Back to Journals » OncoTargets and Therapy » Volume 14
A Novel CD73 Inhibitor SHR170008 Suppresses Adenosine in Tumor and Enhances Anti-Tumor Activity with PD-1 Blockade in a Mouse Model of Breast Cancer
Authors Liu S ![]() , Li D, Liu J, Wang H, Horecny I, Shen R, Zhang R, Wu H, Hu Q, Zhao P, Zhang F, Yan Y, Feng J, Zhuang L, Li J, Zhang L
, Li D, Liu J, Wang H, Horecny I, Shen R, Zhang R, Wu H, Hu Q, Zhao P, Zhang F, Yan Y, Feng J, Zhuang L, Li J, Zhang L ![]() , Tao W
, Tao W
Received 24 June 2021
Accepted for publication 10 August 2021
Published 24 August 2021 Volume 2021:14 Pages 4561—4574
DOI https://doi.org/10.2147/OTT.S326178
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Suxing Liu,1 Di Li,1 Jian Liu,2 Huiyun Wang,1 Ivana Horecny,1 Ru Shen,1 Rumin Zhang,1 Heping Wu,2 Qiyue Hu,3 Peng Zhao,2 Fengqi Zhang,2 Yinfa Yan,2 Jun Feng,4 Linghang Zhuang,2 Jing Li,1 Lianshan Zhang,5 Weikang Tao5
1Department of Biology, Eternity Bioscience Inc., Cranbury, NJ, 08512, USA; 2Department of Chemistry, Eternity Bioscience Inc., Cranbury, NJ, 08512, USA; 3Department of Molecular Modeling, Shanghai Hengrui Pharmaceutical Co. Ltd., Shanghai, 200245, People’s Republic of China; 4Department of Process Chemistry, Shanghai Hengrui Pharmaceutical Co. Ltd., Shanghai, 200245, People’s Republic of China; 5R&D Center, Shanghai Hengrui Pharmaceutical Co. Ltd., Shanghai, 200245, People’s Republic of China
Correspondence: Suxing Liu
Department of Biology, Eternity Bioscience Inc, 6 Cedarbrook Drive, Cranbury, NJ, 08512, USA
Email [email protected]
Introduction: CD73 and adenosine support growth-promoting neovascularization, metastasis, and survival in cells, and promote anti-PD-1 mAb therapy-induced immune escape. Consequently, developing a CD73 inhibitor as monotherapy and a potential beneficial combination partner with immune-checkpoint inhibitors needs investigation.
Methods: CD73 inhibitors were evaluated in vitro with soluble and membrane-bound CD73 enzymes, as well as its PD biomarker responses in human peripheral blood mononuclear cells (PBMC) by flow cytometry and ELISA. The binding modes of the molecules were analyzed via molecular modeling. The anti-tumor activity and synergistic effect of SHR170008 in combination with anti-PD-1 mAb were evaluated in a syngeneic mouse breast cancer model.
Results: SHR170008 was discovered during the initial structural modifications on the link between the ribose and the α-phosphate of AMPCP, which significantly improved the stability of the compound confirmed by the metabolite identification study. Further modifications on the adenine base of AMPCP improved the potency due to forming stronger interactions with CD73 protein. It exhibited potent inhibitory activities on soluble and endogenous membrane-bound CD73 enzymes, and induced IFNγ production, reversed AMP-suppressed CD25+ and CD8+/CD25+ expression, and enhanced granzyme B production on CD8+ T cells in human PBMC. SHR170008 showed dose-dependent anti-tumor efficacy with suppression of adenosine in the tumors in EMT6 mouse breast tumor model. The increase of adenosine in tumor tissue by anti-PD-1 mAb alone was suppressed by SHR170008 in the combination groups. Simultaneous inhibition of CD73 and PD-1 neutralization synergistically enhanced antitumor immunity and biomarkers in response, and exposures of SHR170008 were correlated with the efficacy readouts.
Conclusion: Our findings suggest that CD73 may serve as an immune checkpoint by generating adenosine, which suppresses the antitumor activity of anti-PD-1 mAb, and inhibition of CD73 may be a potential beneficial combination partner with immune-checkpoint inhibitors to improve their therapeutic outcomes in general.
Keywords: small-molecule inhibitor of CD73, adenosine, anti-PD-1 mAb, checkpoint blockade, immunotherapy, combination therapy
Summary
Here we described SHR170008, a novel and potent small-molecule inhibitor of CD73. An outstanding feature of SHR170008 different from AMPCP is that the bisphosphonic acid was replaced by methylenephosphonic acid, which significantly improved the stability of the compound confirmed by the metabolite identification study. Furthermore, SHR170008 not only could rescue the inhibitory effect of adenosine derived from AMP in the activation and proliferation of CD8+ T cells and granzyme B production in human primary immune cells but also exhibited dose-dependent tumor growth inhibition in single agent and significantly enhanced the efficacy in combination with anti-PD-1 mAb in EMT6 mouse breast tumor model. PD-1 blockade resulted in enhanced adenosine levels in tumor as compared with the control group and decreased adenosine levels in combination with SHR170008. Our findings extended the understanding of adenosine generated by CD73 in suppressing antitumor immunity, which contributed to the resistance of PD-1 checkpoint inhibition. Results from further clinical evaluation of this specific mechanism for immunotherapy of cancer, especially in combination with monoclonal antibody therapies targeting immune checkpoints are highly anticipated.
Background
T cell-mediated anti-tumor immune responses are pivotal for effective ablation of primary tumor lesions and for protection against metastases.1 High level of extracellular adenosine in tumor microenvironment (TME) has been described as a protective “halo”, which in turn suppresses antigen-presenting cells, inhibits T cell cytotoxicity and cytokine production, leading to evasion of antitumor immunity and cancer development and progression.2,3 Cluster of differentiation 73 (CD73) enzyme catalyzes the hydrolysis of extracellular AMP into immunosuppressive adenosine, which signals through A1, A2a, A2b and A3 adenosine receptors. Activated A2a and A2b receptors can increase intracellular cAMP levels, which suppress immune cell function, and trigger other signaling pathways, such as PI3K/Akt/mTOR and MAPK/ERK pathways.4 CD73 is expressed on a variety of tumor infiltrating leukocytes (TIL) and overexpressed in human cancer cells because of tissue hypoxia, activation of oncogenes such as KRAS, and epithelial-to-mesenchymal transition.5–8 Expression of CD73 is associated with poor prognosis in many cancer types.4,9,10 Within the TME, upregulation of CD73 expression on cell-surface blocks anti-tumor surveillance by producing extracellular adenosine, leading to tumor immune-escape. Consistent with roles of adenosine, host CD73 promotes tumor growth in a T cell-dependent manner.11 Accordingly, CD73-deficient mice have increased antitumor immunity and significantly suppressed the growth of ovalbumin-expressing MC38 colon cancer, EG7 lymphoma, AT-3 mammary tumors and B16F10 melanoma.12,13 The antitumor effects of CD73 deficiency on primary tumors were dependent on CD8+ T cells and increased antigen-specific IFNγ production.
The emergence of monoclonal antibody (mAb) therapies targeting immune checkpoints, such as programmed cell-death protein 1 (PD-1) or its ligand PD-L1, cytotoxic T-lymphocyte-associated protein 4 (CTLA4), has transformed the core cancer treatments, as they demonstrate good efficacies and less toxicity than conventional chemotherapy and targeted therapies. However, despite all the clinical successes, only a subset of patients for most cancer types effectively respond to these therapies.14 Such less than desirable efficacy is a major clinical problem and one of the most significant challenges in oncology. A variety of tumor-extrinsic factors have been associated with resistance, including increased adenosine production and the expansion of immunosuppressive immune cell population within TME. Combination therapies designed to block adenosine signaling pathways may help to improve the clinical responses.
CD73 expression on tumor cells reduced the immune response evoked by anti-PD-1 mAb therapy.15 In addition, 94 patients who were responders and non-responders to immune checkpoint therapy from five cancer types were evaluated by mass cytometry and single-cell RNA sequencing.16 The results suggested that high expression of CD73 in immune cells persists after anti-PD-1 mAb treatment in glioblastoma multiforme. CD73-deficient mice improved survival in a murine model of glioblastoma multiforme treated with anti-PD-1 mAb and anti-CTLA4 mAb. Anti-CD73 antibodies significantly enhanced the activity of both anti-CTLA4 mAb and anti-PD-1 mAb in preclinical animal models.17,18 Therefore, CD73 expression may constitute a potential biomarker for the efficacy of anti-PD-1 mAb in patients with cancer, and the efficacy of anti-PD-1 mAb can be significantly enhanced by CD73 inhibitors.
There has been a high level of interest in CD73 as a novel immunotherapy target for cancer, including monoclonal antibodies and small-molecule inhibitors against CD73.17,19–23 Small-molecule inhibitors may have advantages versus antibody approaches: for example, the ability of penetration into solid tumor, greater exposure within the TME, and the flexibility of intermittent dosing regarding safety profile, to name a few. Most reports of small-molecule inhibitors of the enzyme were focused on medicinal chemistry efforts and described structure–activity relationships.
Here, we report the discovery of a novel and potent small-molecule CD73 inhibitor SHR170008 in vitro and in vivo. We further investigated whether blockade of CD73 enzyme by SHR170008 could enhance the efficacy of anti-PD-1 mAb. Because tumoral CD73 suppresses antitumor immunity evoked by anti-PD-1 mAb therapy, we hypothesized that CD73 inhibitor and anti-PD-1 mAb may represent a potentially beneficial pairing for dual pathway blockade in cancer therapy.
Materials and Methods
Cell Lines, Antibodies, and Chemicals
Human epithelial melanoma A-375 (CRL-1619) cells, and mouse breast carcinoma EMT6 (CRL-2755) cells were purchased from ATCC. Human peripheral blood mononuclear cells (PBMC) were obtained from STEMCELL Technologies (70025.1). Human soluble His-tag 5ʹ-Nucleotidase/CD73 (Trp27-Lys547) was purchased from R&D Systems (5795-EN). Cytidine 5ʹ-monophosphate (CMP, C1131-1G), adenosine 5ʹ-monophosphate (AMP, A1752) and adenosine 5ʹ-(α, β-methylene) diphosphate (AMPCP, M3763) was purchased from Sigma Aldrich. Malachite Green Phosphate Detection Kit was purchased from R&D Systems (DY996).
Biochemical Assay of Human Soluble CD73 Enzyme
In this assay, 0.34 nM human soluble CD73 enzyme was used in a phosphate-free assay buffer (20 mM HEPES, 137 mM NaCl, 0.001% Tween 20). A serial dilution of SHR170008 or AMPCP was incubated with the CD73 enzyme at 37°C for 20 hours before adding 45 µM CMP (an AMP surrogate substrate). The reactions were incubated at 37°C for 15 minutes, and then phosphate concentrations were measured by Malachite Green Phosphate Detection Kit per manufacturer’s protocol. The positive and negative controls were set up with and without the CD73 enzyme, respectively. The robustness of the biochemical assay was assessed by Z-factor using the equation below.24
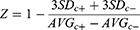
Where AVGc+ and AVGc- are denoted for the averages of positive control signal and negative control signal, respectively. The standard deviations of the signals are denoted as SDc+ and SDc-, respectively.
Data were calculated using signal of the positive control as 0% inhibition and that of the negative control as 100% inhibition. IC50 values were calculated using non-linear regression analysis of GraphPad Prism (RRID:SCR_002798) in all assays.
Enzymatic Activity of Endogenous Cellular Membrane-Bound CD73
A-375 cells and EMT-6 cells were seeded at a density of 2500 and 5000 cells per well in 96 well plates, respectively. Following the wash with phosphate-free assay buffer (20 mM HEPES, 137 mM NaCl, 5.4 mM KCl, 1.3 mM CaCl2, 4.2 mM NaHCO3, and 1 mg/mL Glucose), cells were incubated with a series dilution of SHR170008 and 50 µM CMP for 4 hours at 37°C in duplicates. Phosphate concentrations resulting from CMP hydrolysis were measured using Malachite Green Phosphate Detection Kit per manufacturer’s protocol. Independent experiments were repeated with four batches of SHR170008, respectively.
Molecular Modeling
Binding mode studies for SHR170008 were done using a laptop computer with an Intel i7-7700HQ, 2.8 GHz CPU and 16GB RAM running the Microsoft Windows 10 professional operating system. The software package of Molecular Operating Environment (MOE), from Chemical Computing Group Inc., was used for the modeling study. The structures were downloaded from Protein Data Bank (PDB) and prepared using Structure Preparation within MOE, including adjusting hydrogens and lone pairs using Protonate3D.25 The crystal structure of CD73 in complex with AMPCP (accession code: 4H2I) and small-molecule antagonist (accession code: 6YE2) were downloaded from PDB.26,27 The co-crystal ligand (accession code: 6YE2) was modified to SHR170008. By selecting SHR170008 and the surrounding residues within 4.5Å, iterative energy minimizations with gradually increased tether deviations were performed to relax the clash between the ligand and protein while forming multiple interactions between ligand and protein. Sequence and structures alignments were performed using MOE Align/Superpose function.
Activation and Staining of PBMCs
Human PBMCs (StemCell, 70025.1) in TexMACS medium (Miltenyi Biotec, 130-097-196) were incubated with 100 μM AMP and 1 μM SHR170008 for 2 hours and then were activated by CD3/CD28 dynabeads (Fisher Scientific, 11161D) for 4 days. After 4 days’ incubation, cells were washed and stained using Invitrogen FoxP3 Staining Kit (Thermo Fisher, 00-5521-00) following manufacturer’s protocol. Antibodies used for staining cells were 7AAD (5599255), CD8_APC-H7 (560273), CD25_BV786 (563700), CD73_BV421 (562431) and GranzymeB_FITC (561998) from BD Biosciences, and FoxP3_PE (12-4777-42) from eBioscience.
CD8 Cell Proliferation Assay
Human PBMCs were stained with 5 µM CFSE (BD Biosciences, 565082) for 15 minutes at 37°C, washed twice with PBS, and then incubated with 100 μM AMP and 1 μM SHR170008 for 2 hours. The cells were then stimulated with CD3/CD28 dynabeads and 30 unit/mL human IL-2, incubated for 4 days. After stimulation, cells were collected and stained with CD8-APC-H7 antibody for 20 minutes at room temperature. Then, CSFE positive cells in CD8 cells were analyzed with flow cytometry. The independent experiments were repeated at least twice.
Cell Isolation from Tumor Tissues
Tumor tissues collected from mice at the end of in vivo study were processed on gentleMACS Dissociator (Miltenyi Biotec, 130-096-427) using tumor isolation programs twice, between which was 40 minutes of incubation at 37°C with continuous rotation. Then, cell suspension was applied to 30 µm MACS SmartStrainers (Miltenyi Biotec, 130-098-458) and washed with medium. Collected cells were centrifuged and used for staining as described above. Antibodies CD45-VioGreen (130-123-900) and CD73-APC-Vio770 (130-102-329) were purchased from Miltenyi Biotec.
Flow Cytometric Analysis
For each sample, 20,000 total events were acquired on a Guava EasyCyte instrument. The cells stained with a single antibody were used as controls and compensation was automatically determined by the GuavaSoft software. Data analysis was performed using the FlowJo software (RRID:SCR_008520). Singlets and live cells were first gated out based on the sizes and 7AAD staining results and then gated with CD25+ and CD8+ cells, respectively.
IFNγ Production Assay Using Human PBMC
Human PBMCs (StemCell) in TexMACS medium were incubated with 50 µM AMP and serial diluted SHR170008 in duplicates for 2 hours and then CD3/CD28 dynabeads were added. After the incubation for 72 hours, IFNγ level in supernatant was determined using IFNγ HTRF kit (Cisbio, 62HIFNGPET) following manufacturer’s protocol. The biological independent experiments were repeated nine times.
In vivo Efficacy Study
The in-life portion of the study was carried out at Cephrim Bioscience, Inc. (Woburn, MA). Female BALB/c-inbred mice (RRID: IMSR_ORNL:BALB/cRl) were purchased from Charles River Lab. Upon their arrival, they were kept in SPF environment all the time. Briefly, they were housed on irradiated corncob bedding (ScottPharma) in Innovive caging system on a 12-hour light-dark cycle at 68–74°F and 30–70% humidity. They will be continually fed water and regular rodent diet. Animal handling and procedures were conducted under the protocols and guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of Cephrim Biosciences, Inc.
On the day of inoculation (Day 0), EMT6 mouse breast tumor cells (5 × 105 cells) were injected subcutaneously into the right flank of syngeneic female BALB/c inbred mice. Body weights and tumor volumes were measured twice a week throughout the study, and the measurements were blind to group assignment and outcome assessment. The tumors were measured by a caliper in three dimensions in millimeter (mm), and its volume was then calculated according to the following formula:
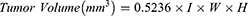
where l = length, W = width, H = height.
When tumors reached approximately 75 mm3 on day 5, mice were randomly assigned to one of eight groups (n = 8 per group). Where indicated, mice were treated with SHR170008 in the vehicle (1.5% DMSO and 98.5% HBSS) daily at the indicated doses, 5 mg/kg anti-mouse-PD-1 mAb (clone: RMP1-14, BioXCell, BP0146) or rat 2A3 isotype control (BioXCell, BE0089, RRID:AB_1107769) were injected once every 3 days for 4 times. The mice in the control group were dosed with vehicle and/or rat 2A3 isotype with the same schedule as SHR170008 and/or anti-mouse-PD-1 mAb. Both were delivered intraperitoneally.
The plasma and tumor tissue samples were collected with three, three and two animals per group at 2-, 6- and 24-hours post dosing at the end of the study, respectively. Tumor tissues were minced into small pieces, homogenized in 1 mL cold DPBS (GIBCO, 14190144) with Halt protease inhibitor cocktail (Thermo Fisher Scientific, 78430) for 30 seconds at 24,000 rpm twice using PRO25D Homogenizer (ProScientific, 01-25100), and then transferred to fresh Eppendorf tubes, spun at the highest speed (17,000 × g) and 4°C for 10 minutes. The supernatants were used for PK analyses of SHR170008 by LC/MS.
Statistical Analyses
The significance of differences between groups was determined by one-way ANOVA (GraphPad Prism), *p < 0.05, **p < 0.01, *** p < 0.001, ****p < 0.0001.
SHR170008 Preparation
SHR170008 ((((2R,3S,4R,5R)-5-(6-chloro-4-(2ʹ,3ʹ-dihydrospiro[azetidine-3,1ʹ-inden]-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)methyl) phosphonic acid was prepared in Eternity Bioscience Inc. according to the following processes. Step 1: 2ʹ,3ʹ-dihydrospirao[azetidine-3,1ʹ-indene] was reacted with (2R,3R,4R,5R)-2-(acetoxymethyl)-5-(4,6-dichloro-1H-pyrazolo[3,4-d]pyrimidin-1-yl)tetrahydrofuran-3,4-diyl diacetate and Et3N in methanol at room temperature to yield ((((2R,3S,4R,5R)-5-(6-chloro-4-(2ʹ,3ʹ-dihydrospiro[azetidine-3,1ʹ-inden]-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxy tetrahydrofuran-2-yl)methoxy)methyl)phosphonic acid (62%). Step 2: The reaction of (2R,3R,4S,5R)-2-(6-chloro-4-(2ʹ,3ʹ-dihydrospiro[azetidine-3,1ʹ-inden]-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol, 2,2-dimethoxypropane and toluenesulfonic acid in acetone at room temperature yielded ((3aR,4R,6R,6aR)-6-(6-chloro-4-(2ʹ,3ʹ-dihydrospiro[azetidine-3,1ʹ-inden]-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3] dioxol-4-yl) methanol (50%). Step 3: ((3aR,4R,6R,6aR)-6-(6-chloro-4-(2ʹ,3ʹ-dihydrospiro[azetidine-3,1ʹ-inden]-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl) methanol was reacted with Mg(OtBu)2 in dimethyl formamide at room temperature, followed by reacting with (diethoxyphosphoryl) methyl nosylate at 40°C to yield diethyl((((3aR,4R,6R,6aR)-6-(6-chloro-4-(2ʹ,3ʹ-dihydrospiro[azetidine-3,1ʹ-inden]-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methoxy)methyl)phosphonate (95%). Step 4: Trimethylsilyl bromide was reacted with diethyl((((3aR,4R,6R,6aR)-6-(6-chloro-4-(2ʹ,3ʹ-dihydrospiro[azetidine-3,1ʹ-inden]-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methoxy) methyl)phosphonate in dichloromethane at 0°C to afford ((((2R,3S,4R,5R)-5-(6-chloro-4-(2ʹ,3ʹ-dihydrospiro[azetidine-3,1ʹ-inden]-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)methyl) phosphonic acid (50%).
1H NMR (500 MHz, Methanol-d4) δ (ppm) 8.05 (s, 1H), 7.58–7.47 (m, 1H), 7.35–7.21 (m, 3H), 6.24 (d, J = 4.0 Hz, 1H), 4.73–4.57 (m, 3H), 4.52–4.40 (m, 3H), 4.22–4.16 (m, 1H), 3.84 (dd, J = 10.9, 3.7 Hz, 1H), 3.76–3.72 (m, 3H), 2.99 (t, J = 7.0 Hz, 2H), 2.60–2.48 (m, 2H). 13C NMR (126 MHz, Methanol-d4) δ (ppm) 159.56, 158.03, 156.47, 147.10, 144.74, 135.15, 128.90, 128.33, 125.52, 123.51, 123.29, 100.41, 90.12, 84.38, 75.12, 72.73, 68.30, 67.01, 65.27, 64.19, 39.74, 31.23. HRMS: calculated for C22H25ClN5O7P (M+H)+ 538.1258, found 538.1260. HPLC purity: 99.55%.
Results
Discovery of a Novel and Potent Small-Molecule Inhibitor of CD73 Enzyme SHR170008
CD73 is a glycosylphosphatidylinositol (GPI)–anchored cell surface ecto-5ʹ-nucleotidase that regulates adenosine signaling by dephosphorylating extracellular AMP to adenosine.6 The enzyme is also present in a soluble active form in serum through cleavage from its GPI linker on cell membranes. AMPCP is known as a competitive inhibitor of CD73 that binds to the active site of the enzyme.26 SHR170008 was discovered during the initial structural modifications on the link between the ribose and the α-phosphate of AMPCP to improve the metabolic stability in rat plasma. Further modifications on the adenine base of AMPCP improved the potency (WO2020047082A1). The chemical structures of AMPCP and SHR170008 are shown in Figure 1A. The NMR spectra, the HR-MS characterization and HPLC spectra of SHR170008 are presented in Supplementary Figure S1 and Supplementary Methods.
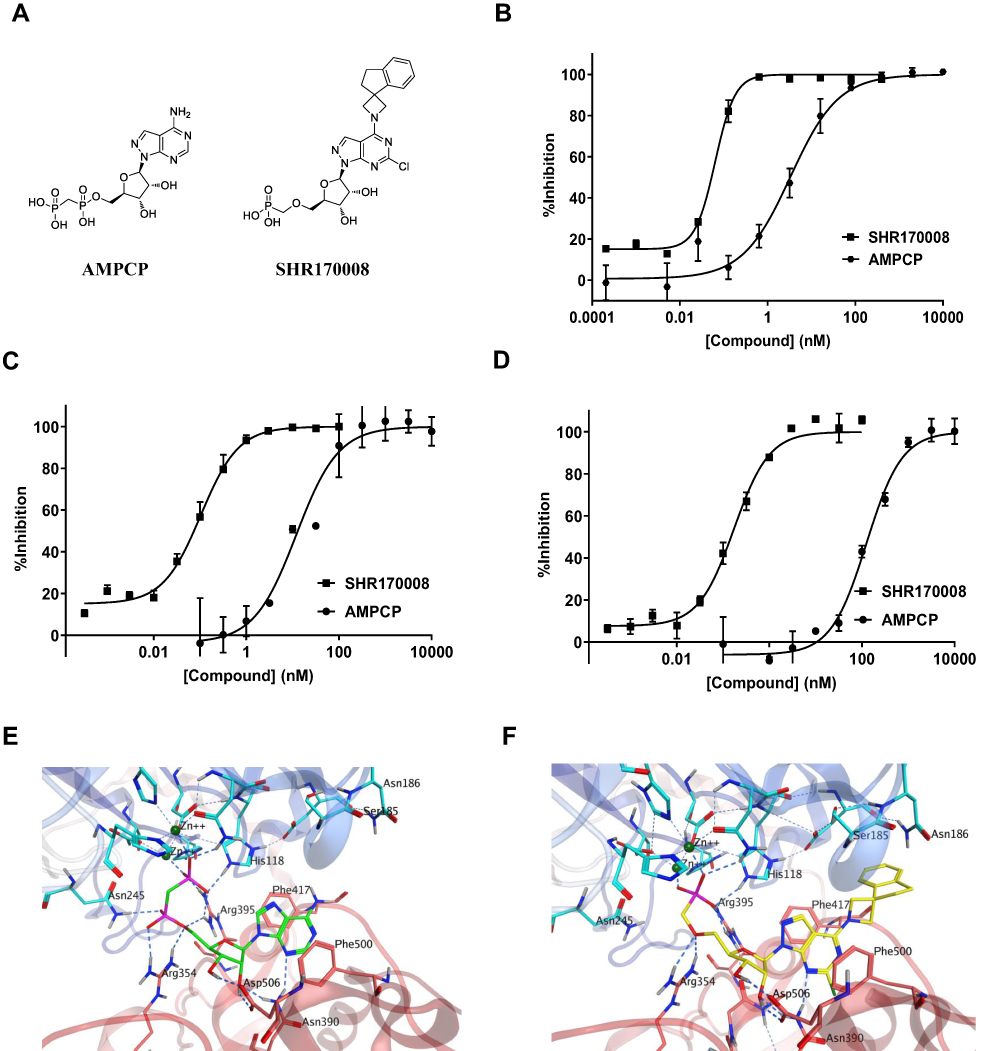 |
Figure 1 Identification of a novel and potent small-molecule inhibitor of CD73 enzyme. (A) Chemical structures of AMPCP and SHR170008. (B–D) Enzymatic inhibition activities of SHR170008 and AMPCP. Representative dose–response curves for SHR170008 and AMPCP against human soluble CD73 enzyme (B), and endogenous cellular surface CD73 in human melanoma cell line A375 (C) and in mouse breast tumor cell line EMT6 (D). Data show mean ± STD. Each data point was performed in duplicate and biological independent experiments were repeated at least three times, respectively. Data were fitted by non-linear regression analysis of GraphPad Prism in all assays. (E and F) Models were generated based on the crystal structures of CD73 (PDB accession code: 4H2I) where AMPCP was in green (E) and (PDB accession code: 6YE2) where SHR170008 in yellow (F), respectively. In both (E) and (F), the CD73 proteins were shown with color ribbons: N-terminal in blue and C-terminal in red. Key residues making contacts with the ligands were labelled accordingly. Zinc ions were colored in deep green. The hydrogen bonds are labeled as dashed lines. |
As expected, the results from the stability study in rat plasma suggested that SHR170008 with the methylenephosphonic acid was metabolically stable (Supplementary Table S1 and Supplementary Methods). The metabolite identification studies of SHR170008 in mouse, rat, dog, monkey, and human liver microsome (in vitro) and in mouse and rat plasma (in vivo) indicated that the parent molecule was intact, and no metabolites were detectable, confirming the notion above (Supplementary Tables S2–S3, Figure S2 and Supplementary Methods).
SHR170008 exhibited excellent potency against human CD73 soluble enzyme (IC50 = 0.050 ± 0.004 nM, n = 3) as compared to AMPCP (IC50 = 3.8 ± 0.8 nM, n = 3). The representative IC50 curves of SHR170008 and AMPCP are shown in Figure 1B. We initially used AMP as a substrate in the CD73 biochemical assay. The Km of AMP was 2.7 μM, consistent with what was reported (Km of AMP = 2.53 μM).29 It was difficult to keep AMP concentration equal to low Km of AMP for a workable assay because the reaction would quickly deplete the substrate. This was probably the reason why the reaction was run for only 5 minutes.28 We replaced AMP with another substrate of CD73, CMP, at its Km of 45 μM and run 15-min reaction to avoid substrate depletion. The quality of the biochemical assay using CMP was assessed using Z-factor (A Z-factor between 0.5 and 1.0 is an excellent assay).24 The Z-factor of the biochemical assay using CMP was satisfactory (Z-factor = 0.88). IC50 of AMPCP was validated in the biochemical assay with CMP vs AMP.
Furthermore, the inhibition of SHR170008 against endogenous membrane-bound CD73 was determined using CD73-expressing human melanoma cell line A375. The resulting IC50 of SHR170008 was 0.051 ± 0.007 nM (n = 4) at incubation with A375 cells in culture, comparable to its potency in biochemical assay (Figure 1C). Since we used the syngeneic mouse breast carcinoma EMT6 model for in vivo efficacy study, the potency of SHR170008 against endogenous membrane-bound CD73 on EMT6 cells was determined (IC50 = 0.133 ± 0.008 nM (n = 4), shown in Figure 1D, approximately 3-fold difference versus its human counterpart. AMPCP was much weaker against endogenous membrane-bound CD73 in both A375 and EMT6 cells (A375, IC50 = 16 ± 5 nM, n =3; EMT6, IC50 = 130 ± 8 nM, n = 3).
The remarkably enhanced potency of SHR170008 as compared with AMPCP was due to forming stronger interactions with CD73 protein. The AMPCP molecule can be divided into three parts, purine ring, ribose ring and bisphosphonic acid (Figure 1A). AMPCP binds into a pocket formed between the N- and C-terminals of CD73 (Figure 1E). The purine motif was sandwiched between the two aromatic residues, Phe417 and Phe500. The two hydroxyl groups of the ribose ring formed hydrogen bonds with Asp506. The bisphosphonic acid group formed charge interactions with Arg354, Arg395 and two Zinc ions, as well as a hydrogen bond with Asn245. Nevertheless, SHR170008 was different from AMPCP in two areas (Figure 1A): one area where the bisphosphonic acid was replaced by methylenephosphonic acid and the other area where the purine ring was substituted by a 6-Cl-pyrazolopyrimidine ring with a bulky spiroindane group substituted on the 4-amino group. As shown in Figure 1F, similar to the bisphosphonic acid, the methylenephosphonic acid of SHR170008 picked up all the key charged interactions, except the hydrogen bond interaction with Asn245. The pyrazolopyrimidine ring of SHR170008 was able to form the same π-π interactions with Phe417 and Phe500 as the purine ring of AMPCP. More importantly, the spiroindane group of SHR170008 occupied a new pocket formed by Ser185 and Asn186 from the N-terminal lobe, which was absent from AMPCP. These interactions adequately explained the significant improvement of potency of SHR170008.
To identify undesirable off-target activity profiles, SHR170008 was tested in SAFETYscan47 panel of seventy-eight functional assays, recommended by major pharmaceutical companies.29 The screening test results of SAFETYscan47 panel, shown in Supplementary Table S4 and Supplementary Methods, indicated that SHR170008 inhibits the targets in the panel at low rates across the board. The hit with the highest percent inhibition is the insulin kinase receptor in two independent measurements, 19% and 17% inhibition, respectively.
SHR170008 Reversed the Inhibitory Effect of Adenosine from AMP in the Activation and Proliferation of CD8+ T Cells and Granzyme B Production
In addition to the inhibition of CD73 enzyme, the functional effects upon T-cells by SHR170008 was examined. An adequate source of T cells with a frequency and functional activity reflective of the true immune response to the immunization is required for in vitro immune analyses. Clearly, human PBMC is the most convenient source of T cells for in vitro immune analysis. We hypothesized that the inhibition of AMP hydrolysis by CD73 enzyme, leading to reduction of adenosine level, would favor activation of T cells. We therefore first stimulated human PBMC with CD3/CD28 dynabeads for 4 days to achieve T cell activation and expansion monitored by T cell activation marker CD25. As shown in Figure 2A, CD25+ cells increased to 76% in cells stimulated with CD3/CD28 dynabeads, compared to those of non-stimulated (1.4%). The percentage of CD25+ cells was reduced by adding 100 µM AMP, substrate of the CD73 enzyme, back to 17%. Co-culture of 1 µM SHR170008 with AMP increased CD25+ cells to 68%, counteracting the inhibitory effect of adenosine from AMP.
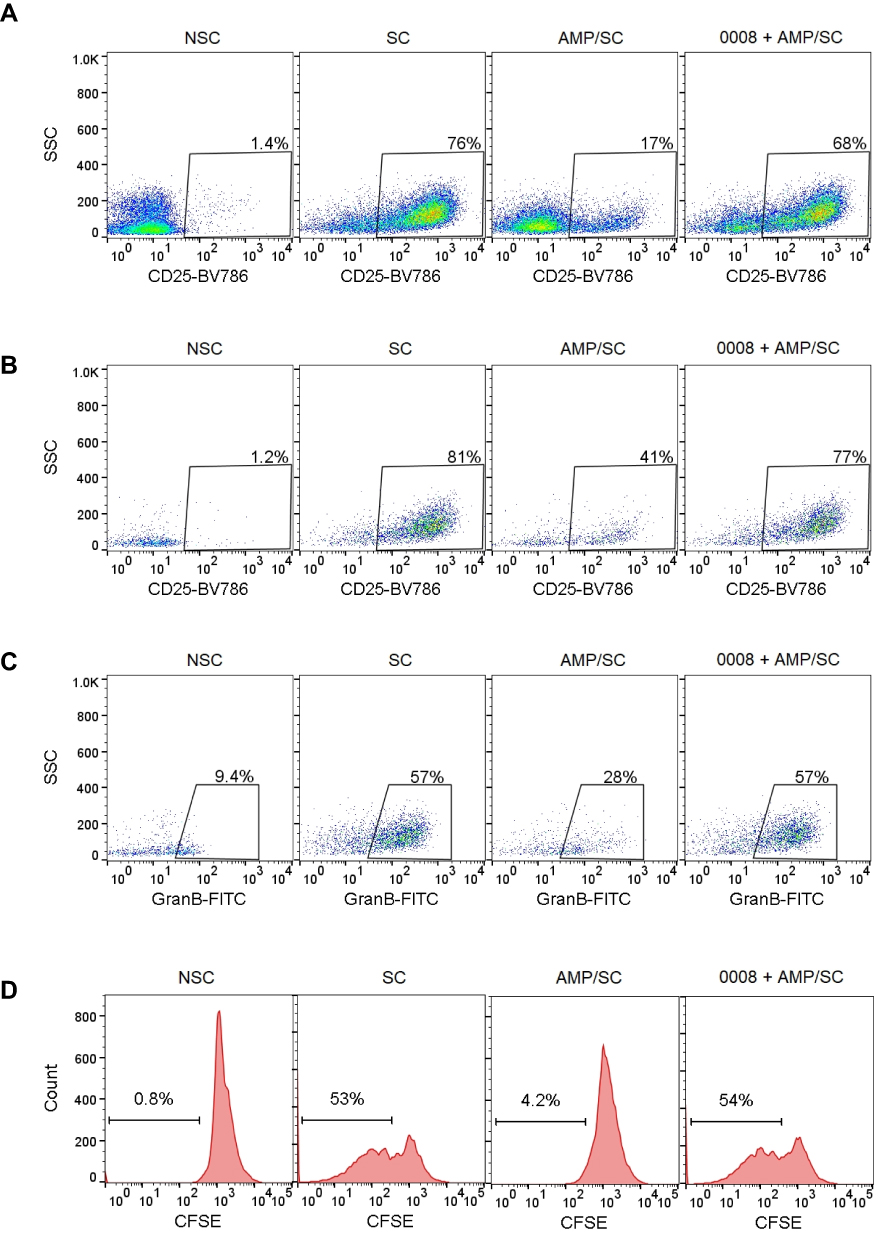 |
Figure 2 SHR170008 reverses the inhibitory effect of adenosine from AMP in the activation and proliferation of CD8+ T cells as well as Granzyme B+ cells in CD8+ cells. (A–C) Representative dot plots showed CD25+ cells in total PBMCs (A) and in CD8+ T cells (B) as well as Granzyme B+ cells in CD8+ cells (C). Human PBMCs were pretreated with AMP (100 µM) ± 1 µM SHR170008 and then stimulated with CD3/CD28 beads for 4 days. The samples were analyzed by flow cytometry. (D) SHR170008 (1 µM) promotes CD8+ T cells proliferation. NSC: non-stimulated control; SC: stimulated control with CD3/CD28 beads. 20,000 total events of each sample were acquired on a Guava EasyCyte instrument. Biological independent experiments were repeated at least twice, and a representative example is shown. 0008 = SHR170008. |
CD8+ cytotoxic T cells directly recognize tumor cells by MHC molecules on the surface of tumor cells as a trigger for direct cytolysis. Increased antitumor immunity of CD73-deficient mice was dependent on CD8+ T cells.12 To analyze the activation of CD8+ cells by SHR170008, cells were co-stained with CD8 and CD25 antibodies. The expression of CD25 in CD8+ cells showed similar trend (Figure 2B). Compared to that of non-stimulated one (1.2%), CD25+ cells in CD8+ cells increased to 81% in cells stimulated with CD3/CD28 dynabeads. In the presence of 100 µM AMP, CD25+ cells in CD8+ cells were decreased to 41%. The addition of 1 µM SHR170008 in the presence of AMP reversed the expression of CD25+ cells in CD8+ cells back to 77%. Furthermore, the expression of Granzyme B+ cells in CD8+ cells was increased after cells were stimulated with CD3/CD28 dynabeads (from 9.4% to 57%) and decreased when co-cultured with 100 µM AMP (28%) (Figure 2C). The addition of SHR170008 in the presence of 100 µM AMP induced the production of granzyme B (57%), completely offsetting the effects of adenosine from AMP.
In addition, SHR170008 completely reversed CD8+ T cell proliferation suppressed by adenosine from AMP (Figure 2D). CD8+ T cells increased after cells were stimulated with CD3/CD28 dynabeads (from 0.8% to 53%) and decreased when co-cultured with 100 µM AMP (4.2%). The addition of 1 µM SHR170008 in the presence of 100 µM AMP completely offset the effects of adenosine from AMP (54% CD8+ T cells). Taken together, the results suggest that SHR170008 could rescue the effects of adenosine produced by CD73 enzyme-mediated hydrolysis of AMP to increase CD25+ expression and CD8+ T cell proliferation as well as granzyme B production in CD8+ T cells.
Effects of Interferon γ (IFNγ) Production in the Presence of Adenosine with or without SHR170008
It was reported that the anti-tumor effects of CD73 blockade was dependent on IFNγ but independent of perforin using gene-targeted mice.18 We investigated the effects of IFNγ production treated with SHR170008 using human PBMC. The cells were stimulated by CD3/CD18 dynabeads for 72 hours, and the concentration of IFNγ in supernatant of the cell culture was then measured (57 ± 4 ng/mL) as compared with the basal level of IFNγ (2 ± 2 ng/mL) without stimulation. To confirm that adenosine might suppress IFNγ secretion in cell culture, human PBMC was pre-treated with various concentrations of adenosine for 2 hours and then stimulated with CD3/CD28 dynabeads for 72 hours. The results indicated that adenosine exhibited a clear dose-dependent suppression of IFNγ production (Figure 3A). Adenosine suppressed IFNγ production by 86% at 50 µM and 96% at 100 µM.
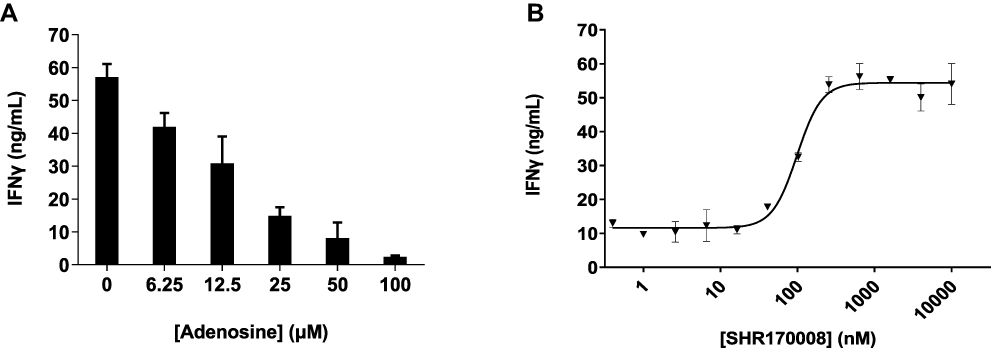 |
Figure 3 IFNγ secretion in response to adenosine or CD73 inhibition in human PBMC. (A) Dose-dependent suppression of IFNγ by adenosine in human PBMC. (B) Induction of IFNγ secretion by SHR170008 as CD73 inhibitor in human PBMC. Each data point was performed in duplicate, and the biological independent experiments were repeated in nine times. A representative curves of dose-dependence inhibition IFNγ production in human PBMC is shown. Data show mean ± STD. Data were fitted by non-linear regression analysis of GraphPad Prism. |
To mimic high concentration of adenosine generated by CD73 in TME, we added 50 µM AMP in the IFNγ production assay. In a typical dose-dependent inhibition (Figure 3B), IFNγ production reached the plateau (54 ± 4 ng/mL) starting from 256 nM of SHR170008, approximately 4.5-fold increase of IFNγ production and a nearly complete reversal of the suppression of IFNγ production caused by adenosine from AMP (12 ± 6 ng/mL). SHR170008 significantly reversed the suppression of IFNγ production with average EC50 = 110 ± 51 nM (n = 9) in human PBMC.
SHR170008 Exhibited in vivo Anti-Tumor Efficacy and Synergistic Effects in Combination with Anti-PD-1 mAb
We used the EMT6 mouse breast tumor model to study the efficacy of SHR170008 and its potential synergy in combination with anti-PD-1 mAb. The tumor-bearing mice were treated with SHR170008 at three different doses (1, 3 and 10 mg/kg, once a day) for 18 days, respectively, and/or injected 5 mg/kg anti-mouse-PD-1 mAb once every 3 days for 4 times.
There were similar body weight changes in all treatment groups as compared to the control group (Figure 4A). Tumor-bearing mice treated with SHR170008 alone showed a dose-dependent tumor growth inhibition (TGI) compared with controls (Table 1). The maximal TGI% of all treatment groups were reached at day 19 and dropped slightly at the end of the study. One-way ANOVA analysis was carried out to compare the significance of differences in tumor volumes of all treatment groups (single and combination) together with those of the control group. The treatment group of SHR170008 at 10 mg/kg, but not other two dosing groups, displayed statistically significant tumor growth inhibition (31% TGI, p < 0.01, day 19) as compared with those of the control group. The mice treated with combination of SHR170008 and anti-PD-1 mAb enhanced tumor growth inhibition in dose-dependent manner (40% TGI, p < 0.01 for 1 mg/kg SHR170008 combination; 51% TGI, p < 0.0001 for 3 mg/kg SHR170008 combination, day 19) compared with those of the control group. Whereas 5 mg/kg anti-PD-1 mAb showed some tumor growth inhibition (29% TGI, p < 0.05, day 19) as compared with the control group.
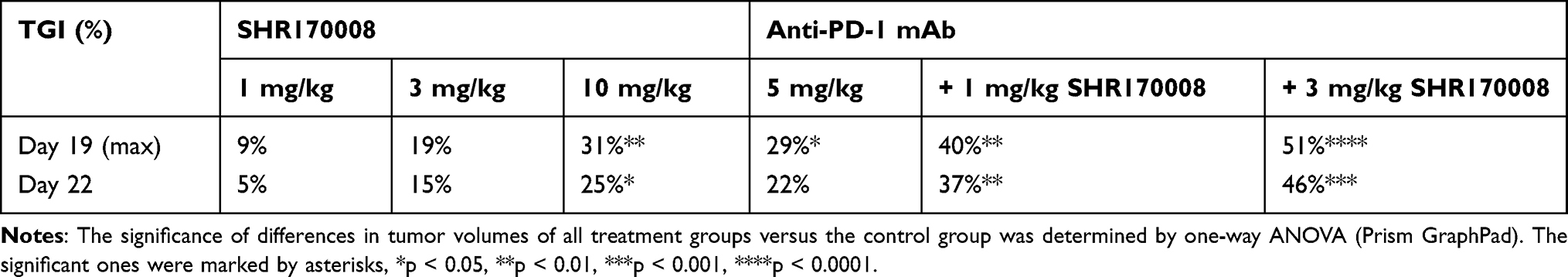 |
Table 1 The Tumor Growth Inhibition (TGI) of CD73 Inhibitor SHR170008 Alone and/or in Combination with Anti-Mouse PD-1 mAb (RMP1-14) in Syngeneic Mouse Breast Tumor EMT6 Model |
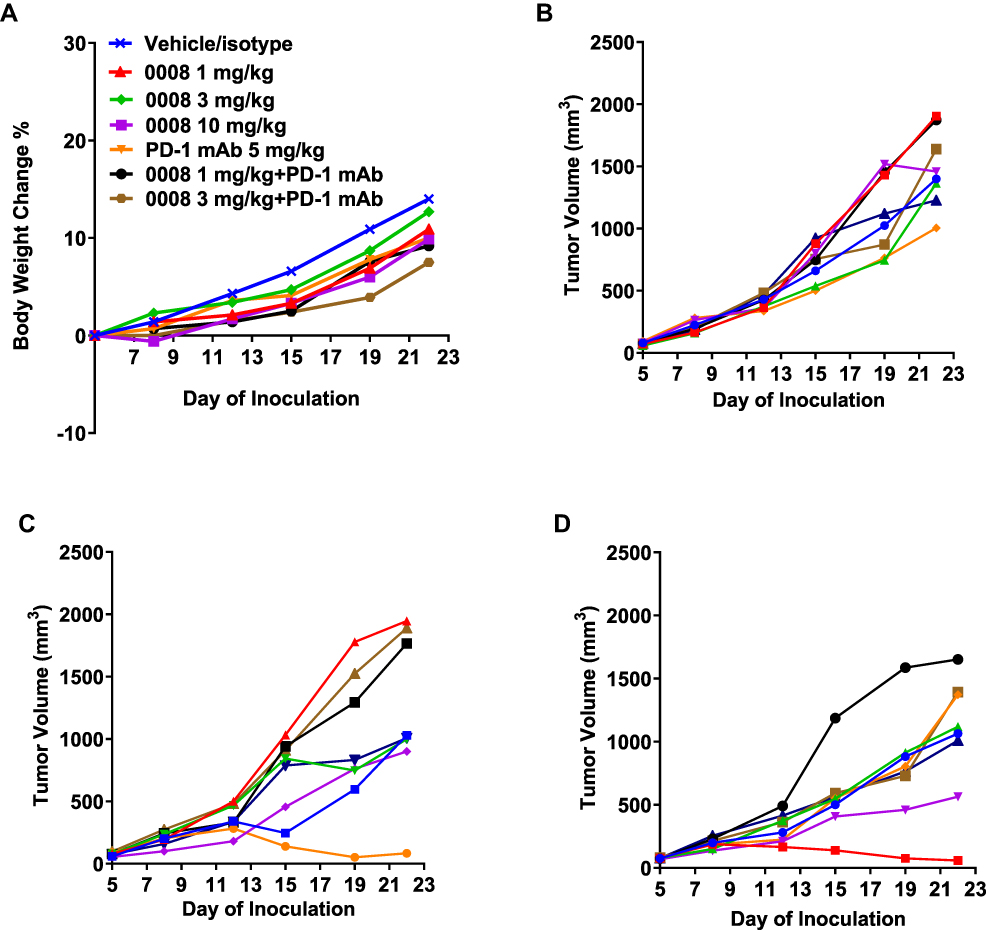 |
Figure 4 Anti-tumor activity of SHR170008 alone and/or in combination with anti-mouse PD-1 mAb in EMT6 mouse syngeneic breast tumor model. (A) Body weight changes of mice during treatment. Body weights were measured twice a week throughout the experiment and shown in average. 0008 = SHR170008. (B) Tumor growth curves of individual mouse bearing EMT6 mouse breast tumor during treatment of 5 mg/kg anti-mouse-PD-1 antibody (RMP1-14) alone. (C) Tumor growth curves of individual mouse bearing EMT6 mouse breast tumor during treatment of 5 mg/kg anti-mouse-PD-1 antibody in combination with 1 mg/kg SHR170008. (D) Tumor growth curves of individual mouse bearing EMT6 mouse breast tumor during treatment of 5 mg/kg anti-mouse-PD-1 antibody in combination with 3 mg/kg SHR170008. Tumor growth curves of individual mouse bearing EMT6 mouse breast tumor during treatment of SHR170008 single-agent groups are shown in Supplementary Figure S3. |
Furthermore, ANOVA analysis was carried out to compare the significance of differences in tumor volumes of the combination group of 1 mg/kg SHR170008 and anti-PD-1 with its corresponding single-agent groups. The reductions of tumor growth in the group of 1 mg/kg SHR170008 in combination with anti-PD-1 were statistically significant as compared with 1 mg/kg SHR170008 single-agent group (p = 0.0304), but not with anti-PD-1 group (p = 0.2808). Stronger statistical significance was observed for the combination group of 3 mg/kg SHR170008 and anti-PD-1 (as compared with the group of 3 mg/kg SHR170008 alone, p = 0.0118, and with anti-PD-1 alone, p = 0.0550, respectively).
Interestingly, we observed that the growth curves of two mice in the combination groups remained nearly flat, suggesting that the tumor growth was significantly curtailed from early days of the study in these two mice (Figure 4C–D). The tumor volumes were 84.6 mm3 on day 22 (versus 89.9 mm3 on day 1) of No. 5 mouse in 1 mg/kg SHR170008 combination group and 58.4 mm3 on day 22 (versus 77.2 mm3 on day 1) of No. 2 mouse in 3 mg/kg SHR170008 combination group, respectively, indicating the tumor regression. These phenomena were not observed in anti-PD-1 mAb (Figure 4B) or SHR170008 single-agent groups (Supplementary Figure S3).
Correlation of Efficacy, PK and PD Biomarkers in vivo
To acquire more insight about the mechanism of the anti-tumor effects of SHR170008 in combination with anti-PD-1 mAb, we analyzed multiple related biomarkers in tumor tissues collected at the end of the study. We first used the cells isolated from tumors collected from in vivo study to analyze TILs and the surface expression of CD73 protein by flow cytometry. As shown in Supplementary Figure S4A, the treatments of SHR170008 in all three dosing groups at 2 hours post dosing increased TIL marker CD45 in tumors (65% ± 6% for 1 mg/kg, 61% ± 5% for 3 mg/kg, and 68% ± 4% for 10 mg/kg) as compared with those in the control group (Control: 46% ± 7%), suggesting the impacts from CD73 inhibition. It was observed that the treatments of SHR170008 in combination with anti-PD-1 mAb exhibited similar extent of TIL changes in tumors (67% ± 4% for 1 mg/kg SHR170008 combination group and 66% ± 2% for 3 mg/kg SHR170008 combination group) as compared to the SHR170008 single-agent groups and anti-PD-1 group (63% ± 3%). No enhancement of TIL levels was observed in the combination groups.
The CD73 enzyme expressed on tumor cells was found to reduce the immune response evoked by anti-PD-1 mAb therapy.15 In this study, the expression levels of CD73 on CD45+ cells in tumor tissues were analyzed. The results indicated that CD73 positive cells in CD45+ cells from tumors were relatively high (85% ± 3% at 2 hours post dosing; 85% ± 4% at 6 hours post dosing) and remained unchanged versus the control group (Supplementary Figures S4B–S4C). Overexpression of CD73 on tumor and immune cells leads to the presence of a high concentration of adenosine in TME and thereby suppression of antitumor immune responses.13,30,31 Adenosine levels in the lysates made from tumor collected at 2 hours and 6 hours post dosing at the end of the study were analyzed by LC/MS. As shown in Figure 5A, adenosine levels at 2 hours post dosing were decreased in SHR170008 single-agent groups (1 and 3 mg/kg) as compared with those in the control group. Notably, PD-1 blockade resulted in enhanced adenosine levels in tumor as compared with the control group and decreased adenosine levels in combination with SHR170008. Similar trends were observed in the data set at 6 hours post dosing (Figure 5B).
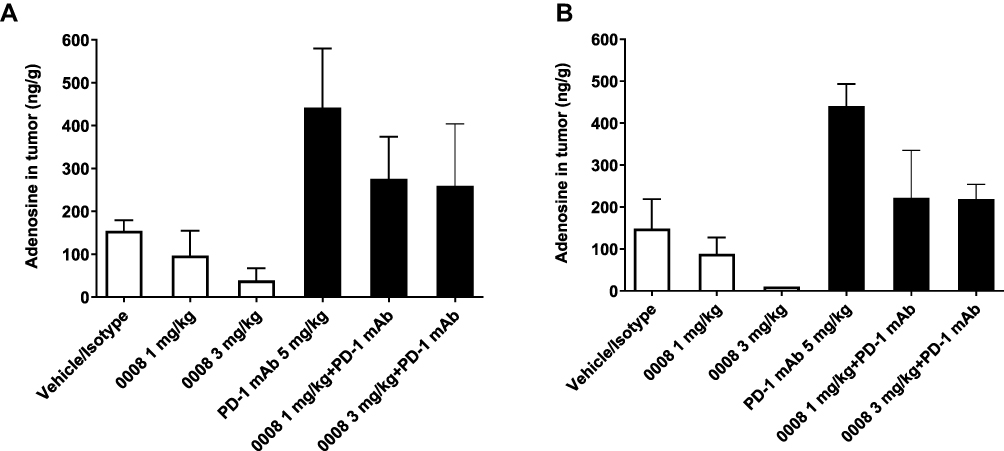 |
Figure 5 Biomarker Responses of SHR170008 in tumor tissues from in vivo efficacy study. (A and B) Adenosine levels in tumor tissues in response to the treatment indicated at 2 hours post dosing (A) and 6 hours post dosing (B) at the end of the study. The samples were below the low limit of quantitation (< 10 ng/g) were plotted as 10 ng/g. Data show mean ± SEM. 0008 = SHR170008. |
The exposures of SHR170008 in plasma and tumor tissues were further analyzed. As shown in Supplementary Figure S5, the exposures of SHR170008 were similar between single-agent and combination groups in both plasma and tumor tissues as expected. In plasma, the average exposures of SHR170008 were 1900 ng/mL at 1 mg/kg and 7150 ng/mL at 3 mg/kg. In tumor tissues, the average exposures of SHR170008 were 197 ng/g at 1 mg/kg and 751 ng/g at 3 mg/kg. SHR170008 showed linear dose-dependency of exposures in both plasma and tumor tissues with approximately exposure ratio of 1 to 10 in tumor tissue versus plasma across the groups. In general, SHR170008 sustained similar exposures at 2 hours and 6 hours post-dosing and decreased at 24 hours post dosing.
Discussion
In this work, we discovered and functionally characterized a novel small-molecule inhibitor SHR170008. One of our goals was to make compounds more metabolically stable We replaced methylenephosphonic acid with the bisphosphonic acid in AMPCP, one of the features that remain in many other CD73 inhibitors.19,20,22,23 As expected, SHR170008 was very metabolically stable (Supplementary Table S1 and Supplementary Methods). The potency of SHR170008 against soluble CD73 enzyme was 50 ± 4 pM, comparable to the reported potency of AB680 against soluble CD73 enzyme (43 pM).20 The remarkably enhanced potency of SHR170008 was due to forming stronger interactions with CD73 protein. The spiroindane group of SHR170008 especially engaged a new pocket formed by Ser185 and Asn186 from the N-terminal lobe, which was absent from AMPCP (Figure 1C). In addition to its biochemical potency against CD73 enzymes, SHR17008 could rescue the inhibitory effect of adenosine from AMP in the activation and proliferation of CD8+ T cells and granzyme B production in human primary immune cells (Figure 2).
Although immune checkpoint blockade with antibodies, such as anti-PD-1 mAbs, have revolutionized the cancer therapy, their success is still limited to a subset of patients.14 Therefore, further work is needed to enhance the therapeutic benefit of anti-PD-1 mAb. CD73 is a negative prognostic marker in many cancer types.4,9,10 Within the TME, high expression of CD73 on cell-surface blocks anti-tumor surveillance by generating extracellular adenosine, leading to tumor immune-escape.4,9,10 The CD73:A2a pathway can limit the efficacy of anti-PD-1 therapy.15 This may be alleviated with concomitant treatment with CD73 inhibitor. In EMT6 mouse breast tumor model, SHR170008 in combination with anti-PD-1 mAb enhanced tumor growth inhibition as compared with the control group (Table 1). A close look at the tumor growth curves of individual mice revealed that two mice in combination groups reached tumor regression, which were not observed in any groups of single agents, indicating that the tumor growth was significantly delimited from early days of the study (Figure 4). CD73 protein was expressed relatively high (85%) on CD45+ cell surface in tumor and remained unchanged versus those in the control group (Supplementary Figures S4B–S4C and Supplementary Methods). Remarkably, we observed that PD-1 blockade increased adenosine expression in tumor and the combination with CD73 inhibitor led to suppression of adenosine generation and greater efficacy, suggesting that highly surface expressed CD73 enzyme in tumor and immune cells led to the presence of a high concentration of adenosine in TME and thereby suppression of antitumor immune responses (Figure 5 and Table 1). The results supported the hypothesis that tumoral CD73 and adenosine suppress antitumor immunity evoked by anti-PD-1 mAb therapy.13,30,31 The findings from in vivo combination study extended understanding of adenosine generated by CD73 in suppressing antitumor immunity that contributed to the resistance of PD-1 checkpoint inhibition and small-molecule inhibitor of CD73 can enhance tumor growth inhibition when combining with anti-PD-1 mAb. This finding was further supported by a recent demonstration that gut microbiota-derived inosine, whose production depends on adenosine, significantly enhances the efficacy of immune checkpoint inhibitors via A2a receptor.32
In conclusion, here we introduce a novel and potent small-molecule inhibitor of CD73 SHR170008 that is suitable for the study that provides new insight into the role of CD73 within the TME. Results from further clinical evaluation of this specific mechanism for immunotherapy of cancer, especially in combination with monoclonal antibody therapies targeting immune checkpoints are highly anticipated.
Data Sharing Statement
All data needed to evaluate the conclusions in the paper are presented in the paper and the Supplementary Materials and Methods.
Acknowledgments
We thank Dr Yuchang Mao for analyses of metabolic pathway identification and rat plasma stability, Dr Qiyun Shao for analytical and spectral determinations, and Hongli Chen for making Supplementary Figure S1.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by Hengrui Pharmaceutical Co. Ltd.
Disclosure
Suxing Liu, Di Li, Jian Liu, Huiyun Wang, Ivana Horecny, Ru Shen, Rumin Zhang, Heping Wu, Peng Zhao, Fengqi Zhang, Yinfa Yan, Linghang Zhuang, and Jing Li are employees of Eternity Bioscience Inc. Qiyue Hu, Jun Feng, Lianshan Zhang, and Weikang Tao are employees of Shanghai Hengrui Pharmaceutical Co. Ltd. Dr Suxing Liu, Dr Jian Liu, Dr Huiyun Wang, Miss Ivana Horecny, and Mr Heping Wu report a patent WO2020047082A1 pending to Eternity Bioscience Inc.
The authors report no other conflicts of interest in this work.
References
1. Escors D. Tumor immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J Sci. 2014;734515. doi:10.1155/2014/734515
2. Antoioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. 2013;13(12):842–857. doi:10.1038/nrc3613
3. Ghirnghelli E, Bruchard M, Chalmin F, Rebe C. Production of adenosine by ectonucleotidases: a key factor in tumor immunoescape. J Biomed Biotechnol. 2012;2012:473712. doi:10.1155/2012/473712
4. Allard D, Allard B, Gaudreau P-O, Chrobak P, Stagg J. CD73-adenosine: a next-generation target in immune-oncology. Immunotherapy. 2016;8(2):145–163. doi:10.2217/imt.15.106
5. Antonioli L, Pacher P, Vizi S, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19(6):355–367. doi:10.1016/j.molmed.2013.03.005
6. Zimmermann H. 5ʹ-nucleotidase: molecular structure and functional aspects. Biochem J. 1992;285(Pt2):345–365. doi:10.1042/bj2850345
7. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276(1):121–144. doi:10.1111/imr.12528
8. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immune-oncology. Nat Rev Clin Oncol. 2020;17:611–629. doi:10.1038/s41571-020-0382-2
9. Inoue Y, Yoshimura K, Kurabe N, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget. 2017;8(5):8738–8751. doi:10.18632/oncotarget.14434
10. Zhu J, Zeng Y, Li W, et al. CD73/NT5E is a target of miR-30a-5p and plays an important role in the pathogenesis of non-small cell lung cancer. Mol Cancer. 2017;16(1):34. doi:10.1186/s12943-017-0591-1
11. Wang L, Fan J, Thompson LF, et al. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest. 2011;121(6):2371–2382. doi:10.1172/JCI45559
12. Stagg J, Divisekera U, Duret H, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011;71(8):2892–2900. doi:10.1158/0008-5472
13. Yegutkin GG, Marttila-Ichihara F, Karikoski M, et al. Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur J Immunol. 2011;41(5):1231–1241. doi:10.1002/eji.201041292
14. Chocarro de Erauso L, Zuazo M, Arasanz H, et al. Resistance to PD-L1/PD-1 Blockade Immunotherapy. A Tumor-intrinsic or tumor-extrinsic phenomenon? Front Pharmacol. 2020;11:441. doi:10.3389/fphar.2020.00441
15. Beavis PA, Milenkovski N, Henderson MA, et al. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol Res. 2015;3(5):506–517. doi:10.1158/2326-6066.CIR-14-0211
16. Goswami S, Walle W, Cornish AE, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. 2020;26(1):39–46. doi:10.1038/s41591-019-0694-x
17. Hay CM, Sult E, Huanga Q, et al. Targeting CD73 in the tumor microenvironment with MED19447. Oncoimmunology. 2016;5(8):e1208875. doi:10.1080/2162402X.2016.1208875
18. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. 2013;19(20):5626–5635. doi:10.1158/1078-0432.CCR-13-0545
19. Junker A, Renn C, Dobelmann C, et al. Structure-activity relationship of purine and pyrimidine nucleotides as ecto-5ʹ-nucleotidase (CD73) inhibitors. J Med Chem. 2019;62(7):3677–3695. doi:10.1021/acs.jmedchem.9b00164
20. Lawson KV, Kalisiak J, Lindsey EA, et al. Discovery of AB680: a potent and selective inhibitor of CD73. J Med Chem. 2020;63(20):11448–11468. doi:10.1021/acs.jmedchem.0c00525
21. Du X, Moore J, Blank BR, et al. Orally bioavailable small molecule CD73 inhibitor (OP-5244) reverses immunosuppression through blockade of adenosine production. J Med Chem. 2020;63(18):10433–10459. doi:10.1021/acs.jmedchem.0c01086
22. Bowman CE, Silva RG, Pham A, Yong SW. An exceptionally potent inhibitor of human CD73. Biochemistry. 2019;58(31):3331–3334. doi:10.1021/acs.biochem.9b00448
23. Gong Y-P, Wan R-Z, Liu Z-P. Evaluation of WO2017098421: GSK’s benzothiazine compounds as CD73 inhibitor filings. Expert Opin Ther Pat. 2018;28(2):167–171. doi:10.1080/13543776.2018.1407756
24. Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi:10.1177/108705719900400206
25. Labute P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins. 2009;75(1):187–205. doi:10.1002/prot.22234
26. Knapp K, Zebisch M, Pippel J, et al. Crystal structure of the human ecto-5ʹ-nucleotidase (CD73): insights into the regulation of purinergic signaling. Structure. 2012;20(12):2161–2173. doi:10.1016/j.str.2012.10.001
27. Sharif EU, Kalisiak J, Lawson KV, et al. Discovery of potent and selective methylenephosphonic acid CD73 inhibitors. J Med Chem. 2021;64(1):845–860. doi:10.1021/acs.jmedchem.0c01835
28. Goueli S, Hsiao K. Monitoring and characterizing soluble and membrane-bound ectonucleotidases CD73 and CD39. PLoS One. 2019;14(10):e0220094. doi:10.1371/journal.pone.0220094
29. Bowes J, Brown AJ, Hamon J, et al. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov. 2012;11(12):909–922. doi:10.1038/nrd3845
30. Stagg J, Divisekera U, McLaughlin N, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A. 2010;107(4):1547–1552. doi:10.1073/pnas.0908801107
31. Antonioli L, Yegutkin GG, Pacher P, Blandizzi C, Hasko G. Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends Cancer. 2016;2(2):95–109. doi:10.1016/j.trecan.2016.01.003
32. Mager LF, Burkhard R, Pett N, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. 2020;369(6510):1481–1489. doi:10.1126/science.abc3421
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.