regeneration by Activating the Shh/Ptch-1/Gli-1 Signaling Pathway
Our data thus far suggested that CH1 is a promising therapeutic agent that can enhance poststroke axonal plasticity and neurological recovery. We then investigated the mechanism by which CH1 increases the outgrowth of axons. Shh signaling plays a critical role in axonal outgrowth.12 The Shh signaling pathway is composed of sonic hedgehog (Shh), the transmembrane receptor proteins Ptch-1, smo and Gli family transcription factors and downstream target genes. Thus, shh and its downstream molecules Ptch-1, smo and Gli-1 were examined for their involvement in the observed protective effect of CH1.
As in our previous study, the shh signaling pathway was activated after ischemic stroke. Treatment with CH1 significantly increased the relative expression of shh, Ptch-1, smo and Gli-1 compared with the vehicle group at 28 days after stroke (Figure 6A and B), demonstrating that the shh pathway was further activated in the presence of CH1. Activation of the Shh signaling pathway triggers the transcription factor Gli-1 to enter the nucleus from the cytoplasm to control its target genes.33,34 To corroborate this result, we explored the distribution of Gli-1 with an immunofluorescence assay. As expected, after dMCAO, Gli-1 transferred to the nuclei from the cytoplasm. More Gli-1 accumulated in the nuclei in the CH1 group than in the vehicle group (Figure 6C). These results demonstrated that CH1 sensitively targets the Shh/Ptch/Gli-1 signaling pathway to exert its beneficial effect.
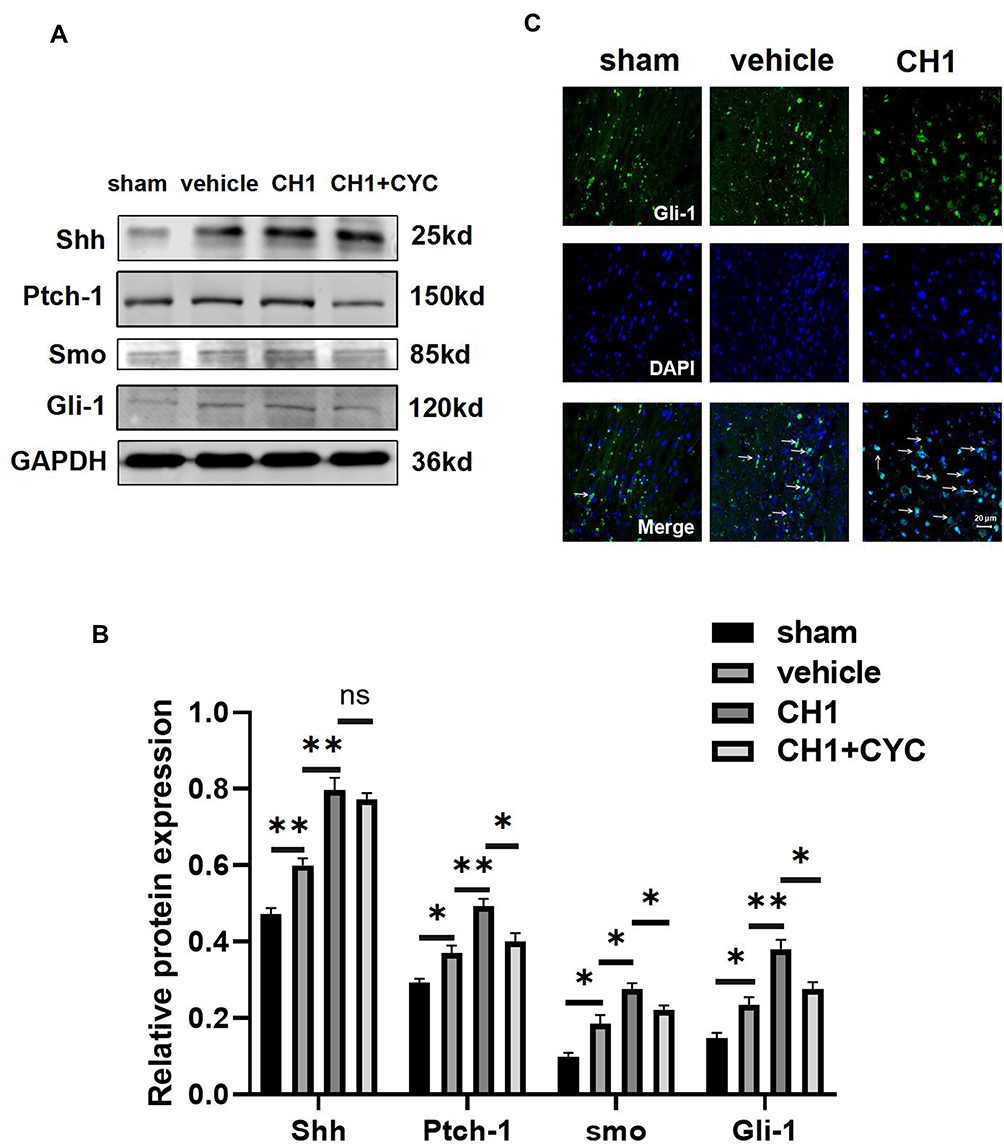 |
Figure 6 CH1 activates the shh signaling pathway after stroke. (A and B) Representative Western blot images (A) and quantitative data (B) of proteins extracted from the ischemic penumbra of brains. *P< 0.05, **P< 0.01 by one-way ANOVA, n =4 mice per group. (C) Representative image of Gli-1 location in the ischemic penumbra. The white arrows indicate translocation of Gli-1 from cytoplasm to nucleus. Scale bar = 20μm. |
Blockade of the Shh Pathway Reversed CH1-Induced Improvements in Neurological Outcomes
To confirm whether there was a causal link between CH1 and the Shh pathway, we introduced cyclopamine (CYC), a small-molecule selective inhibitor of smo, to study the role of Shh signaling in CH1-induced neurological improvements. CYC decreased the expression of Gli-1 and Ptch-1 but not shh compared with the CH1 group, demonstrating that CYC can effectively block Shh signaling in the presence of CH1 (Figure 6A). Shh was not affected by CYC because shh is upstream of smo. As displayed in Figure 7A, the latency to fall off the rotarod was significantly reduced in the CH1+CYC group compared with the CH1 group, showing that CYC reversed the improvements in neurological outcomes induced by CH1 (Figure 7A). However, there was no significant difference between the vehicle group and the CH1+CYC group. Meanwhile, the effects of CH1 on the adhesive removal test and mNSS results were also blocked by the administration of CYC (Figure 7B–D). We then measured the expression of GAP43 after the application of CYC. As expected, the effect of CH1 on GAP43 expression was reversed by the application of CYC (Figure 7E and F). These data indicated that CYC blocked the effect of CH1 on neurological outcomes in dMCAO mice.
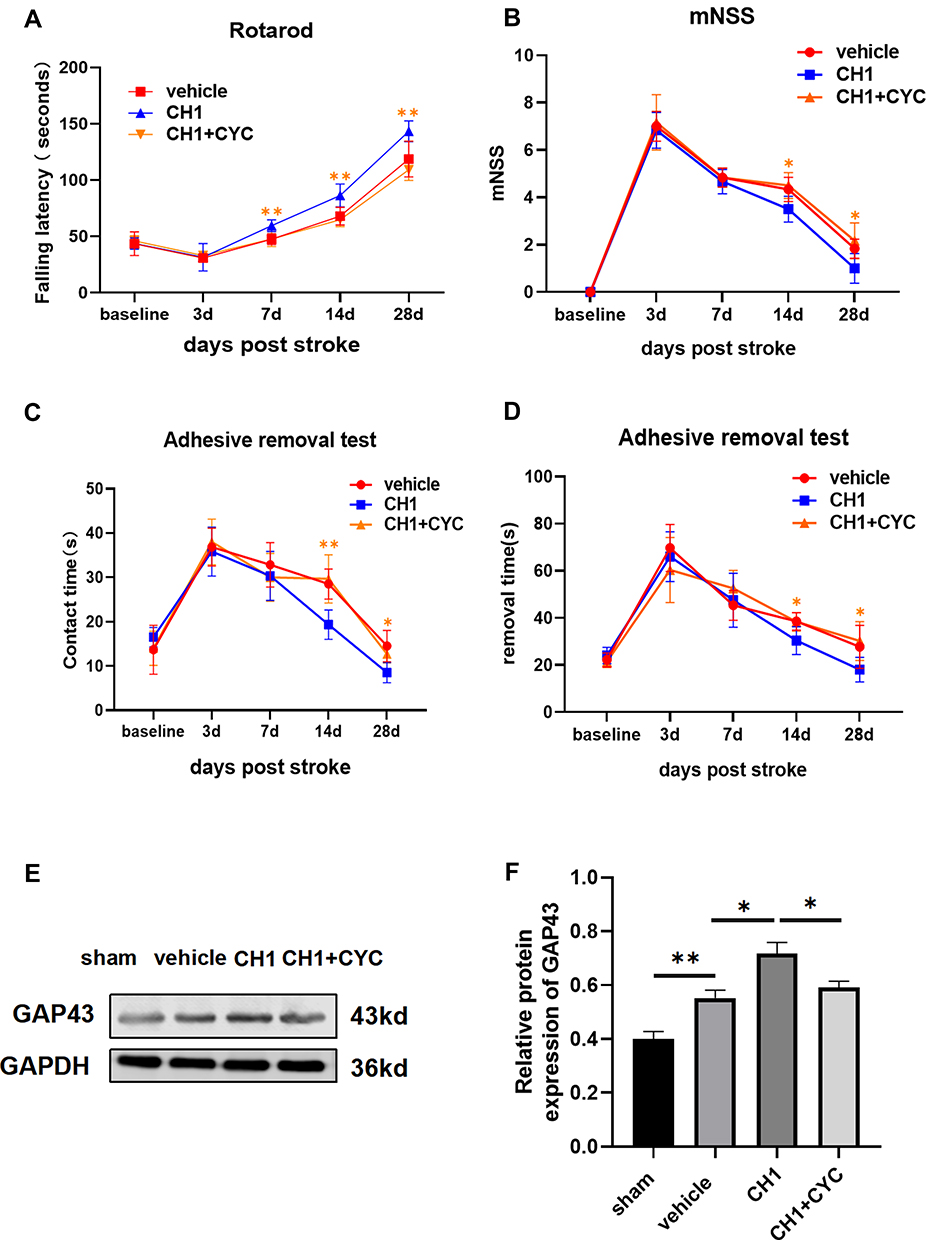 |
Figure 7 Blockade of shh signaling pathway reversed the effect of CH1 on neurological outcomes. (A and D) Sensorimotor functions were once again assessed before (baseline) and up to 28 days in the absence or presence of CYC by the rotarod (A), mNSS evaluation (B) and adhesive removal test (C and D). (E and F) Representative Western blot images (E) and quantitative data (F) of GAP43 in the absence or presence of CYC. *P< 0.05, **P< 0.01 by one-way ANOVA, n =5–6 mice per group. |
Taken together, these data provide strong evidence that CH1 plays a protective role in stroke-induced neurological improvement by activating the Shh signaling pathway.
Discussion
The present study investigated the therapeutic effect of CH1 on long-term neurological deficits and poststroke axonal plasticity. Our results demonstrated that intraperitoneal injection of CH1 mitigated stroke-induced white matter injury, facilitated axonal sprouting, and promoted long-term neurological function during the late phase of ischemic injury.
CH is a mixture containing abundant bioactive neuropeptides and free amino acids derived from the porcine brain. It mimics the effect of neurotrophic factors such as brain-derived neurotrophic factor (BDNF), glial-derived neurotrophic factor (GDNF), ciliary neurotrophic factor (CNF), and nerve growth factor (NGF) and treats stroke in multiple ways.35,36 Hence, CH is clearly a potential neuroprotective drug for the treatment of ischemic stroke in the clinic and is widely used worldwide.
In the present study, we observed that 10 mg/kg CH1 effectively improved neurological function at various times. However, the infarct volume and cortex width index in the CH1 group were comparable to those in the vehicle group at 28 days after stroke. To explore the cause of the asymmetry of neurological function recovery and neuropathological outcome at 28 days poststroke, we assessed the microstructure of the gray matter through immunostaining for NeuN (a marker of neurons), which has been considered an indicator of gray matter intensity. As expected, we still found comparable viable neuron densities between the CH1 group and the vehicle group at 28 days after stroke.
White matter intensity is known to be strongly correlated with sensorimotor functional outcomes in stroke patients.37 It is vital for communication between widely separated neurons and axons. We therefore speculated that the neurological functional improvement due to CH1 treatment did not result from gray matter restoration but from a long-term beneficial effect on WMI. White matter restoration is not accompanied by changes in the number of cortical somata but plays a vital role in signal transduction across brain regions and is critical for functional prognosis. Our data revealed a fundamental impact of CH1 on white matter integrity after ischemic stroke. We observed an increase in the SMI/MBP ratio, which indicates the exacerbation of white matter injury encompassing both axons and myelin in mice after dMCAO; this injury was mitigated by CH1 treatment.
Ischemic brain injury engages endogenous repair mechanisms that attempt to reestablish the integrity of white matter by axonal outgrowth. Axonal plasticity is a hallmark of regenerative plasticity associated with reactivation of the intrinsic neuronal growth program. Therefore, we measured axonal plasticity in two specific regions. One was the BDA-positive area in the peri-infarct CTX, reflecting the in situ endogenous axonal regeneration of denervated neurons. The other was the number of midline-crossing fibers in the CC from the contralesional (left) cortex to the denervated neurons in the peri-infarct CTX, reflecting regenerated intracortical projections. After dMCAO, we observed a robust increase in BDA-labeled fibers at CTX and CC levels in the CH1 group compared with vehicle controls, suggesting that the endogenous repair process increased after brain injury. CH1 further facilitated the in situ endogenous axonal regeneration and tract-tracing assessment of axons projecting from the contralesional motor cortex at 28 days poststroke. GAP43, expressed in terminal axonal growth cones, is a reliable marker of axonal sprouting.38 GAP43 is induced shortly after ischemic stroke, leading to axonal regeneration and remodeling, and expressed for at least 28 days in mice.39 Our results found that the expression of GAP43 is induced at 28 days after stroke and further increased with the application of CH1.
Mechanistically, CH1 treatment induced increased expression of shh, Ptch-1 and Gli-1 at 28 days after stroke, as well as translocation of Gli-1 to the nucleus from the cytoplasm, demonstrating activation of the shh pathway by CH1. Moreover, shh was not affected by CYC, as the inhibitor mainly acts upon the Smo receptor and blocks Smo and its downstream molecules. However, it is not clear why CYC inhibited the Ptch-1 receptor in this study. As previously reported, the expression of Ptch-1 is downregulated by cyclopamine.12,40,41 The involvement of the Shh signaling pathway was substantiated by the demonstration that CYC treatment blocked these beneficial effects of CH1.
Conclusion
In conclusion, our findings demonstrated that the beneficial effect of CH1 on neurological outcomes was caused not by the preservation of viable neurons but by the restoration of white matter integrity and axonal plasticity in the late stage of ischemic stroke. We have unveiled a previously unappreciated role for CH in white matter repair and brain plasticity after stroke. Our findings also emphasize the importance of white matter restoration and axonal plasticity for the treatment of ischemic stroke. More neuroprotective strategies need to emphasize the effects of treatment on endogenous long-term brain repair processes and not merely the neuroprotective effect of treatment in the hyperacute phase of ischemia.
Abbreviations
CH, cerebroprotein hydrolysate; Shh, sonic hedgehog; Ptch-1, Patched-1; Smo, smoothened; dMCAO, distal middle cerebral artery occlusion; SPF, specific-pathogen-free; CCA, common carotid artery; MCA, middle cerebral artery; CBF, cerebral blood flow; mNSS, modified neurological severity score; TTC, 2,3,5-triphenyltetrazolium chloride; CWI, cortical width index; MBP, myelin basic protein; WMI, white matter intensity; CTX, cortex; STR, striatum; CC, corpus callosum; CYC, cyclopamine.
Acknowledgments
This work was generously supported by grants from the National Natural Science Foundation of China (grant No.81974184), and the Health Commission of Hebei Province (20190061).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics-2018 update: a report from the american heart association. Circulation. 2018;137(12):e67–67e492. doi:10.1161/CIR.0000000000000558
2. Amani H, Habibey R, Shokri F, et al. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci Rep. 2019;9(1):6044. doi:10.1038/s41598-019-42633-9
3. Kastrup A, Gröschel K, Ringer TM, et al. Early disruption of the blood-brain barrier after thrombolytic therapy predicts hemorrhage in patients with acute stroke. Stroke. 2008;39(8):2385–2387. doi:10.1161/STROKEAHA.107.505420
4. Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359(13):1317–1329. doi:10.1056/NEJMoa0804656
5. Shi L, Rocha M, Leak RK, et al. A new era for stroke therapy: integrating neurovascular protection with optimal reperfusion. J Cereb Blood Flow Metab. 2018;38(12):2073–2091. doi:10.1177/0271678X18798162
6. Kinnunen KM, Greenwood R, Powell JH, et al. White matter damage and cognitive impairment after traumatic brain injury. Brain. 2011;134(Pt 2):449–463. doi:10.1093/brain/awq347
7. Wooten DW, Ortiz-Terán L, Zubcevik N, et al. Multi-modal signatures of tau pathology, neuronal fiber integrity, and functional connectivity in traumatic brain injury. J Neurotrauma. 2019;36(23):3233–3243. doi:10.1089/neu.2018.6178
8. Song D, Zhang X, Chen J, et al. Wnt canonical pathway activator TWS119 drives microglial anti-inflammatory activation and facilitates neurological recovery following experimental stroke. J Neuroinflammation. 2019;16(1):256. doi:10.1186/s12974-019-1660-8
9. Kissela B, Lindsell CJ, Kleindorfer D, et al. Clinical prediction of functional outcome after ischemic stroke: the surprising importance of periventricular white matter disease and race. Stroke. 2009;40(2):530–536. doi:10.1161/STROKEAHA.108.521906
10. Rost NS, Cougo P, Lorenzano S, et al. Diffuse microvascular dysfunction and loss of white matter integrity predict poor outcomes in patients with acute ischemic stroke. J Cereb Blood Flow Metab. 2018;38(1):75–86. doi:10.1177/0271678X17706449
11. Stokowska A, Atkins AL, Morán J, et al. Complement peptide C3a stimulates neural plasticity after experimental brain ischaemia. Brain. 2017;140(2):353–369. doi:10.1093/brain/aww314
12. He W, Cui L, Zhang C, et al. Sonic hedgehog promotes neurite outgrowth of cortical neurons under oxidative stress: involving of mitochondria and energy metabolism. Exp Cell Res. 2017;350(1):83–90. doi:10.1016/j.yexcr.2016.11.008
13. Sullivan GM, Armstrong RC. Transplanted adult neural stem cells express sonic hedgehog in vivo and suppress white matter neuroinflammation after experimental traumatic brain injury. Stem Cells Int. 2017;2017:9342534. doi:10.1155/2017/9342534
14. Zhang Y, Chopp M, Gang Zhang Z, et al. Prospective, randomized, blinded, and placebo-controlled study of Cerebrolysin dose-response effects on long-term functional outcomes in a rat model of mild traumatic brain injury. J Neurosurg. 2018;129(5):1295–1304. doi:10.3171/2017.6.JNS171007
15. Zhang C, Chopp M, Cui Y, et al. Cerebrolysin enhances neurogenesis in the ischemic brain and improves functional outcome after stroke. J Neurosci Res. 2010;88(15):3275–3281. doi:10.1002/jnr.22495
16. Yang Y, Zhang Y, Wang Z, et al. Attenuation of acute phase injury in rat intracranial hemorrhage by cerebrolysin that inhibits brain edema and inflammatory response. Neurochem Res. 2016;41(4):748–757. doi:10.1007/s11064-015-1745-4
17. Heiss W-D, Brainin M, Bornstein NM, Tuomilehto J, Hong Z. Cerebrolysin in patients with acute ischemic stroke in Asia: results of a double-blind, placebo-controlled randomized trial. Stroke. 2012;43(3):630–636. doi:10.1161/STROKEAHA.111.628537
18. Ziganshina LE, Abakumova T, Hoyle CH. Cerebrolysin for acute ischaemic stroke. Cochrane Database Syst Rev. 2020;7(7):CD007026. doi:10.1002/14651858.CD007026.pub6
19. Khalili H, Niakan A, Ghaffarpasand F. Effects of cerebrolysin on functional recovery in patients with severe disability after traumatic brain injury: A historical cohort study. Clin Neurol Neurosurg. 2017;152:34–38. doi:10.1016/j.clineuro.2016.11.011
20. Chen -C-C, Wei S-T, Tsaia S-C, Chen -X-X, Cho D-Y. Cerebrolysin enhances cognitive recovery of mild traumatic brain injury patients: double-blind, placebo-controlled, randomized study. British J Neurosurgery. 2013;27(6):803–807. doi:10.3109/02688697.2013.793287
21. Alvarez XA, Cacabelos R, Sampedro C, et al. Efficacy and safety of Cerebrolysin in moderate to moderately severe Alzheimer’s disease: results of a randomized, double-blind, controlled trial investigating three dosages of Cerebrolysin. Eur J Neurol. 2011;18(1):59–68. doi:10.1111/j.1468-1331.2010.03092.x
22. Cui L, Murikinati SR, Wang D, Zhang X, Duan W-M, Zhao L-R. Reestablishing neuronal networks in the aged brain by stem cell factor and granulocyte-colony stimulating factor in a mouse model of chronic stroke. PLoS One. 2013;8(6):e64684. doi:10.1371/journal.pone.0064684
23. Gao C, Qian Y, Huang J, et al. A three-day consecutive fingolimod administration improves neurological functions and modulates multiple immune responses of CCI mice. Mol Neurobiol. 2017;54(10):8348–8360. doi:10.1007/s12035-016-0318-0
24. Freret T, Bouet V, Leconte C, et al. Behavioral deficits after distal focal cerebral ischemia in mice: usefulness of adhesive removal test. Behav Neurosci. 2009;123(1):224–230. doi:10.1037/a0014157
25. Caballero-Garrido E, Pena-Philippides JC, Lordkipanidze T, et al. In vivo inhibition of mir-155 promotes recovery after experimental mouse stroke. J Neurosci. 2015;35(36):12446–12464. doi:10.1523/JNEUROSCI.1641-15.2015
26. Shi Y, Jiang X, Zhang L, et al. Endothelium-targeted overexpression of heat shock protein 27 ameliorates blood–brain barrier disruption after ischemic brain injury. Proc Natl Acad Sci U S A. 2017;114(7):E1243–1243E1252. doi:10.1073/pnas.1621174114
27. Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke. 1986;17(6):1304–1308.
28. Wang Y, Zhao Z, Rege SV, et al. 3K3A-activated protein C stimulates postischemic neuronal repair by human neural stem cells in mice. Nat Med. 2016;22(9):1050–1055. doi:10.1038/nm.4154
29. Xia Y, Pu H, Leak RK, et al. Tissue plasminogen activator promotes white matter integrity and functional recovery in a murine model of traumatic brain injury. Proc Natl Acad Sci U S A. 2018;115(39):E9230–9230E9238. doi:10.1073/pnas.1810693115
30. Wang R, Pu H, Ye Q, et al. Transforming growth factor beta-activated kinase 1-dependent microglial and macrophage responses aggravate long-term outcomes after ischemic stroke. Stroke. 2020;51(3):975–985. doi:10.1161/STROKEAHA.119.028398
31. Pu H, Shi Y, Zhang L, et al. Protease-independent action of tissue plasminogen activator in brain plasticity and neurological recovery after ischemic stroke. Proc Natl Acad Sci U S A. 2019;116(18):9115–9124. doi:10.1073/pnas.1821979116
32. Zhao L, Biesbroek JM, Shi L, et al. Strategic infarct location for post-stroke cognitive impairment: A multivariate lesion-symptom mapping study. J Cereb Blood Flow Metab. 2018;38(8):1299–1311. doi:10.1177/0271678X17728162
33. Breunig JJ, Sarkisian MR, Arellano JI, et al. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci U S A. 2008;105(35):13127–13132. doi:10.1073/pnas.0804558105
34. Cheng W, Yu P, Wang L, et al. Sonic hedgehog signaling mediates resveratrol to increase proliferation of neural stem cells after oxygen-glucose deprivation/reoxygenation injury in vitro. Cell Physiol Biochem. 2015;35(5):2019–2032. doi:10.1159/000374009
35. Zhào H, Wang R, Zhang Y, Liu Y, Huang Y. Neuroprotective effects of troxerutin and cerebroprotein hydrolysate injection on the neurovascular unit in a rat model of Middle cerebral artery occlusion. Int J Neurosci. 2020;1–15. doi:10.1080/00207454.2020.1738431
36. Zhang L, Chopp M, Wang C, et al. Prospective, double blinded, comparative assessment of the pharmacological activity of Cerebrolysin and distinct peptide preparations for the treatment of embolic stroke. J Neurol Sci. 2019;398:22–26. doi:10.1016/j.jns.2019.01.017
37. Oksala NK, Oksala A, Pohjasvaara T, et al. Age related white matter changes predict stroke death in long term follow-up. J Neurol Neurosurg Psychiatry. 2009;80(7):762–766. doi:10.1136/jnnp.2008.154104
38. Gao X, Zhang X, Cui L, et al. Ginsenoside rb1 promotes motor functional recovery and axonal regeneration in post-stroke mice through cAMP/PKA/CREB signaling pathway. Brain Res Bull. 2020;154:51–60. doi:10.1016/j.brainresbull.2019.10.006
39. Carmichael ST, Archibeque I, Luke L, Nolan T, Momiy J, Li S. Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol. 2005;193(2):291–311.
40. Ji H, Miao J, Zhang X, et al. Inhibition of sonic hedgehog signaling aggravates brain damage associated with the down-regulation of Gli1, Ptch1 and SOD1 expression in acute ischemic stroke. Neurosci Lett. 2012;506(1):1–6. doi:10.1016/j.neulet.2011.11.027
41. Yu P, Wang L, Tang F, et al. Resveratrol pretreatment decreases ischemic injury and improves neurological function via sonic hedgehog signaling after stroke in rats. Mol Neurobiol. 2017;54(1):212–226. doi:10.1007/s12035-015-9639-7
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.