Back to Journals » International Journal of Nanomedicine » Volume 15
A Non-Lipolysis Nanoemulsion Improved Oral Bioavailability by Reducing the First-Pass Metabolism of Raloxifene, and Related Absorption Mechanisms Being Studied
Authors Ye JY, Chen ZY, Huang CL ![]() , Huang B, Zheng YR, Zhang YF, Lu BY, He L, Liu CS
, Huang B, Zheng YR, Zhang YF, Lu BY, He L, Liu CS ![]() , Long XY
, Long XY
Received 26 April 2020
Accepted for publication 3 August 2020
Published 26 August 2020 Volume 2020:15 Pages 6503—6518
DOI https://doi.org/10.2147/IJN.S259993
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Jing-Yi Ye,1 Zhong-Yun Chen,1 Chuan-Li Huang,1 Bei Huang,2 Yu-Rong Zheng,2 Ying-Feng Zhang,1 Ban-Yi Lu,2 Lin He,2 Chang-Shun Liu3 ,* Xiao-Ying Long1,4, *
1School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, People’s Republic of China; 2School of Chinese Medicine, Guangdong Pharmaceutical University, Guangzhou 510006, People’s Republic of China; 3School of Traditional Chinese Medicine, Southern Medical University, Guangzhou, 510515, People’s Republic of China; 4Guangdong Engineering & Technology Research Center of Topical Precise Drug Delivery System, Guangdong Pharmaceutical University, Guangzhou 510006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chang-Shun Liu
Southern Medical University, No. 1023-1063 of Shatai South Road, Guangzhou, Guangdong 510515, People’s Republic of China
Tel +86 13430301554
Email [email protected]
Xiao-Ying Long
Guangdong Pharmaceutical University, No. 280 of Waihuan East Road, Guangzhou, Guangdong 510006, People’s Republic of China
Tel +86 13798171092
Fax +86 2039352174
Email [email protected]
Objective: A non-lipolysis nanoemulsion (NNE) was designed to reduce the first-pass metabolism of raloxifene (RAL) by intestinal UDP-glucuronosyltransferases (UGTs) for increasing the oral absorption of RAL, coupled with in vitro and in vivo studies.
Methods: In vitro stability of NNE was evaluated by lipolysis and the UGT metabolism system. The oral bioavailability of NNE was studied in rats and pigs. Finally, the absorption mechanisms of NNE were investigated by in situ single-pass intestinal perfusion (SPIP) in rats, Madin-Darby canine kidney (MDCK) cells model, and lymphatic blocking model.
Results: The pre-NNE consisted of isopropyl palmitate, linoleic acid, Cremophor RH40, and ethanol in a weight ratio of 3.33:1.67:3:2. Compared to lipolysis nanoemulsion of RAL (RAL-LNE), the RAL-NNE was more stable in in vitro gastrointestinal buffers, lipolysis, and UGT metabolism system (p < 0.05). The oral bioavailability was significantly improved by the NNE (203.30%) and the LNE (205.89%) relative to the suspension group in rats. However, 541.28% relative bioavailability was achieved in pigs after oral NNE intake compared to the suspension and had two-fold greater bioavailability than the LNE (p < 0.05). The RAL-NNE was mainly absorbed in the jejunum and had high permeability at the intestine of rats. The results of both SPIP and MDCK cell models demonstrated that the RAL-NNE was absorbed via endocytosis mediated by caveolin and clathrin. The other absorption route, the lymphatic transport (cycloheximide as blocking agent), was significantly improved by the NNE compared with the LNE (p < 0.05).
Conclusion: A NNE was successfully developed to reduce the first-pass metabolism of RAL in the intestine and enhance its lymphatic transport, thereby improving the oral bioavailability. Altogether, NNE is a promising carrier for the oral delivery of drugs with significant first-pass metabolism.
Keywords: non-lipolysis nanoemulsion, raloxifene, first-pass metabolism, stability, bioavailability, endocytosis
Introduction
Oral preparations are the mainstay drug formulations as they are convenient and result in better patient compliance. However, their complex physical and biological barriers result in several challenges during their absorption, regarding their solubility, permeability, and stability.1 Solid dispersions, inclusion technologies, nanoparticle formulations, such as nanoemulsions (NEs), as well as self-nanoemulsifying drug delivery systems and nanoparticles have been employed to improve the oral absorption of Biopharmaceutics Classification System (BCS) class II drugs (poorly water-soluble and highly permeable drugs) by promoting drug dissolution and inhibiting drug efflux in the gastrointestinal tract (GIT).2–4 Some BCS class II drugs, including isoproterenol, testosterone, berberine, and raloxifene (RAL), undergo significant first-pass metabolism in the GIT.5–8 Furthermore, improvements in drug dissolution and solubilization via formulation development are insufficient to solve the issue of poor oral absorption.
In the traditional NE, such as the lipolysis nanoemulsion (LNE}, the highly lipophilic BCS class II drugs are encapsulated in the lipid cores of the NE globules, and the oils in the NE are digested in the same manner as food lipids. Bile salts, lecithin, and related endogenous surfactants are secreted, and the lipid cores are emulsified and hydrolyzed by lipases in order to form fatty acids and monoglycerides.9–12 Through the application of an in vitro lipolysis model to examine some of the drugs loaded into NEs, studies have shown that when the oils in the NEs are digested by lipase, the NE collapses and the loaded drugs are released and exposed during lipolysis. In this way, the drug undergoes the first-pass metabolism after the digestion of NE in the GIT.13–15 In contrast, only a small part of NNEs can be hydrolyzed and of the majority of non-lipolysis nanoemulsion (NNE) are absorbed as the globules or mixed micelles which maybe differ with LNE. Therefore, in this study, we attempted to develop a NNE that could not only enhance the solubility and permeability of drugs but also avoid the first-pass metabolism of drugs in the GIT by the different absorption mechanisms.
RAL is a second-generation estrogen receptor modulator that is used as a first-line drug for the prevention and treatment of postmenopausal osteoporosis.16 However, owing to its poor aqueous solubility (solubility: 0.25 μg/mL; log P: 5.2) and the significant first-pass metabolism, the oral bioavailability of RAL is only 2% in humans.16–18 In the intestine, RAL is mainly metabolized by UDP-glucuronosyltransferase (UGT) 1A8 and UGT1A10.19 Therefore, the oral bioavailability of RAL was surmised to be improved via loading into the NNE, which could enhance its solubility and reduce the intestinal first-pass metabolism.
The present study aimed to develop and characterize a NNE formulation. The NNE was systematically improved by in vitro lipolysis. Thereafter, its solubility for RAL, emulsification efficiency, as well as drug-loading properties were derived to screen the required components, optimize the ratio of the components, and confirm the NNE formulation. After characterizing the morphology and globule size of the RAL-NNE, its tolerance in the intestine, namely, its intestinal stability was evaluated by dilution in GIT buffer, followed by in vitro lipolysis and UGT metabolism studies. The protective effect of the NNE on RAL was demonstrated via bioavailability studies in rats and pigs. The absorption mechanism of NNE was investigated by the Madin-Darby canine kidney (MDCK) cells model, situ single-pass intestinal perfusion (SPIP) in rats, as well as the chylomicron flow blocking approach.
Materials and Methods
Materials
RAL (99.8%), RAL-6-β-glucuronide (M1, 95%), and RAL-4′-β-glucuronide (M2, 97.4%) were purchased from Toronto Research Chemicals (North York, Canada). Cycloheximide (CHX; 99.86%) was obtained from MedChemExpress (New Jersey, USA). Soybean oil (SO; 98.0%; triglyceride), isopropyl palmitate (IP, 97%; monoester), and alamethicin (98%) were obtained from Aladdin Reagent Inc. (Shanghai, China). Linoleic acid (LA, 99.0%) and Tween 80 (98.0%) were obtained from AIKE Reagent (Chengdu, China). Cremophor RH40 (99.0%), Cremophor EL (99.0%), Pluronic F68 (99.0%), and Pluronic F-127 (99.0%) were purchased from BASF (Ludwigshafen, Germany). Labrafac PG (diester), Labrafil M 1944 CS, Maisine 35–1 (monoester), Peceol (monoester), Plurol oleique CC 497, and Transcutol HP (99.9%) were obtained from Gattefossé (Lyon, France). Medium-chain triglyceride (MCT) was purchased from CNAC Pharma Co. (Beijing, China). Lipoid E 80 (PC, egg phospholipids with 80% phosphatidylcholine) was obtained from Lipoid GmbH (Ludwigshafen, Germany). Sodium taurodeoxycholate hydrate (NaTDC, 95%), Trizma maleate (95%), porcine pancreatin (from porcine pancreas, 8×USP), saccharolactone, berberine hydrochloride (BH, 98%), testosterone, and undine 5′-diphosphoglucuronil acid were purchased from Sigma-Aldrich (St. Louis, MO, USA). Tris-HCl was procured from Beyotime Institute of Biotechnology (Shanghai, China). Human UGT1A10 and UGT1A8 were purchased from Corning (New York, USA). Nystatin (4500 units/mg), chlorpromazine (98%), and amiloride (99%) were obtained from Solarbio Life Sciences (Beijing, China). Dulbecco’s Modified Eagle Medium (DMEM, high glucose), Trypsin-EDTA (0.25%), fetal bovine serum (FBS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), phosphate buffer saline (PBS), BCA protein assay kit, and MDCK cells were obtained from GIBCO (New York, USA). Transwell plates (96 well and 24 well) were purchased from Corning (New York, USA). Ethanol and 1.2-propanediol (AR) were supplied by Damao Chemical Reagent Factory (Tianjin, China). Other reagents were of high-performance liquid chromatography (HPLC) or analytical grade.
Methods
The Extent of Lipolysis of Excipients
The excipients (Figure 1) were emulsified and screened by in vitro lipolysis. Briefly, the oil phase comprised of oil mixed with LA (1:1, w/w). The oils, Cremophor RH40, and ethanol were emulsified in a weight ratio of 3:5:2 in order to form the pre-nanoemulsion (pre-NE). The pre-NE was titrated with water at 50°C and stirred at 1000 rpm to obtain the NE (15 mL). The NE was then used to explore the extent of the lipolysis of the oils. A water-soluble surfactant (0.5 g) and co-surfactant (0.2 g) were respectively dissolved in water to explore the extent of the lipolysis of the surfactants and co-surfactants. The in vitro lipolysis study was conducted in accordance with a previously applied method.20 The digestion buffer consisted of 50 mM Trizma maleate and 150 mM NaCl; while 5 M NaOH was also used to adjust the pH to 6.8. The NaTDC/PC solution was prepared by adding digestion buffer, with 5mM and 1.25 mM concentration of NaTDC and PC, respectively. The prepared NE and NaTDC/PC solution were mixed together to obtain a total volume of 46 mL and the pH was adjusted to 6.8 using 1 M NaOH. After the addition of 4 mL of pancreatin, lipolysis was initiated at 37°C with stirring at 100 rpm. After the induction of the lipolysis, a 0.25 M Ca2+ solution was added at a rate of 34 μL/min. The temperature and the pH of the lipolysis system were maintained at 37°C and pH 6.8 (by 1 M NaOH). NaOH consumption was recorded and lipolysis was allowed to proceed for 60 min. To assess the extent of lipolysis, the titration volume of NaOH was used to calculate fatty acid production.
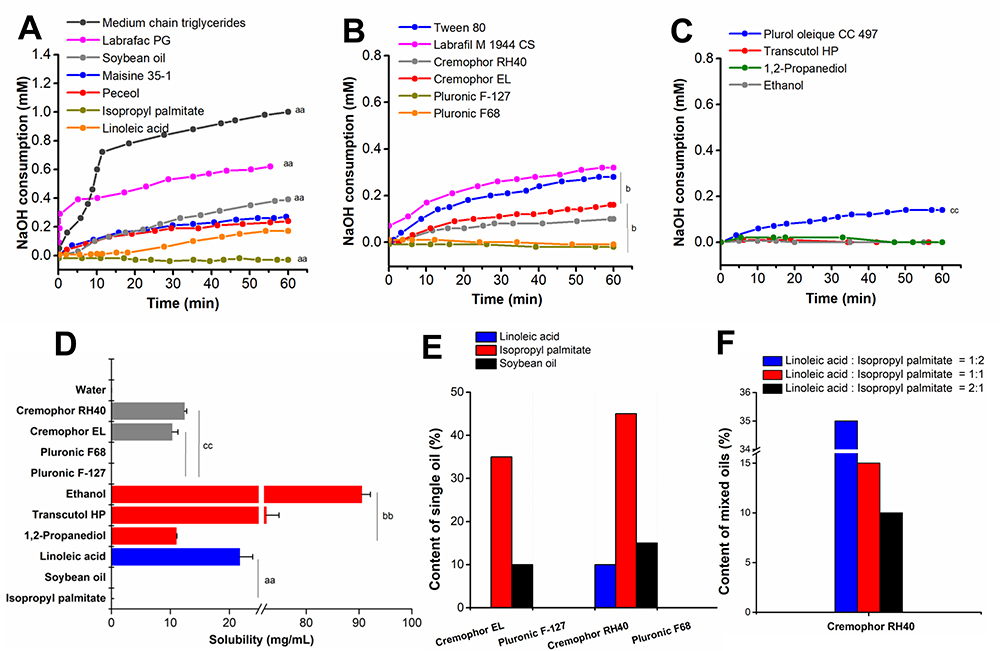 |
Figure 1 Excipient screening for NNEs. Consumption of NaOH by (A) oils, (B) surfactants, and (C) co-surfactants during the lipolysis; (D) Solubility of RAL in the excipients; (E) Emulsification efficiencies of surfactants; (F) Emulsification efficiencies of RH40 with different ratios of LA and IP. |
The Solubility of RAL in Excipients
The solubility of RAL was tested in the excipients (oils, surfactants, and co-surfactants) that had a low extent of lipolysis. Excess RAL was added into the excipients, ultrasonically mixed for 30 min, and agitated in a shaking bath (SPH-200B, Shanghai ShiPing Laboratory Equipment Co., Ltd., China) at 37°C for 72 h. Thereafter, the samples were centrifuged at 37°C for 10 min at 10,142 × g to obtain the supernatant. RAL was quantified at 289 nm with a UV-Vis spectrophotometer (UV-1600; Mapada, Shanghai, China) after appropriate dilution.
Emulsification Efficiency of Surfactants and Co-Surfactants
After lipolysis, the emulsification efficiencies of the surfactants with oils which had low extent of lipolysis were investigated. The surfactant and oil were mixed in different ratios (1:9, 1.5:8.5, 2:8, etc., up to 9:1) and stirred at 50°C and 1000 rpm to prepare the pre-NE. Water was added to the pre-NE to obtain the NE (15 mL). The emulsification efficiency of the optimal surfactant was assessed using the amount of oil emulsified, globule size (≤ 100 nm), and the polydispersity index (PDI < 0.3) after 24 h. Globule size and PDI were measured with a Malvern Zetasizer (Nano ZS90, Malvern, Worcestershire, UK). When the RAL loading dosage was considered, different ratios of LA and IP (1:2, 1:1 and 2:1, w/w) were used as a mixed oil phase of pre-NE to determine the emulsification efficiency of mixed oils. The optimal mixed oil ratio was chosen to prepare the pre-NNEs in two proportions (oily mixtures: surfactants: co-surfactants = 3:5:2 and 5:3:2, w/w) and then select the optimal co-surfactant (Transcutol HP, ethanol, and 1.2-propanediol) according to the indexes of no phase separation and the smaller globule size.
Optimization and Characterization of the RAL-NNE
For comprehensive consideration on RAL loading dose and emulsification efficiency, three pre-NNE composed of mixed oils (LA:IP = 1:2 or 2:1), Cremophor RH40, and ethanol at weight ratios of 3:5:2 (NNE1 and NNE2) and 5:3:2 (NNE3) were used to select the optimal RAL-NNE formulation. The optimized NNE achieved a high drug-loading dose, small globule size, and did not precipitate within 24 h.
The pre-NE of the optimal RAL-NNE (1.0 g) was prepared by mixing RAL (33.75 mg) and LA. After stirring and heating the mixture to dissolve the components, the remaining excipients were added. The pre-NE was emulsified with water titrated at 37°C and 1000 rpm. The final concentration of RAL in NNE was 2.25 mg/mL (15 mL). The IP was replaced with SO to prepare the RAL-LNE.
RAL-NNE and RAL-LNE were diluted 50-fold with water to measure the globule size and PDI. After staining with a phosphotungstic acid solution for 30 s, NNE and LNE were diluted 500-fold with water to determine their morphology with a transmission electron microscope (JEOL JEM-1400, Japan).
Stability of the RAL-NNE in Different pH Buffers at Various Dilutions
RAL-NNE was diluted 10-, 50-, 100-, and 200-folds in water, hydrochloric acid buffer (pH 1.2), and phosphate buffer (pH 5.0 and 6.8), respectively. The globule size and PDI of RAL-NNE were measured to evaluate its pH and dilution stability.
Stability of RAL-NNE in the in vitro Lipolysis System
RAL-NNE (15 mL) was added to the in vitro lipolysis system (total 50 mL) at 37°C and stirred at 100 rpm for 60 min. The lipolysis experiment was performed as described above. Briefly, after lipolysis, the mixture was stirred at 100 rpm and maintained at 37°C for 9 h to determine the changes in RAL content. Before (0 h) and after lipolysis (1, 2, 3, 5, 7, and 9 h), 1 mL was retrieved from the lipolysis reaction for sampling and centrifuged at 37°C for 10 min at 10,142 × g. After appropriate dilution, the content of RAL in the NNE and lipolytic fluid was detected by HPLC.
Stability of RAL-NNE in the Intestinal UGT Metabolism System
After 60 min of lipolysis, RAL-NNE (6 μM) was used in the UGT-mediated metabolic experiment. UGT1A8 (200 μg protein/mL) or UGT1A10 (400 μg protein/mL), MgCl2 (10 mM), alamethicin (60 μg/mL), saccharolactone (4.4 mM), undine 5ʹ-diphosphoglucuronil acid (5 mM), and Tris-HCl (pH 7.4, 50 mM) were mixed and stored at 0°C for 20 min to activate the UGTs. The mixture was pre-incubated at 37°C for 5 min with 200 rpm of stirring; thereafter, RAL-NNE (before or after lipolysis) was added to the mixture. By adding undine 5ʹ-diphosphoglucuronyl acid, the reaction was initiated and allowed to proceed for 120 min. The reaction was terminated by the addition of 400 μL cold acetonitrile (containing 3 μM testosterone as an internal standard) and stored at −20°C. Similarly, the metabolism of the RAL solution (suspension) and RAL-LNE was evaluated. The content of RAL and its metabolites (M1 and M2) was analyzed by HPLC. The metabolic rate of RAL was calculated by determining the content of the metabolized drug as a percentage of the total drug content. The production rate of M1 and M2 was calculated as the content of transferred drugs based on the percentage of the metabolized drug.
Bioavailability in Rats
Female Sprague–Dawley rats (220 ± 20 g) were purchased from the Animal Research Center of Guangzhou University of Chinese Medicine (Guangzhou, China). All procedures in this study were approved by and performed in compliance with the regulations of the Animal Ethics Committee of Guangdong Pharmaceutical University (NO. SPF2017127). Rats were allowed to acclimatize for 1 week in a standardized lab environment with free access to food and water. Rats were fasted for 12 h before the experiments and given free access to water.
Twenty rats were randomly divided into four groups (n = 5). Three normal groups, namely RAL suspension (RAL-control), RAL-NNE, and RAL-LNE, were orally administered 45 mg/kg, respectively. The fourth group, RAL solution (ethanol:1,2-propanediol: dimethyl sulfoxide: normal saline = 1:2:2:10, v/v), was intravenously injected 7.5 mg/kg. Blood samples were collected from the caudal vein at 0, 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 h after administration and stored in tubes containing heparin.
Plasma (150 μL) was extracted in four volumes of extraction solvent (methanol: acetonitrile = 1:1, v/v) containing 70 μM BH (internal standard). The mixtures were vortexed for 2 min and centrifuged at 4°C for 15 min at 10,142 × g. The extracts were evaporated to dryness under a nitrogen atmosphere at 60°C. The residue was reconstituted with 100 μL of the mobile phase and the concentration of RAL was quantified by HPLC.
The bioavailability parameters (Cmax, Tmax, and AUC) were calculated by non-compartmental analysis of the drug concentration-time profiles using the Drug and Statistics (DAS) 3.0 software (Chinese Mathematical Pharmacology Society, Beijing, China). The relative bioavailability (Fr) and absolute bioavailability (Fa) of RAL were calculated according to Equation (1) and Equation (2), respectively.
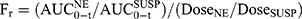
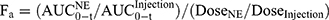
Bioavailability in Pigs
Female Wuzhishan pigs (25 ± 2 kg) were purchased from the Feed Research Institute (Guangzhou, China), as permitted by the Animal Ethics Committee of South China Agricultural University (NO. 2018B038). Pigs were allowed to acclimatize for 1 week with free access to food and water, and fasted for 12 h before the experiments but given free access to water.
Three pigs were administered each formulation in a randomized crossover design, with a washout period of 1 week between administration. Before administration, the pigs were anesthetized with atropine sulfate, Zoletil 50, and xylazine hydrochloride. The oral and intravenous administration dosages were 10 mg/kg and 1.7 mg/kg, respectively. Blood samples were collected from the superior vena cava at 0, 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, 24, and 36 h after drug administration. The plasma (0.5 mL) was used and the sample was prepared as described in “Bioavailability in rats”. The concentration of RAL was quantified by HPLC and the pharmacokinetics data was analyzed as described above.
Absorption Site and Endocytosis of RAL-NNE in situ SIPI
An in situ SIPI investigation was performed following an established method.21 Briefly, rats were intraperitoneally anesthetized with ethyl carbamate (1.5 mg/kg). Thereafter, the tested intestinal segments (the duodenum, jejunum, ileum, and colon) were identified, cannulated with plastic tubing, and ligated for perfusion. The segment was attached to an infusion pump (Longerpump, Baoding, China) and rinsed with saline (37°C) to remove residues. All perfusates contained the non-absorbable marker, phenol red (30 M), to correct the water flux. The perfusate containing RAL-NNE was perfused at a flow rate of 0.2 mL/min for 20 min to ensure steady-state. Similarly, the perfusates containing RAL-NNE and endocytosis inhibitors, nystatin (30 M), chlorpromazine (30 M), and amiloride (100 M), were respectively injected into the jejunum to assess their impact on the absorption of RAL-NNE.
Perfusion was then performed and the perfusate was collected into microtubes at the predetermined time intervals (0, 10, 20, 30, 40, 50, 60, and 70 min). The length and internal diameter of the perfused segment were measured at the last collection and the rats were killed. The perfusate was extracted using 1-fold methanol containing 70 μM BH (internal standard). The mixtures were vortexed for 2 min, centrifuged (10,142 × g, 15 min) and filtrated. The samples (10 μL) were collected and analyzed by HPLC.
The absorption rate constant (Ka) and effective permeability (Peff) of RAL across the rat gut wall were determined using Equation (3) and Equation (4), respectively.21
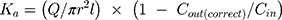
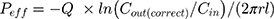
where Q was the perfusion speed (0.2 mL/min); Cin and Cout (correct) were the concentrations of RAL in the perfusate in and out of the intestinal segment, respectively; and r and l were the radius and length of the test intestinal segment, respectively.
Viability of MDCK Cells
The endocytosis mechanisms of RAL-NNE were further investigated using MDCK cells. The RAL-NNE or free RAL was diluted by EMEM to prepare a series of RAL-EMEM (0.47, 0.94, 1.88, 3.75, 7.50, 15.00, 30.00, and 60.00 μg/mL). For investigating cell toxicity of RAL-NNE, 200 μL of EMEM was added into plates (n=6) and cultured at 37°C for 24 h. The MTT method (5mg/mL) was used to detect the viability of MDCK cells and cell viability was calculated by Supplementary Equation (1) (Supplementary data).
Uptake and Transport of RAL-NNE by MDCK Cells
The endocytosis inhibitors, nystatin (30 M), chlorpromazine (30 M), and amiloride (100 M), are the inhibitors of caveolin, clathrin, and macropinocytosis, respectively.22 The inhibitors were respectively added into well and incubated for 30 min before the uptake experiment for establishing the blocking model. The MDCK cells were rinsed 3× with PBS and EMEM contained RAL-NNE was added and further cultured for 3 h. Then the uptake was stopped by rinsing with PBS. The cells were lysed and centrifuged at 10,142 × g, 4°C, for 15 min. The supernatant was withdrawn and 3-fold acetonitrile was mixed to extract RAL twice. The supernatants were combined and dried then reconstituted with 100 μL mobile phase. RAL content and the protein amount were determined by HPLC and BCA protein assay kit, respectively. The amount of cell uptake was calculated by the ratio of the concentration of RAL and protein (Supplementary data).
When the transepithelial electrical resistance of MDCK cells was > 180 Ω·cm2, the cells were used for a transport experiment.23 Prior to the transport experiment, the cells at the apical side were rinsed 3× with PBS. The endocytosis inhibitors used in the uptake experiment were applied to block the transport of RAL-NNE and the cells were rinsed before EMEM (containing RAL-NNE) adding. RAL content was determined by HPLC. The cumulative transport amount (Q) was calculated by the Supplementary Equation (2) (Supplementary data).
Lymphatic Transport of RAL-NNE in Rats
The rats were purchased and fed as “Bioavailability in rats”. CXH has been used to inhibit the secretion of chylomicrons from the enterocytes and without causing damage to other active and passive absorption pathways of drugs.4,24-26 For investigating the lymphatic transport of RAL, the RAL-NNE and RAL-LNE (45 mg/kg) were respectively orally administered after intraperitoneal injection of 1 mg/mL CHX (1 mg/kg) for 30 min as blocking groups (n = 5). The rat was orally given of RAL-NNE or RAL-LNE but without CHX injection as the normal groups. Blood was collected and treated as described in “Bioavailability in rats”.
HPLC Analysis
RAL concentration was detected using the Waters HPLC System (Waters Corp., Milford, MA, USA), which comprised a 2707 autosampler, a 1525 binary pump, and a 2489 UV-VIS detector. RAL was eluted from a Luna polar omega C18 column (4.60 mm × 250 mm, 5 μm; Phenomenex, Torrance, USA) at a flow rate of 1.0 mL/min.
In the UGT metabolism system, the RAL, M1, and M2 were separated by gradient elution and the mobile phase consisted of ammonium acetate buffer (mobile phase A, pH 4.0) and acetonitrile (mobile phase B). The gradient elution process as follows, 0–16 min, 78% A; 16–32 min, 78–51% A; 32–38 min, 51% A; 38–42 min, 51–78% A; 42–43 min 78% A. The testosterone as internal standard and it was detected at 254 nm. The column temperature, detection wavelength, and injection volume were 30°C, 289 nm, and 10 μL, respectively.
The RAL content in lipolysis, uptake and transport experiments of MDCK cells, SIPI experiment, plasma of rats and pigs were quantitated using isocratic elution. The mobile phase was in a ratio of 30:70 (B:A, v/v). However, in the SIPI experiment, in vivo absorption of rats and pigs, the internal standard, BH was detected at 352 nm. Other separation conditions were the same as previously described but the injection volume of in vivo sample was 20 μL.
Statistical Analysis
Statistical analysis was conducted using SPSS 22.0. Data are presented as mean ± SD. Data were analyzed by one-way ANOVA, and p values < 0.05 were considered to indicate statistical significance.
Results
Screening the Components of NNE
The extent of lipolysis for the NE excipients is shown in Figure 1. Among the tested oils, unsaturated fatty acid (LA) displayed the lowest NaOH consumption, followed by the monoester (Peceol ≈ Maisine 35–1), long-chain triglycerides (SO), medium-chain diester (Labrafac PG), and MCT (Figure 1A, p <0.01). NaOH consumption by the oil, LA, was lower than that by Peceol and Maisine 35–1; however, the oil, IP, did not consume any NaOH. NaOH consumption of the surfactants was found to occur in the following descending order: Tween 80 and Labrafil M 1944 CS > Cremophor EL ≈ Cremophor RH40 (Figure 1B, p <0.05). Furthermore, Pluronic F-127 and Pluronic F68 did not consume any NaOH. Similarly, the co-surfactants, Transcutol HP, 1.2-propanediol, and ethanol did not consume NaOH, whereas Plurol Oleique CC 497 could consume NaOH (Figure 1C, p <0.01).
The solubility of RAL was measured in the excipients with a low degree of lipolysis including the oils (IP and LA), surfactants (Cremophor EL, Cremophor RH40, Pluronic F-127, and Pluronic F68) and co-surfactants (Transcutol HP, 1.2-propanediol, and ethanol). In Figure 1D, RAL was most soluble in LA, Cremophor RH40, and ethanol, respectively.
Emulsification Efficiency of the Surfactants and Co-Surfactants
Based on the extent of lipolysis and RAL solubility, the emulsification efficiency of the surfactants for LA and IP was screened. From the results, the surfactant Cremophor RH40 had the highest capability to emulsify a single oil; this was followed by Cremophor EL (Figure 1E). At the conditions used in this study, the oils could not be emulsified by Pluronic F-127 and Pluronic F68. Furthermore, Cremophor RH40 led to a higher emulsification efficiency when a higher ratio of IP was used in the oil mixture (Figure 1F). Thus, this mixture of oils (LA:IP = 1:2, w/w) and Cremophor RH40 were used to select the co-surfactant. As shown in Table 1, the characteristics of the NNE prepared with the three co-surfactants were not significantly different in the 3:5:2 weight ratio of oil, surfactant, and co-surfactant. However, ethanol displayed excellent emulsification efficiency at the weight ratio of 5:3:2. Phase separations were apparent in formulations containing Transcutol HP and 1.2-propanediol. Also, their globule size was larger than that of the NNE containing ethanol. From these results, LA, IP, Cremophor RH40, and ethanol were selected as the components of the NNE.
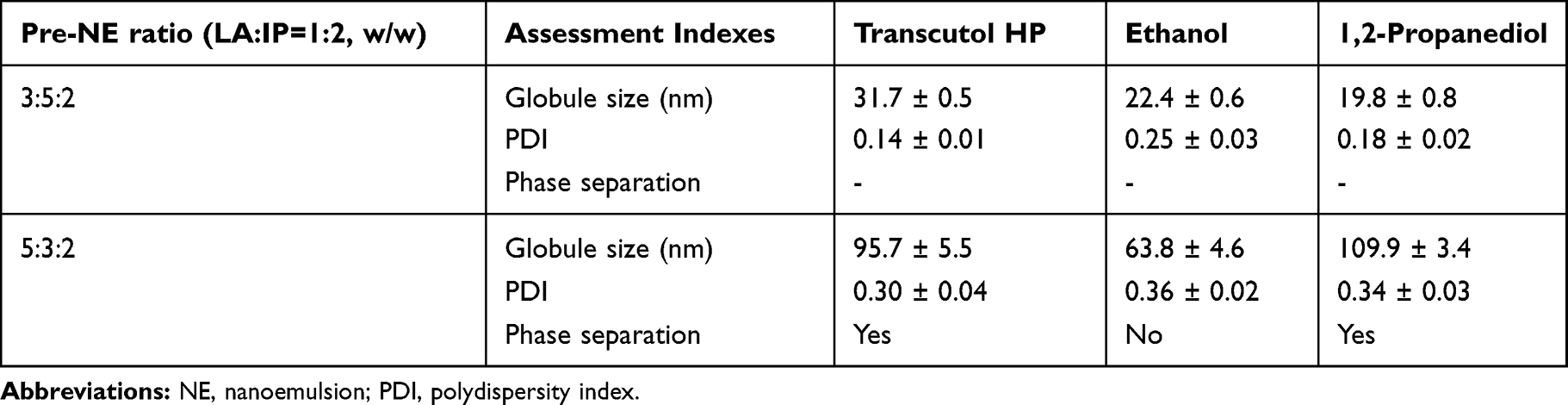 |
Table 1 Emulsification Efficiency of the Co-Surfactants (n = 3) |
Optimization and Characterization of the RAL-NNE
Although the higher ratio of IP could enhance emulsification efficiency, the solubility of RAL in LA was significantly higher than IP (Figure 1D). As shown in Table 2, the loading dose of the RAL in NNE3 was higher than that in NNE1 and NNE2; however, its globule size and PDI were smaller. Besides, RAL precipitation did not occur in NNE3. Taking account for the emulsification efficiency and the loading dose of RAL, NNE3 was the optimal formulation for RAL. Because of the lipolytic nature of SO, IP was replaced with SO to prepare the LNE for comparison to the NNE.
 |
Table 2 Composition and Characteristics of NEs in the Different Formulations |
The globule size and PDI of RAL-NNE and RAL-LNE are presented in Table 2 and Figure 2A. The images obtained from transmission electron microscopy (Figure 2B) demonstrate the spherical shape of NNE and LNE.
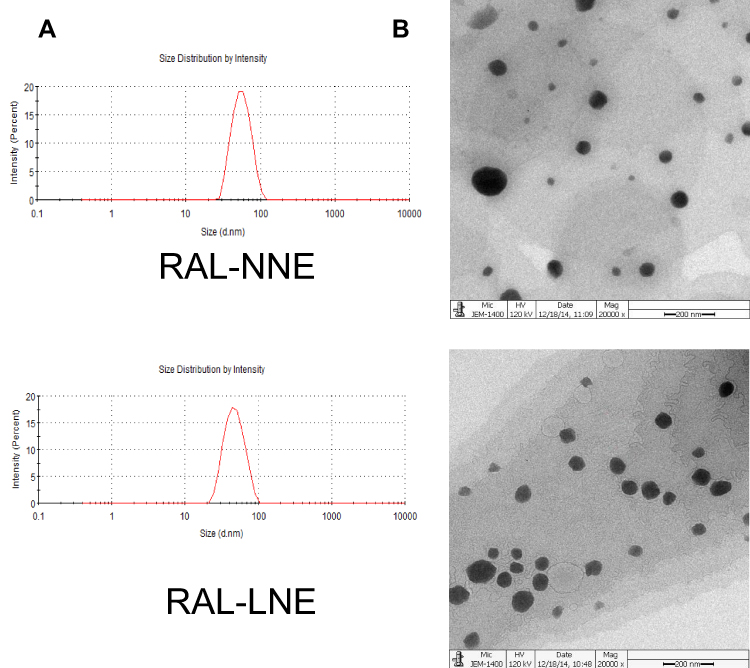 |
Figure 2 Characterizations of RAL-NNE and RAL-LNE. Globule sizes (A), morphology (B) of RAL-NNE and RAL-LNE (× 15,000). |
Stability of the RAL-NNE in the Buffers and Lipolysis System
RAL-NNE was stable in water and buffers with a pH from 1.2 to 6.8. However, there were no significant changes in the globule size and PDI of the RAL-NNE with the different dilutions, thereby indicating its good stability in the buffers (Figure 3A).
 |
Figure 3 Stability of RAL-NNE in gastrointestinal situation. (A) Stability in different media after dilution; (B) Lipolysis curves of RAL-NNE and RAL-LNE; (C) Content change of RAL in the NNE and LNE during and after the lipolysis; (D) UGT-mediated metabolism rates of RAL solution, RAL-NNE, and RAL-LNE. |
During the 60 min lipolysis period, although NaOH consumption of the RAL-LNE was significantly higher than that of the RAL-NNE (Figure 3B), RAL content was similar between RAL-NNE (77.18%) and RAL-LNE (76.23%). However, after completion of lipolysis, RAL content in LNE was markedly lower than that in the NNE after 3 h. Over time, the difference was found to generally increase (Figure 3C).
Stability of the RAL-NNE in the Intestinal UGT Metabolism System
As shown in Figure 3D, 99.29 ± 2.83% and 57.41 ± 0.86% of the RAL in solution were metabolized by UGT1A10 and UGT1A8, respectively. The respective production ratios of M1 and M2 were 4.66 ± 1.74% and 63.41 ± 1.89% by UGT1A10 and 20.61 ± 0.33% and 36.71 ± 1.07% by UGT1A8, respectively. In the RAL-LNE, no evident change in the RAL level was observed before lipolysis. However, after lipolysis, the ratios of the RAL metabolized by UGT1A10 and UGT1A8 were 51.83 ± 2.95% and 46.00 ± 0.83%, respectively. These values were 4-fold and 38-fold higher than those for the RAL-LNE without lipolysis. In RAL-NNE, only a small amount of RAL was metabolized by intestinal UGT1A10 and UGT1A8 (without lipolysis: UGT1A10, 5.69 ± 1.34%; UGT1A8, 0.50 ± 0.49%; after lipolysis: UGT1A10, 10.84 ± 3.13%; UGT1A8, 5.70 ± 1.45%), independent of whether lipolysis was performed (Figure 3D). These results indicate that RAL-NNE was stable following lipolysis and UGT metabolism in the intestinal environment.
Oral Bioavailability of RAL-NNE in Rats
After oral administration to rats, RAL exhibited a similar bioavailability both in the LNE and NNE groups, but the oral bioavailability of NEs was two-fold than that of the RAL-control group (Figure 4A). The AUC of RAL in the LNE and NNE groups was 4.31 ± 0.57 h·μg/mL and 4.36 ± 0.93 h·μg/mL, respectively; these values were significantly higher than that of the RAL-control group (p < 0.01). The Fr and Fa of RAL in the NE group were 200% and 14%, respectively. No significant differences in Tmax and AUC were found between the NNE and LNE groups (Table 3), but the Cmax of LNE was significantly higher than that of the other group (p < 0.05).
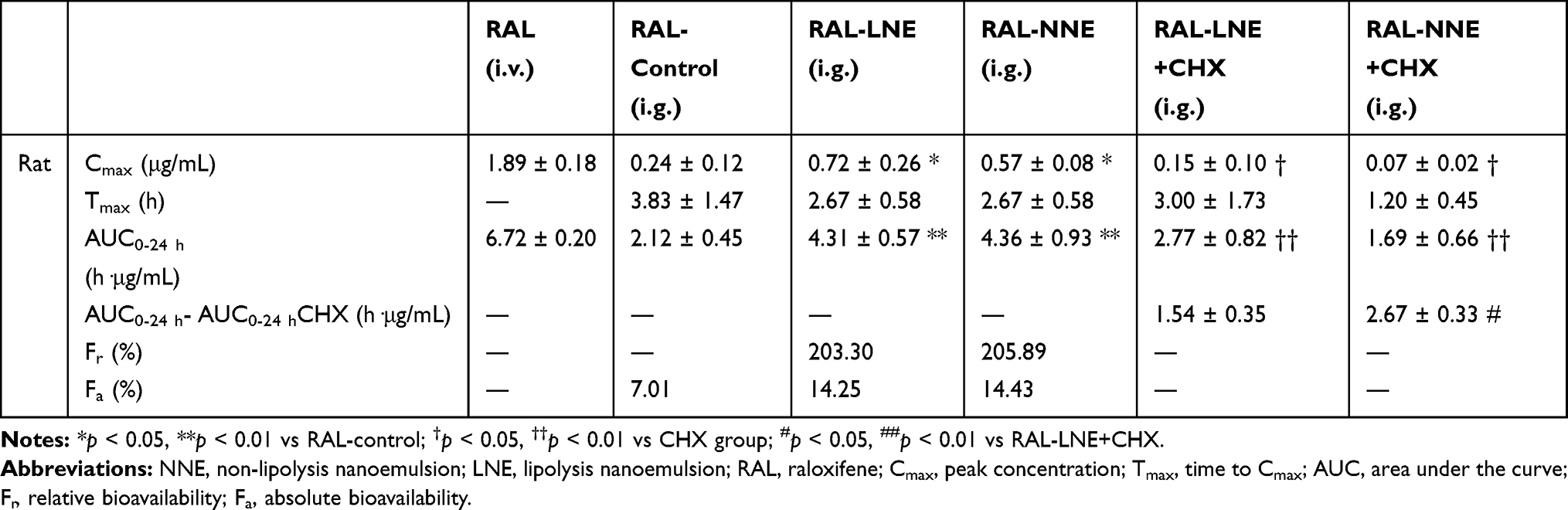 |
Table 3 Bioavailability Parameters and Lymphatic Transport of RAL in Rats (n = 5) |
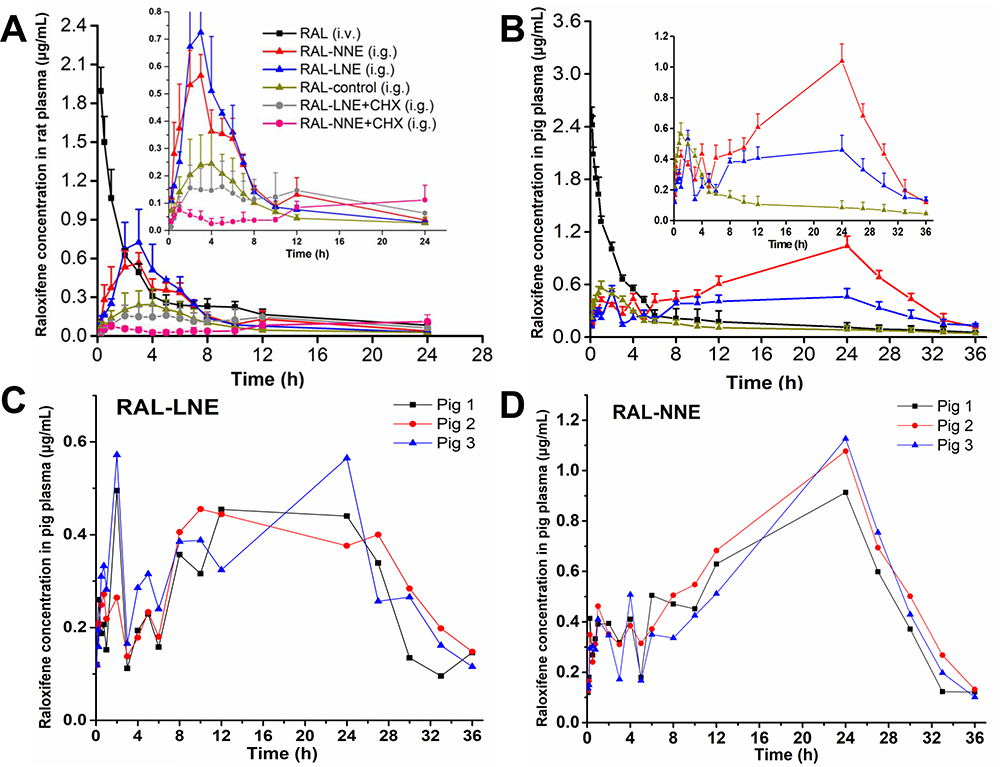 |
Figure 4 Plasma concentration-time profiles of RAL-LNE and RAL-NNE in rats (A) and pigs (B) after oral administration. Plasma concentration-time profiles of RAL-LNE (C) and RAL-NNE (D) in pigs. |
Oral Bioavailability of RAL-NNE in Pigs
In pigs (Table 4 and Figure 4B–D), the Fr of RAL-NNE was 541.28% while Fa was 23.34% relative to the RAL-control group. Furthermore, Fr and Fa of RAL were 230.23% and 9.93%, respectively, in the LNE group. Unlike in rats, two Cmax values were found in pigs after RAL-NE was orally administered. Although the C1max did not significantly differ between NNE and LNE, the C2max of NNE was higher than its C1max and the Cmax of LNE (p < 0.05). Similarly, there was no significant difference in T1max but at 24 h, the T2max of the NNE and LNE groups was prolonged relative to the control group. The large standard deviation for the T2max of RAL-LNE was due to the inter-individual differences for animals. In contrast, the inter-individual differences in the absorption of RAL-NNE were much smaller (Figure 4C and D).
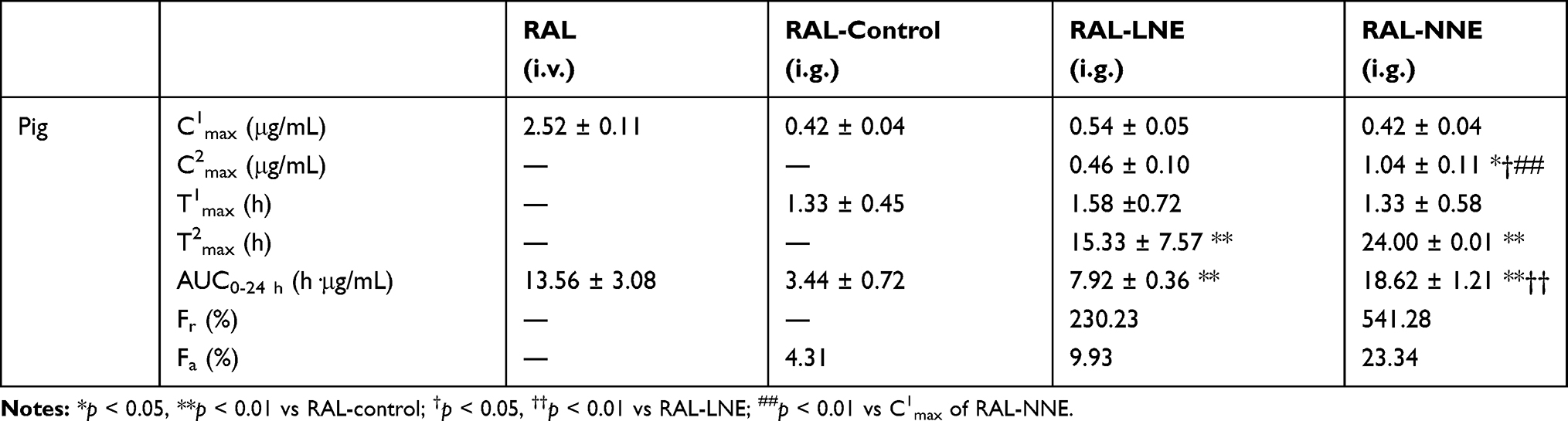 |
Table 4 Bioavailability Parameters of RAL in Pigs (n = 3) |
Absorption of RAL-NNE in situ SIPI in Rats
RAL loaded into NNEs could be absorbed in different parts of the gut. However, absorption was found to predominantly occur in the jejunum (Table 5). In the jejunum, when RAL-NNE was perfused with amiloride, chlorpromazine, and nystatin, the Ka and Peff values of RAL were significantly decreased. The Peff of RAL-NNE was reduced by 3–10-fold while Ka was reduced by 2–7-fold. The inhibition efficiency followed the order nystatin (caveolin) ≈ chlorpromazine (clathrin) > amiloride (macropinocytosis). These findings suggest that the intestinal absorption of RAL-NNE was significantly inhibited by the endocytosis inhibitors (p < 0.05).
 |
Table 5 Intestinal Absorption Parameters (Ka and Peff) of the RAL-Loaded NNE Administered to Rats (n = 3) |
Cell Viability as Well as Uptake and Transport of RAL-NNE in MDCK Cells
The cell viability of free RAL and RAL-NNE was compared in Supplementary Figure 1 (A) and the cell viability > 80% when the concentration of RAL < 3.75 μg/mL. The uptake amounts of RAL were significantly inhibited by nystatin and amiloride (p < 0.05, Supplementary Figure 1 (B)). The cumulative transport amount of the RAL-NNE was decreasing but there were no significant differences compared with the absence of inhibitors.
Lymphatic Transport of RAL-NNE in Rats
After injecting CHX (Table 3), the Cmax and AUC0-24 h of RAL-LNE and RAL-NNE decreased significantly (p < 0.05) compared with the normal groups (CHX absence). In Figure 4A, the plasma concentrations of RAL-LNE+CHX and RAL-NNE+CHX were lower than the normal group. The values of AUC0-24 h-AUC0-24 hCHX were above zero and that of NNE was higher than LNE (p < 0.05).
Discussion
RAL-NNE Development
In the present study, RAL-NNE was developed according to the resistance of the excipients to lipolysis, the higher loading of RAL in the pre-NE, and good particle morphology. As shown in Figure 1, chain length and steric hindrance of the oils and the number of ester bonds in the excipients can affect the extent of lipolysis. Of the oils, MCT exhibited the highest NaOH consumption over a 60 min lipolysis period. Comparatively, the NaOH consumption by SO (long-chain triglycerides) displayed an intermediate extent of lipolysis, with a rate lower than that by Labrafac PG and propylene glycol dicaprylate (medium-chain diester). This is because the long-chain triglycerides have a slower lipolysis rate than the medium-chain diester, which aligns with the results of previous studies.27–29 Maisine 35–1 and Peceol consumed less NaOH than Labrafac PG due to the fewer ester bonds. Interestingly, the isopropyl ester of the monoester, IP, did not consume NaOH and was not digested (Figure 1A). The steric hindrance of the isopropyl group against the active pocket of the porcine pancreatic lipase resulted in resistance to lipolysis.30 LA also consumed a low amount of NaOH. Therefore, the oils, IP and LA, were resistant to lipolysis and the extent of lipolysis in the other oils followed the order, medium-chain triglycerides > medium-chain diglycerides > long-chain triglycerides > long-chain monoglycerides.
As the surfactants, Pluronic F-127 and Pluronic F68 lacked ester bonds, they were resistant to lipolysis. Although Cremophor EL was highly variable in the composition of poly-oxyethylene glycerol triricinoleate, it contained polyethylene glycol and polyethylene glycol ether, which could affect its combination with pancreatin and cause a lower extent of lipolysis than Tween 80 or Labrafil M 1944 CS (containing a lot of ester bonds).31 Cremophor RH40 has a similar composition to Cremophor EL, and thus, underwent a similar extent of lipolysis. Therefore, the excipients without esters might be resistant to lipolysis. The co-emulsifiers, Transcutol HP, ethanol, and 1.2-propylene glycol, did not consume NaOH, whereas Plurol oleique CC 497 did. After the emulsification capacity of the surfactants for oils (Table 2) and the solubility of RAL in these excipients were considered, IP, LA, Cremophor RH40, and ethanol were selected as the components of RAL-NNE.
The LA ratio in the oil phase can increase RAL loading in NNE. As shown in Table 2, the proportion of the oil phase was increased while that of Cremophor RH40 was reduced; these changes not only caused higher drug-loading but also led to a smaller globule size. This may have been because the greater proportion of Cremophor RH40 increased the viscosity of the pre-NE, which formed more lamellar liquid crystals and gels, owing to the absence of thorough mixing, and larger globule size formation during the emulsification process.32 Furthermore, the intermolecular forces of the ionic bond between RAL and LA were enhanced, resulting in smaller globule sizes.15,33 Although RAL was most soluble in ethanol, which could efficiently improve its emulsification efficiency, the weight ratio of ethanol in the pre-NE was limited to 10%-20% as ethanol did not retain its solubilization capacity after dispersion in an aqueous system because of its water solubility, which may lead to drug precipitation.34,35 Therefore, we identified that IP:LA (1:2, w/w), Cremophor RH40, and ethanol in a 5:3:2 weight ratio in pre -NNE was used to prepare the optimal RAL-NNE.
Improvement of the in vitro Stability of RAL-NNE
The stability results from in vitro lipolysis support those from the in vitro metabolism studies. RAL-NNE was found to be stable in buffers of different pH, and the globule size did not change with different dilutions (Figure 3A). The unchanged globule size of NNE demonstrated the successful preparation of the NE and its stability in the GIT environment.36 After lipolysis of NNE and LNE for 60 min, an equal decrease in RAL content was found in the aqueous phase (Figure 3C), a finding related to the 7% lipolysis of Cremophor RH40 in the NNE formulation. A small amount of RAL was also released from the NNE.37 However when lipolysis was terminated by halting the addition of NaOH and Ca2+ to the lipolytic system, and continuously stirring the system for 8 h, the RAL content of the LNE was significantly reduced from 76.23% to 39.45% (p < 0.01), while that of the NNE in the aqueous phase was only decreased from 77.18% to 70.75%. Such findings indicate that most of the LNE droplets collapsed during the lipolysis period. Also, the much higher consumption of NaOH indicated that the RAL-LNE globules were broken after the lipolysis, whereas those of the RAL-NNE remained intact.
The results of the in vitro metabolism study supported those of in vitro lipolysis. As demonstrated by the in vitro metabolism data (Figure 3D), the significant reduction in the rate of RAL metabolism from the RAL-NNE (< 11%) revealed that RAL was encapsulated in the NNE globule and could avoid metabolism by UGTs. Conversely, the rate of the RAL metabolism was much higher after the lipolysis of the RAL-LNE. This is because the LNE was hydrolyzed by pancreatin and the released RAL was metabolized by UGTs. Therefore, the NNE was deemed stable, and the globule remained intact during the lipolysis and metabolism processes in vitro.
The in vivo Absorption of RAL-NNE
The in vivo oral bioavailability results were found to support the in vitro stability data, thereby confirming that the stability and protection of RAL in the GIT was improved by the NNE. After lipolysis, RAL was rapidly released and the chylomicrons were formed by lipolytic products of LNE and then uptake into the blood or lymphatic system, resulting in a higher Cmax than those of SUSP and NNE.15,38 In contrast, the RAL encapsulated in NNE was slowly released and resulted in the lower Cmax. Although no evident difference between the oral bioavailability of NNE and LNE in rats (Table 3), the bioavailability of NNE in pigs was significantly higher than that of LNE (Table 4). Unlike humans or pigs, rats do not have UGT1A10, which is the most important enzyme in RAL metabolism. UGT1A10 also causes a higher rate of RAL metabolism than UGT1A8 (Figure 4).39,40 Therefore, the protective effect of NNE on RAL was not completely demonstrated or could not be distinguished from that of LNE in rats.
In pigs, the C1max was relatively higher than the C2max of LNE, thereby demonstrating that most RAL was exposed at T1max, and the T2max occurred between 10 and 24 h (Figure 4C). These findings may be related to the saturated absorption of RAL after LNE lipolysis and the massive formation of the RAL precipitate. Unlike RAL-LNE, the C2max of RAL-NNE was significantly higher than its C1max (p < 0.01), which indicated that RAL was encapsulated in the NNE and transported by endocytosis, ultimately delaying T2max.
The prolonged delay of the T2max in pigs may be owing to their anesthetic status after oral administration (Table 4 and Figure 4B). In this study, the persistent movement and struggling by pigs led to persistent diarrhea, which may affect RAL absorption seriously. The oral absorption of RAL in pigs occurred while animals were under the influence of the anesthetic, thereby causing a slow RAL absorption rate in pigs. The slow absorption rate intensified the protective effect of RAL-NNE, thereby prolonging Tmax and causing a slower elimination rate. In addition, the safety of NNE had been checked by the experiment of cell viability (Supplementary Figure 1 (A)), which indicated that the RAL-NNE was safe to enterocytes when the concentration of RAL was <3.75 μg/mL. Thus, oral RAL-NNE administration may not have caused diarrhea in pigs. The findings indicate that oral absorption of RAL in pigs could be significantly improved by NNE because of its intactness when transported via blood circulation. However, whether the improvement in oral absorption induced by NNE was indeed superior to that induced by LNE requires further verification under the normal physiological state.
Different from NNE, LNE had been used to enhance the bioavailability of the RAL and it was improved by 2–4-fold compared with SUSP, as previously reported.41–43 However, although the concept of NNE had been proposed, the NNE never be designed for RAL or other poorly soluble drugs to increase their oral absorption.44 Above all, NNE was designed, through a series of systemic studies, the oral bioavailability of RAL in pigs was significantly enhanced by NNE (5 folds) when compared with RAL-SUSP. Moreover, the AUC of NNE was also significantly higher than that of RAL-LNE (Table 4), indicating that NNE was much stable in the GIT and protected RAL from enzymatic (pancreatin and UGT) metabolism, which showed a better oral absorption on RAL-NNE.
Other interesting results include that the plasma concentration profiles in Figure 4C and D revealed the large inter-individual differences in the absorption of RAL-LNE in pigs. However, only a small difference was found in the absorption profiles of RAL-NNE. Previously, large inter-individual differences were reported to exist for the enhancement of oral bioavailability by NEs.45 By loading vitamin K1, which has variable bioavailability, into self-nanoemulsifying lyophilized tablets, El-Say et al found that the bioavailability of vitamin K1 increased and its inter-individual differences in humans decreased relative to that of commercial tablets post-dosing.46 As the LNE is digested in the GIT, the released RAL may form a precipitate with various components, leading to different in vivo profiles.47 However, when RAL was encapsulated in the NNE, it can be absorbed via a more regular route in humans, thereby reducing the inter-individual differences. Such finding indicates that NNE may be a promising drug delivery tool, withminifying inter-individual differences in humans. However, as the oral absorption was only tested in three pigs, further data are required to support the conclusions. Moreover, owing to the complicated GIT environment, the intestinal disposition of RAL-NNE must be investigated.
The Absorption Mechanisms of RAL-NNE
As shown in Table 5 and Supplementary Figure 1 (B), NNE was uptake by endocytosis which mediated by caveolin and clathrin and that was in agreement with previous research.48–51 The chylomicron flow blocking approach has been used to study the intestinal lymphatic transport of drugs in lipid-based formulations.24,52,53 The AUC0-24 h-AUC0-24 hCHX of the LNE and NNE was 1.54 ± 0.35 h·μg/mL and 2.67 ± 0.33 h·μg/mL, respectively, which confirmed the lymphatic transport of RAL in NE. Compared with the LNE, a small part of the NNE was hydrolyzed by lipase and the rest of NNE was uptake by the way of endocytosis. The higher AUC0-24 h-AUC0-24 hCHX of NNE than that of LNE demonstrated more RAL was transported into lymph by NNE. The inhibition effect of CHX on M cell has been demonstrated by Sun et al.54 Moreover, lymphatic transport of the drugs mainly occurred in the jejunum, such as chylomicron formation.55 These results indicated that the RAL-NNE was absorbed by endocytosis, which was mediated by caveolin and clathrin into the blood system, and by chylomicrons or M cells into the lymphatic system.
Conclusions
In the present study, the RAL-NNE was developed successfully and its in vitro GIT stability and UGT enzyme metabolism demonstrated better stability than the LNE. The in vitro stability data were in agreement with the oral bioavailability results obtained from pigs and the oral absorption of RAL was markedly enhanced by NNE. Through uptake and transport in MDCK cells and in situ SPIP study, RAL-NNE was found to be mainly absorbed via endocytosis in the jejunum. The lymphatic transport of RAL was significantly improved by NNE than that of LNE in rats. In conclusion, NNE is a promising method for enhancing oral administration of BCS class II drugs that undergo significant first-pass intestinal metabolism. Through encapsulation with intact NNE, small drug molecules, such as RAL, and macromolecules, such as insulin, may be protected by the NNE through a multiple emulsion system. Therefore, NNE may have numerous applications in the pharmaceutical field.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Oshima T, Miwa H. Gastrointestinal mucosal barrier function and diseases. J Gastroenterol. 2016;51(8):768–778. doi:10.1007/s00535-016-1207-z
2. Goke K, Lorenz T, Repanas A, et al. Novel strategies for the formulation and processing of poorly water-soluble drugs. Eur J Pharm Biopharm. 2018;126:40–56. doi:10.1016/j.ejpb.2017.05.008
3. Kumar S, Bhargava D, Thakkar A, Arora S. Drug carrier systems for solubility enhancement of BCS class II drugs: a critical review. Crit Rev Ther Drug Carrier Syst. 2013;30(3):217–256. doi:10.1615/CritRevTherDrugCarrierSyst.2013005964
4. Gao F, Zhang Z, Bu H, et al. Nanoemulsion improves the oral absorption of candesartan cilexetil in rats: performance and mechanism. J Control Release. 2011;149(2):168–174. doi:10.1016/j.jconrel.2010.10.013
5. Conway WD, Minatoya H, Lands AM, Shekosky JM. Absorption and elimination profile of isoproterenol. 3. The metabolic fate of dl-isoproterenol-7-3H in the dog. J Pharm Sci. 1968;57(7):1135–1141. doi:10.1002/jps.2600570710
6. Crocker JM, Munday KA. Specific metabolism of testosterone during intestinal transport in the rat. J Endocrinol. 1970;48(4):52–153.
7. Liu YT, Hao HP, Xie HG, et al. Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats. Drug Metab Dispos. 2010;38(10):1779–1784. doi:10.1124/dmd.110.033936
8. Balasubramani S, Rajendhiran T, Moola AK, Diana RKB. Development of nanoemulsion from Vitex negundo L. essential oil and their efficacy of antioxidant, antimicrobial and larvicidal activities (Aedes aegypti L.). Environ Sci Pollut R. 2017;24(17):15125–15133. doi:10.1007/s11356-017-9118-y
9. Kalantzi L, Persson E, Polentarutti B, et al. Canine intestinal contents vs. simulated media for the assessment of solubility of two weak bases in the human small intestinal contents. Pharm Res. 2006;23(6):1373–1381. doi:10.1007/s11095-006-0207-8
10. Lowe ME. The triglyceride lipases of the pancreas. J Lipid Res. 2002;43(12):2007–2016. doi:10.1194/jlr.r200012-jlr200
11. Shiau YF. Mechanisms of intestinal fat absorption. Am J Physiol. 1981;240(1):
12. van Tilbeurgh H, Sarda L, Verger R, Cambillau C. Structure of the pancreatic lipase-procolipase complex. Nature. 1992;359(6391):159–162. doi:10.1038/359159a0
13. Persson EM, Nilsson RG, Hansson GI, et al. A clinical single-pass perfusion investigation of the dynamic in vivo secretory response to a dietary meal in human proximal small intestine. Pharm Res. 2006;23(4):742–751. doi:10.1007/s11095-006-9607-z
14. Salvia-Trujillo L, Qian C, Martin-Belloso O, McClements DJ. Influence of particle size on lipid digestion and beta-carotene bioaccessibility in emulsions and nanoemulsions. Food Chem. 2013;141(2):1472–1480. doi:10.1016/j.foodchem.2013.03.050
15. Ye J, Wu H, Huang C, et al. Comparisons of in vitro Fick’s first law, lipolysis, and in vivo rat models for oral absorption on BCS II drugs in SNEDDS. Int J Nanomed. 2019;14:5623–5636. doi:10.2147/IJN.S203911
16. Thorn HA, Yasin M, Dickinson PA, Lennernas H. Extensive intestinal glucuronidation of raloxifene in vivo in pigs and impact for oral drug delivery. Xenobiotica. 2012;42(9):917–928. doi:10.3109/00498254.2012.683497
17. Teeter JS, Meyerhoff RD. Environmental fate and chemistry of raloxifene hydrochloride. Environ Toxicol Chem. 2002;21(4):729–736. doi:10.1002/etc.5620210407
18. Jeong EJ, Liu Y, Lin H, Hu M. Species- and disposition model-dependent metabolism of raloxifene in gut and liver: role of UGT1A10. Drug Metab Dispos. 2005;33(6):785–794. doi:10.1124/dmd.104.001883
19. Kemp DC, Fan PW, Stevens JC. Characterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearance. Drug Metab Dispos. 2002;30(6):694–700. doi:10.1124/dmd.30.6.694
20. Larsen AT, Ohlsson AG, Polentarutti B, et al. Oral bioavailability of cinnarizine in dogs: relation to SNEDDS droplet size, drug solubility and in vitro precipitation. Eur J Pharm Sci. 2013;48(12):339–350. doi:10.1016/j.ejps.2012.11.004
21. Cummins CL, Salphati L, Reid MJ, Benet LZ. In vivo modulation of intestinal CYP3A metabolism by P-glycoprotein: studies using the rat single-pass intestinal perfusion model. J Pharmacol Exp Ther. 2003;305(1):306–314. doi:10.1124/jpet.102.044719
22. Miao X, Li Y, Wang X, Lee SM, Zheng Y. Transport mechanism of coumarin 6 nanocrystals with two particle sizes in MDCKII monolayer and larval zebrafish. ACS Appl Mater Interfaces. 2016;8(20):12620–12630. doi:10.1021/acsami.6b01680
23. Khatri P, Shao J. Transport of lipid nano-droplets through MDCK epithelial cell monolayer. Colloids Surf B Biointerfaces. 2017;153:237–243. doi:10.1016/j.colsurfb.2017.02.024
24. Dahan A, Hoffman A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur J Pharm Sci. 2005;24(4):381–388. doi:10.1016/j.ejps.2004.12.006
25. Lind ML, Jacobsen J, Holm R, Mullertz A. Intestinal lymphatic transport of halofantrine in rats assessed using a chylomicron flow blocking approach: the influence of polysorbate 60 and 80. Eur J Pharm Sci. 2008;35(3):211–218. doi:10.1016/j.ejps.2008.07.003
26. Zhang Z, Gao F, Bu H, Xiao J, Li Y. Solid lipid nanoparticles loading candesartan cilexetil enhance oral bioavailability: in vitro characteristics and absorption mechanism in rats. Nanomedicine. 2012;8(5):740–747. doi:10.1016/j.nano.2011.08.016
27. Giang TM, Gaucel S, Brestaz P, et al. Dynamic modeling of in vitro lipid digestion: individual fatty acid release and bioaccessibility kinetics. Food Chem. 2016;194:1180–1188. doi:10.1016/j.foodchem.2015.08.125
28. Benito-Gallo P, Franceschetto A, Wong JC, et al. Chain length affects pancreatic lipase activity and the extent and pH-time profile of triglyceride lipolysis. Eur J Pharm Biopharm. 2015;93:353–362. doi:10.1016/j.ejpb.2015.04.027
29. Zhu X, Ye A, Verrier T, Singh H. Free fatty acid profiles of emulsified lipids during in vitro digestion with pancreatic lipase. Food Chem. 2013;139(14):398–404. doi:10.1016/j.foodchem.2012.12.060
30. Brockerhoff H. Substrate specificity of pancreatic lipase. Influence of the structure of fatty acids on the reactivity of esters. Biochim Biophys Acta. 1970;212(1):92–101. doi:10.1016/0005-2744(70)90181-6
31. Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–1598. doi:10.1016/s0959-8049(01)00171-x
32. Alany RG, Tucker IG, Davies NM, Rades T. Characterizing colloidal structures of pseudoternary phase diagrams formed by oil/water/amphiphile systems. Drug Dev Ind Pharm. 2001;27(1):31–38. doi:10.1081/DDC-100000125
33. Khan J, Rades T, Boyd BJ. Lipid-based formulations can enable the model poorly water-soluble weakly basic drug cinnarizine to precipitate in an amorphous-salt form during in vitro digestion. Mol Pharm. 2016;13(11):3783–3793. doi:10.1021/acs.molpharmaceut.6b00594
34. Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60(6):625–637. doi:10.1021/acs.molpharmaceut.6b00594
35. Larsen AT, Ogbonna A, Abu-Rmaileh R, Abrahamsson B, Ostergaard J, Mullertz A. SNEDDS containing poorly water soluble cinnarizine; development and in vitro characterization of dispersion, digestion and solubilization. Pharmaceutics. 2012;4(4):641–665. doi:10.3390/pharmaceutics4040641
36. Anton N, Vandamme TF. Nano-emulsions and micro-emulsions: clarifications of the critical differences. Pharm Res. 2011;28(5):978–985. doi:10.1007/s11095-010-0309-1
37. Cuine JF, McEvoy CL, Charman WN, et al. Evaluation of the impact of surfactant digestion on the bioavailability of danazol after oral administration of lipidic self-emulsifying formulations to dogs. J Pharm Sci. 2008;97(2):995–1012. doi:10.1002/jps.21246
38. Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. doi:10.1038/nrd2197
39. King CD, Rios GR, Green MD, Tephly TR. UDP-glucuronosyltransferases. Curr Drug Metab. 2000;1(2):143–161. doi:10.2174/1389200003339171
40. Vaessen SF, van Lipzig MM, Pieters RH, Krul CA, Wortelboer HM, van de Steeg E. Regional expression levels of drug transporters and metabolizing enzymes along the pig and human intestinal tract and comparison with Caco-2 cells. Drug Metab Dispos. 2017;45(4):353–360. doi:10.1124/dmd.116.072231.41
41. Jain A, Kaur R, Beg S, Kushwah V, Jain S, Singh B. Novel cationic supersaturable nanomicellar systems of raloxifene hydrochloride with enhanced biopharmaceutical attributes. Drug Deliv Transl Res. 2018;8:670–692. doi:10.1007/s13346-018-0514-8.
42. Murthy A, Ravi PR, Kathuria H, Malekar S. Oral bioavailability enhancement of raloxifene with nanostructured lipid carriers. Nanomaterials. 2020;10(6):E1085. doi:10.3390/nano10061085
43. Izgelov D, Cherniakov I, Aldouby Bier G, Domb AJ, Hoffman A. The effect of piperine pro-nano lipospheres on direct intestinal Phase II metabolism: the raloxifene paradigm of enhanced oral bioavailability. Mol Pharm. 2018;15(4):1548–1555. doi:10.1021/acs.molpharmaceut.7b01090
44. Porter CJ, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–691. doi:10.1016/j.addr.2007.10.014
45. McClements DJ. Nanoemulsion-based oral delivery systems for lipophilic bioactive components: nutraceuticals and pharmaceuticals. Ther Deliv. 2013;4(7):841–857. doi:10.4155/tde.13.46
46. El-Say KM, Ahmed TA, Ahmed OAA, Hosny KM, Abd-Allah FI. Self-nanoemulsifying lyophilized tablets for flash oral transmucosal delivery of vitamin K: development and clinical evaluation. J Pharm Sci. 2017;106(9):2447–2456. doi:10.1016/j.xphs.2017.01.001
47. Alskar LC, Keemink J, Johannesson J, Porter CJH, Bergstrom CAS. Impact of drug physicochemical properties on lipolysis-triggered drug supersaturation and precipitation from lipid-based formulations. Mol Pharm. 2018;15(10):4733–4744. doi:10.1021/acs.molpharmaceut.8b00699
48. Banerjee A, Berezhkovskii A, Nossal R. Kinetics of cellular uptake of viruses and nanoparticles via clathrin-mediated endocytosis. Phys Biol. 2016;13(1):016005. doi:10.1088/1478-3975/13/1/016005
49. Harush-Frenkel O, Debotton N, Benita S, Altschuler Y. Targeting of nanoparticles to the clathrin-mediated endocytic pathway. Biochem Biophys Res Commun. 2007;353(1):26–32. doi:10.1016/j.bbrc.2006.11.135
50. Ho YT, Kamm RD, Kah JCY. Influence of protein corona and caveolae-mediated endocytosis on nanoparticle uptake and transcytosis. Nanoscale. 2018;10(26):12386–12397. doi:10.1039/c8nr02393j
51. Ravi PR, Aditya N, Kathuria H, Malekar S, Vats R. Lipid nanoparticles for oral delivery of raloxifene: optimization, stability, in vivo evaluation and uptake mechanism. Eur J Pharm Biopharm. 2014;87(1):114–124. doi:10.1016/j.ejpb.2013.12.015
52. Makwana V, Jain R, Patel K, Nivsarkar M, Joshi A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: elucidation of mechanism of uptake using chylomicron flow blocking approach. Int J Pharm. 2015;495(1):439–446. doi:10.1016/j.ijpharm.2015.09.014
53. Garg B, Beg S, Kaur R, Kumar R, Katare OP, Singh B. Long-chain triglycerides-based self-nanoemulsifying oily formulations (SNEOFs) of darunavir with improved lymphatic targeting potential. J Drug Target. 2018;26(3):252–266. doi:10.1080/1061186X.2017.1365875
54. Sun M, Zhai X, Xue K, et al. Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with oral bioavailabilities in rats. Eur J Pharm Sci. 2011;43(3):132–140. doi:10.1016/j.ejps.2011.04.011
55. Mak KM, Trier JS. Lipoprotein particles in the jejunal mucosa of postnatal developing rats. Anat Rec. 1979;194(4):491–506. doi:10.1002/ar.1091940403
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.