")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
A new look at an old drug: neuroprotective effects and therapeutic potentials of lithium salts
Authors Dell'Osso L, Del Grande C, Gesi C, Carmassi C , Musetti L
Received 16 February 2016
Accepted for publication 22 April 2016
Published 11 July 2016 Volume 2016:12 Pages 1687—1703
DOI https://doi.org/10.2147/NDT.S106479
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Liliana Dell’Osso, Claudia Del Grande, Camilla Gesi, Claudia Carmassi, Laura Musetti
Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy
Abstract: Increasing evidence highlights bipolar disorder as being associated with impaired neurogenesis, cellular plasticity, and resiliency, as well as with cell atrophy or loss in specific brain regions. This has led most recent research to focus on the possible neuroprotective effects of medications, and particularly interesting findings have emerged for lithium. A growing body of evidence from preclinical in vitro and in vivo studies has in fact documented its neuroprotective effects from different insults acting on cellular signaling pathways, both preventing apoptosis and increasing neurotrophins and cell-survival molecules. Furthermore, positive effects of lithium on neurogenesis, brain remodeling, angiogenesis, mesenchymal stem cells functioning, and inflammation have been revealed, with a key role played through the inhibition of the glycogen synthase kinase-3, a serine/threonine kinase implicated in the pathogenesis of many neuropsychiatric disorders. These recent evidences suggest the potential utility of lithium in the treatment of neurodegenerative diseases, neurodevelopmental disorders, and hypoxic–ischemic/traumatic brain injury, with positive results at even lower lithium doses than those traditionally considered to be antimanic. The aim of this review is to briefly summarize the potential benefits of lithium salts on neuroprotection and neuroregeneration, emphasizing preclinical and clinical evidence suggesting new therapeutic potentials of this drug beyond its mood stabilizing properties.
Keywords: bipolar disorder, GSK-3, neurodegeneration, neurogenesis, neurodevelopmental disorders
Introduction
Lithium is still considered a first-line therapy for both the acute and long-term treatment of bipolar disorder (BD),1 due to its well-documented antimanic,2 antisuicidal,3–5 and prophylactic properties6,7 and the adjunctive treatment of major depression.8 Although all potential mechanisms underlying lithium mood-stabilizing properties have not yet been completely clarified, a growing body of evidence has recently suggested the potential benefits on neuroprotection and neuroplasticity of this drug.
There is now evidence that BD is related to progressive impairment in neurocognitive functioning, as well as to regional impairments in neuroplasticity, cellular resilience, and adult neurogenesis.9–15 Morphometric brain-imaging studies, in fact, have documented cell atrophy or loss in several brain areas, especially prefrontal regions (eg, anterior cingulated and subgenual prefrontal cortex), as well as hippocampal volume decrease and smaller amygdalar volumes, in both adult and pediatric BD patients compared with healthy control subjects.16–20 Such regional brain abnormalities have been suggested to play a role in the pathophysiology of BD, giving rise to the debate as to whether they derive from neurodevelopmental, neurodegenerative, or combined deficits.10,21,22
Consistently, most recent research studies have progressively focused on the possible neuroprotective effects of psychotropic medications for BD, and interesting results have particularly emerged for lithium salts.
This review aims to briefly summarize the most recent evidence on lithium benefits beyond its mood-stabilizing properties, with particular attention to its potential effects on neuroprotection and neuroregeneration, in both preclinical and clinical studies, highlighting possible new therapeutic potentials of this old drug.
Methodology
MEDLINE/PubMed (1985–2015) articles published in English were identified using the following keyword combinations: lithium, bipolar disorder, neuroprotection, neurogenesis, neurodegenerative disorders, neurodevelopmental disorders, autism, and glycogen synthase kinase-3 (GSK-3).
Neuroimaging outcomes of lithium treatment in BD
Several neuroimaging studies in BD patients have shown lithium treatment to increase cortical gray matter volumes, especially in the anterior cingulate and in the paralimbic cortices that are implicated in attention, motivation, and emotional modulation, as well as the hippocampus volume.18,19,23–30 The association between lithium treatment and gray matter volume increase has been demonstrated regardless of the mood state, the diagnostic subtype, and presence or absence of concomitant medications.31
However, the nature and mechanisms of this positive association remain to be clarified in terms of whether these may depend on a direct effect of lithium on brain structures, through its action on the biochemical pathways involved in neurogenesis and apoptosis, or whether these are an epiphenomenon of the acute or prophylactic lithium efficacy. In a neuroimaging study using voxel-based morphometry, Hajek et al32 recently demonstrated that the positive association between lithium treatment and increase in hippocampal volume seems to be independent of long-term treatment response, as occurred also in those BD patients on lithium who reported episodes of illness while on treatment. Additional evidence comes from studies using quantitative proton magnetic resonance spectroscopy to measure cortex levels of N-acetyl-aspartate (NAA), a marker of neuronal viability and/or functioning, in BD patients. Chronic lithium treatment was found to increase NAA levels in both BD patients and healthy controls.33,34 In a study comparing two groups of BD patients with a similar illness burden but differing lithium levels of exposure with respect to healthy control subjects, prefrontal NAA levels were found to be lower in patients without or with limited lifetime exposure to lithium compared with those under ongoing lithium treatment or controls.35 Furthermore, these two latter groups of participants did not differ in terms of NAA levels; a negative correlation between prefrontal NAA and duration of illness was found only in the group of patients not exposed or with limited exposure to lithium. This lithium effect on NAA levels was suggested to be related to the expansion of neuropil content, accordingly with the evidence of lithium-induced increase in gray matter volumes.
Since the neuroprotective effects of lithium on brain structures have been shown even in healthy subjects and appear to be independent from prophylactic treatment response, Hajek and Weiner31 recently hypothesized that lithium treatment might help maintain brain health even in patients without BD and could possibly demonstrate disease-modifying properties in neurodegenerative disorders. In this regard, Forlenza et al36 evaluated whether the neurobiological properties of lithium reflect in increased regional brain glucose metabolism with the aid of 18 fluorine-fluoro-2-deoxy-d-glucose positron emission tomography in a sample of 19 older adults with amnestic mild cognitive impairment. The exposure to subtherapeutic levels of lithium carbonate (0.25–0.5 mEq/L) for 4 years in 12 of these patients was associated with a significant reduction in the glucose uptake in several clusters of the cerebellum and in both hippocampi, in comparison with matched controls. These results do not support the hypothesis that lithium might increase regional brain metabolism as a consequence of its neurotrophic effects. However, none of the patients in the sample reported any clinically relevant signs of lithium toxicity. Future studies with appropriate methodology are needed to address the clinical implications of these findings, particularly in light of the potential use of lithium as a disease-modifying treatment approach for certain neurodegenerative disorders.
Neuroprotective effects of lithium: preclinical evidence and molecular mechanisms
A number of preclinical in vitro and in vivo studies have documented a neuroprotective effect of lithium against neuronal injury caused by various noxious insults, mainly acted by preventing apoptosis and increasing neurotrophins excretion.9,13,37–40 Additional neuroprotective mechanisms include the modulation of autophagy and oxidative stress and the upregulation of mitochondrial function.41 Furthermore, lithium appears to reduce proinflammatory status, by modulating inflammatory activity.42–46
In particular, lithium has been shown to inhibit GSK-3α and GSK-β; upregulate neurotrophins and cell survival molecules (eg, B-cell lymphoma 2 [Bcl-2], brain-derived neurotrophic factor [BDNF]/tropomyosin receptor kinase B [TrkB], cyclic adenosine monophosphate-responsive element-binding protein [CREB], heat shock protein 70 [Hsp70], and β-catenin); downregulate proapoptotic activities (eg, excitotoxicity, p53, Bcl-2-associated X protein, caspase, cytochrome c release, β-amyloid peptide production, and tau hyperphosphorylation); inactivate N-methyl-D-aspartate (NMDA) receptors; inhibit inositol monophosphatase (IMP); and activate the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt) cell survival pathway.38,47–56 Such a wide range of intracellular responses involved in the neuroprotective action of lithium may be secondary to its inhibitory effect on two key targets, namely, GSK-3β and IMP. The modification of these intracellular pathways is relevant to the understanding of the pathogenesis of certain neuropsychiatric and neurodegenerative diseases (Figure 1).41
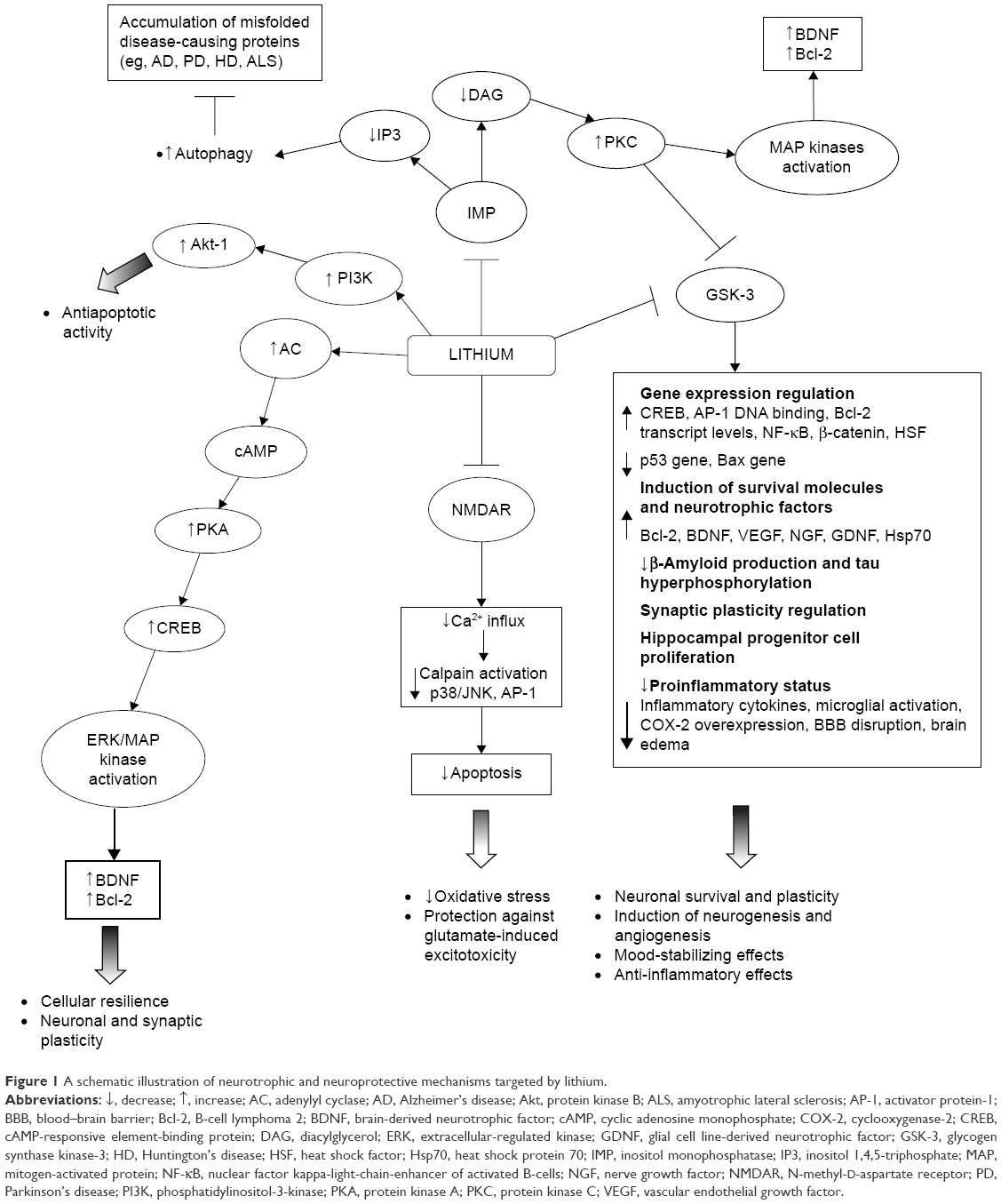 | Figure 1 A schematic illustration of neurotrophic and neuroprotective mechanisms targeted by lithium. |
Lithium inhibition of the GSK-3, both direct binding to the magnesium-sensitive site of the enzyme and indirect, enhancing the phosphorylation of specific N-terminal serine residues, has been demonstrated to play a major role for lithium neuroprotective functions.53,57 There is also evidence of lithium modulation of GSK-3β expression at the transcriptional level in a dose-dependent manner, as a consequence of the inhibition of mRNA transcription.58 The GSK-3 is a serine/threonine kinase that regulates several cellular processes, such as glycogen synthesis, gene transcription, synaptic plasticity, cell apoptosis, cellular structure and resilience, and the circadian cycle.53 Its activation functionally inhibits CREB, β-catenin, and other survival-promoting transcription factors. Conversely, its inactivation has been suggested to promote neuronal survival and to improve cell structural stability directly influencing gene transcription.50,59–61 In addition, GSK-3 directly regulates several neurotransmitter systems, including serotonergic, dopaminergic, cholinergic, and glutamatergic.62,63 Because of changes in these neurotransmitter systems, dysregulated GSK-3 has been linked to depression, BD, and schizophrenia.
The GSK-3 is also implicated in the pathogenesis of many neuropsychiatric disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), spinocerebellar ataxia type 1 (SCA1), multiple sclerosis (MS), fragile X syndrome (FXS), Down syndrome (DS), traumatic brain injury (TBI), and ischemic stroke.40,64–72 It represents the predominant kinase in the brain responsible for the aberrantly hyperphosphorylation of the tau protein and also plays a major role in the amyloid deposition affecting the core pathological features of AD.68,73,74 Therapeutic concentrations of lithium, through the inhibition of the GSK-3, have been demonstrated to significantly lower the levels of the tau phosphorylation in living cells and neurons, thus reducing the levels of aggregated, insoluble tau.75,76 Further, they have been demonstrated to block both the production of amyloid-beta peptides, by interfering with the amyloid precursor protein cleavage, and the accumulation of amyloid-beta peptides in the brain of mice that overproduce amyloid precursor protein.76
Both the downregulation of GSK-3β and the elevated β-catenin, two components of the canonical Wnt signaling pathway, were implicated in adult hippocampal progenitor cell proliferation triggered by lithium treatment.77 The GSK-3 may regulate cognitive functions by influencing the components of synaptic plasticity that are long-term depression (LTD) and long-term potentiation (LTP), involved in the regulation of learning and memory.78,79 This kinase is a key regulator of the balance between pro- and anti-inflammatory cytokine production in both the peripheral and the central nervous system and influences T-cell proliferation, differentiation and survival, so that its inhibition determines anti-inflammatory effects.42 Administration of GSK-3 inhibitors in mice has been shown to control several inflammatory and immune conditions in both the periphery and the central nervous system.42 The inhibition of GSK-3, as well as its genetic inactivation, has also produced antidepressive-like behaviors and strong anti-manic-like effects in a wide range of animal models.80–87 According to this, a deficient inhibition of GSK-3 has been linked to an increased susceptibility to mood disorders.87 The finding that transgenic mice overexpressing β-catenin demonstrate behavioral changes comparable to those observed after lithium administration gives further support to the notion that the therapeutic effects of lithium are mediated by the inhibition of GSK-3 and the consequent increase in β-catenin levels.88
As mentioned earlier, chronic inhibition of GSK-3 may mediate the neuroprotective effects of lithium in the long term by stimulating the gene and protein expression of antiapoptotic factors (eg, CREB, nuclear factor kappa-light-chain-enhancer of activated B-cells [NF-κB], Bcl-2, BDNF, β-catenin).81,89 In particular, preclinical studies in rat brains and human neurons showed chronic lithium administration to induce prominent neuroprotective and neurotrophic proteins, such as Bcl-2 and BDNF.48,51,90,91 The Bcl-2 not only is a major antiapoptotic protein but also stimulates axonal regeneration following injury.55,92 The upregulation of Bcl-2 by lithium has been postulated to account for the neuropil expansion manifesting as an increase in gray matter volume in the human brain.18,55 Interestingly, also the chronic administration of low lithium doses, producing plasma levels of ~0.35 mM, showed to robustly increase Bcl-2 levels in the frontal cortex and hippocampus of rats.93 The BDNF is a neurotrophin playing a critical role in cortical development, synaptic plasticity, neuronal differentiation, and survival.21,94 It has been demonstrated to be involved in the pathophysiology of mental disorders, with decreased peripheral levels being reported during the acute manic, mixed and depressive episodes of BD,95–99 as well as in patients with chronic post-traumatic stress disorder (PTSD)100 or schizophrenia.101 Treatment with therapeutic concentrations of lithium and valproic acid is known to selectively increase the levels of exon IV-containing BDNF mRNA and the activity of BDNF promoter IV,94 as well as to induce the subsequent activation of BDNF receptor (TrkB) in cultured rat cortical neurons.38,51 Consistently, elevated BDNF levels were reported in the hippocampus and in the frontal and temporal cortex of rat brains after chronic treatment with lithium and valproate.90,102–104 Clinical studies showed lithium to restore and even increase BDNF levels in patients with BD,105 and a relationship between lithium prophylactic efficacy and such levels was reported in a sample of euthymic bipolar patients.106 Lithium has also been shown, in in vivo and in vitro preclinical studies, to increase the expression of other neurotrophins involved in neuronal survival and plasticity, such as the nerve growth factor, the glial cell line-derived neurotrophic factor, and the vascular endothelial growth factor (VEGF).107–112
Some evidence demonstrates that lithium produces neurotrophic effects affecting cyclic adenosine monophosphate-mediated signal transduction. This action is mainly exerted elevating basal adenylyl cyclase activity, but also reducing receptor-stimulated responses, as demonstrated in both preclinical and clinical studies.60,113,114 The physiologic effects of cyclic adenosine monophosphate are primarily mediated by the activation of protein kinase A, one of whose major targets in the central nervous system is CREB, a transcription factor that plays a major role in mediating adaptive responses at glutamatergic synapses, as well as in the neuroprotective effects of neurotrophins and in long-term neuroplasticity.60 Indeed, the activation of the extracellular-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) pathway via CREB initiates the transcription of BDNF and also induces Bcl-2 expression. At therapeutically relevant concentrations, both lithium and valproate have been shown to activate the ERK/MAPK cascade in human neuroblastoma SH-SY5Y cells in vitro,115 as well as in hippocampus and frontal cortex areas of the rodent brain,103 thus producing neurotrophic effects.
Experimental studies also found chronic lithium administration to provide nearly complete protection against glutamate-induced, NMDA receptor (NMDAR)-mediated excitotoxicity in primary cerebellar, cerebral cortical, and hippocampal neuronal cultures.52,116,117 Excessive NMDA throughput has been, in fact, hypothesized to be involved in stress-induced hippocampal atrophy and implicated in the pathogenesis of various neurodegenerative diseases.38,116,118 This neuroprotective effect was obtained also with low lithium doses (0.1–0.6 mM) and was associated with the inhibition of NMDAR-mediated calcium influx.38,52 Additional evidence indicates that the lithium mechanism of action results from the attenuation of constitutive phosphorylation of the N-methyl D-aspartate receptor subtype 2B (NR2B) subunit of the NMDAR, which is catalyzed by Fyn, a member of the Src tyrosine kinase family.52,119 Brain ischemia is known to increase Src-mediated tyrosine phosphorylation of NR2A, as well as the interaction of NR2A with Src and Fyn, which is mediated by postsynaptic density protein 95.120,121 Lithium was shown to reduce NR2A phosphorylation and its postsynaptic density protein 95-mediated interactions with Src and Fyn in rat hippocampus following cerebral ischemia.122 Furthermore, chronic lithium treatment antagonizes glutamate-induced activation of c-Jun-N-terminal kinase, p38 kinase, and activator protein-1 transcription factor binding, which has a major role in cytotoxicity, and suppresses glutamate-induced decrease of the CREB phosphorylation in cultured cerebellar granule neurons.38,54
According to the inositol depletion hypothesis,123 the phosphatidylinositol (PI) pathway has also been proposed as a major target through which lithium could exert its therapeutic effects. As with GSK-3, lithium directly inhibits IMP and inositol polyphosphate-1 activity by the competitive displacement of Mg2+ from the catalytic site of the enzyme.56 The inhibition of IMP and inositol polyphosphate-1 prevents the reuptake of inositol, leading to depletion of intracellular levels and subsequent inhibition of the phosphoinositol cycle.41 The decrease in free-myoinositol levels and, subsequently in the production of diacylglycerol induced by the inhibition of IMP, has the downstream effect of decreasing protein kinase C levels and activity.60 The reduction of protein kinase C levels and activity was demonstrated in cells as well as in rodents after chronic lithium treatment, with implication on neuroplastic events.124 Moreover, the inhibition of protein kinase C has been suggested as a mechanism producing antimanic effects.60,125
More recently, the ability of lithium to inhibit the activity of IMP and subsequently decrease inositol 1,4,5-triphosphate levels was identified as a novel mechanism for inducing autophagy.126–129 Autophagy, a physiological process for degrading cytoplasmic proteins or organelles, has been recognized as a principal response to cellular stress and an important regulator of neuronal function and survival.129 It is believed to be particularly beneficial as a “quality control” process in neurodegenerative disorders characterized by the accumulation of misfolded disease-causing proteins (eg, AD, PD, Huntington’s disease [HD], amyotrophic lateral sclerosis [ALS]).130–132
The inhibition of the two main targets of lithium, IMP, and GSK-3β, shows opposite effects on autophagy, that is modulated in a dose-dependent manner: lithium induces inhibition of IMP at lower doses (Ki ≈0.8 mM), enhancing autophagy,126 while it induces inhibition of GSK-3β at higher doses (Ki ≈2 mM), downregulating autophagy via activation of mammalian target of rapamycin (mTOR).118,133
The inhibition of PI3K, a kinase involved in cell survival pathways, has been found to block the activity of the antiapoptotic serine-threonine Akt-1 kinase causing neuronal death, and this apoptotic pathway was prevented by chronic lithium exposure increasing PI3K activity in primary cerebellar neurons.47 Acute lithium exposure also protected cortical neurons via a PI3K-mediated increase in intracellular calcium through the phospholipase Cγ pathway.134 Furthermore, in cultured human neuroblastoma SHSY5Y cells, lithium treatment ameliorated HIV-gp120-mediated toxicity via the PI3K/Akt pathway.135
Recent evidence suggests that lithium treatment enhances the mitochondrial respiratory rate, reduces oxidative stress, protects DNA against damage from oxidative stress, and modulates calcium influx in the mitochondria.60,136–139 Both lithium treatment of acute mania and clinical improvement of depressive symptoms after lithium administration were associated with a reduction of pro-oxidative stress markers.12,140 In addition, lithium showed to affect oxidative stress in healthy subjects increasing antioxidative and reducing pro-oxidative markers.141 Nitric oxide (NO) is one of the most important intracellular second messenger activated by oxidative stress status. NMDAR activation can increase the activation of NO synthase, the enzyme responsible for NO production, and lithium treatment has recently been suggested to regulate this molecular mechanism.142
Besides promoting neuronal survival after damage, lithium has been reported to promote hippocampal neurogenesis. In mammals, new neurons continue to be produced throughout adult life in the subventricular zone and the hippocampal dentate gyrus (DG).143 Compared with preexisting mature dentate granule cells, newborn neurons exhibit peculiar electrophysiological features (ie, increased intrinsic excitability, enhanced synaptic plasticity, reduced sensitivity to GABAergic inhibition, and lower threshold for the induction of LTP) that showed to be essential for hippocampal function in the DG, an area primarily involved in cognitive processes.144–147 Impaired adult neurogenesis has been observed in various models relevant to neuropsychiatric disorders (ie, BD, major depression, schizophrenia, and AD) and neurodevelopmental disorders (ie, FXS and DS).143,148–152 Recent data have correlated low proliferation and differentiation capacities of adult hippocampal stem cells with memory dysfunction,152 suggesting adult neurogenesis might be a promising therapeutic target for disease-associated cognitive impairment. Several preclinical studies showed lithium to stimulate the proliferation and differentiation of neuronal progenitor cells, as well as their maturation and functioning.153–158
Lithium in neurodegenerative diseases
The risk of cognitive impairment and dementia has been reported to be higher in patients with BD than in the general population, correlating with the duration of illness, as well as with the number of illness episodes, especially the depressive ones, and the degree of impairment.159,160 Both preclinical and clinical studies have produced new evidence of lithium benefits in terms of preventing dementia through its action on GSK-3,66,161 leading to its investigation also in neurodegenerative disorders.
In a case–control study that compared AD prevalence rates among elderly euthymic bipolar patients on chronic lithium therapy with respect to those of similar patients who had not been treated with lithium recently, Nunes et al162 showed lithium to be able to reduce AD prevalence to levels comparable with those of the general elderly population. Since the number of previous affective episodes was equivalent between the two patient groups, this putative neuroprotective effect was related to the intrinsic biological brain effects of lithium. Findings from two other observational studies corroborated the reduced risk of AD in BD patients on long-term lithium treatment.163,164 Conversely, this positive effect was not shown in patients taking anticonvulsants, antidepressants, or antipsychotics.164
Evidence of benefits of lithium in preventing cognitive impairment in nonbipolar AD patients is mixed. A small, open-label study found no cognitive benefits of lithium carbonate administration for up to 1 year to 25 patients with mild to moderate AD.165 In a randomized, single-blind, placebo-controlled, 10-week study of 71 patients with a mild form of AD, treatment with lithium at therapeutic levels (0.5–0.8 mmol/L) showed no significant benefits on either cognitive performance or cerebrospinal fluid concentrations of disease-related biomarkers.166 On the contrary, in a subset of a greater sample recruited for a randomized, single-blind, placebo-controlled, parallel-group 10-week multicenter study, investigating the efficacy of lithium treatment in early AD patients, a significant increase of BDNF serum levels was shown.167 In a 12-month, single-center, randomized, double-blind, placebo-controlled study, carried out on subjects with amnestic mild cognitive impairment,168 chronic lithium administration was associated with a significant decrease in cerebrospinal fluid concentrations of P-tau and better performances on the cognitive subscale of the AD assessment scale and in attention tasks. The reduction of the cerebrospinal fluid P-tau was found in subjects with amnestic mild cognitive impairment who did not convert to AD, suggesting that this disease-modifying effect of lithium may be a useful parameter for predicting its property to prevent, or at least to delay, the progression from pre-dementia stage to clinical dementia. Lithium was administered at subtherapeutic levels (serum levels of 0.2–0.4 mmol/L), which resulted safe and well tolerated. In contrast with previous studies that failed to find a significant effect of lithium either on cognition or on AD-related biomarkers,165,166 this study included individuals without a manifest form of dementia. Finally, a more recent intriguing trial found beneficial effects of a lithium microdose of 300 μg administered once daily to AD patients for 15 months in preventing cognitive loss.169 Overall, findings from clinical samples highlight preferential benefits of lithium in dementia prevention in patients with mood disorders and mild cognitive impairment, suggesting that long-term lithium treatment may have disease-modifying properties on the core pathophysiologic features of AD, mostly if started at the earlier stages of the disease process.41,168,170
Lithium inhibition of the GSK-3 has also been hypothesized to produce neuroprotective effects in other neurodegenerative disease models, such as PD, tauopathies, HD, ALS, SCA1, and MS.10,40,41,65,171–173 Whether these pharmacodynamic properties of lithium can be translated into neurodegenerative diseases modifying effects in human subjects is a critical question.174
Several preclinical studies have suggested that lithium can prevent neurodegeneration in vitro and in vivo models of PD.67,175–177 Therapeutic concentrations of lithium have been shown to facilitate clearance of the mutant form of α-synuclein, one of the major components of Lewy bodies, inducing autophagic processes.126,177 Other preliminary data suggest that the antioxidant effects of lithium may be beneficial for PD by preventing free radical damage of dopaminergic neurons.171 In a mouse model of PD, mice were fed with a diet containing 1.1, 2.2, 3.3, and 4.4 g/kg lithium chloride for 4 weeks prior to the injection of the parkinsonism neurotoxin N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).178 The 3.3 g/kg lithium diet gave serum level value very similar to what is observed in lithium therapy in man and the 4.4 g/kg diet gave serum level values well above this. Results of this study showed chronic lithium diet to prevent the decrease of dopamine levels, as well as of tyrosine hydroxylase activity and protein levels in striatum. A direct relationship was established between serum lithium concentration and the prevention of MPTP-induced depletion of striatal dopamine. MPTP-treated mice showed significantly decreased striatal Bcl-2 levels, as opposed to increased Bcl-2-associated X protein levels, and lithium treatment was found to prevent such changes. Chen et al179 demonstrated that 6-hydroxydopamine (6-OHDA), a PD mimetitc, activates GSK-3β in cultured human neuroblastoma SH-SY5Y cells, as well as in cultures of rat cerebellar granule neurons, inducing neuronal death. Lithium and other specific GSK-3β inhibitors showed to protect SH-SY5Y cells and cerebellar granule neurons from 6-OHDA-induced apoptosis. However, other studies have presented conflicting results, showing lithium to fail in the protection of dopaminergic neurons from 6-OHDA-induced degeneration in the substantia nigra of rats.41,180 To date, no clinical trials have been conducted in human patients to test these findings.
HD is a progressive, autosomal dominant neurodegenerative disorder caused by an expanded CAG repeats at exon 1 of the huntingtin (Htt) gene resulting in the aggregate formation of mutant huntingtin protein (mHtt).181 Clinical presentation of HD includes psychiatric disturbances such as significant changes in personality or mood, cognitive decline that may gradually lead to dementia, and motor dysfunction characterized by chorea. Pathological processes and pathways involved in the development of HD include selective death/atrophy of medium-sized spiny neurons within the striatum and, to a lesser extent, loss of neuros in the cortex; dysregulation of cellular autophagy; mitochondrial dysfunction; decreased neurotrophic and growth factor levels; and transcriptional dysregulation and aberrant epigenetic modifications.181 Presently no cure exists to treat or halt the progression of this devastating disease. On the basis of accumulating evidence indicating the promising role of lithium for the treatment of neurodegenerative diseases, its use has been investigated in cellular and animal HD models. Pre- and posttreatment with lithium chloride (2.5–5.0 mM) protected nonneuronal and neuronal cells transfected with mutant Htt exon 1 fragment containing 74 CAG repeats, increased cell survival, and decreased aggregate formation compared with nontreated group in both the cell types.65 In a rat model of HD induced by quinolinic acid (QA) unilateral infusion into the striatum, subcutaneous injection of lithium chloride for 16 days prior to QA infusion significantly reduced the size of the excitotoxicity-induced striatal lesion. The lithium-elicited neuroprotective action was associated with an increase in Bcl-2 protein levels.182 A subsequent study assessed the ability of lithium to reduce neurodegeneration and to stimulate cell proliferation in a rat model of HD induced by QA. LiCl (0.5–3.0 mEq/kg) was injected subcutaneously 24 hours before and 1 hour after QA infusion. Seven days after QA injection, lithium significantly diminished the loss of neurons, prevented apoptosis by attenuating QA-induced DNA damage and caspase-3 activation, upregulated Bcl-2 levels, and promoted neuronal and astroglial progenitor cell proliferation.183 Combined treatment with therapeutic levels of lithium (1.0 mM) and valproate (0.5 mM) was found to be more effective than treatment with either drug alone in upregulating mRNA and protein levels of BDNF, as well as other neuroprotective proteins such as Hsp70 in the striatum and cortex, in two models of transgenic HD mice. Cotreatment with lithium and valproate also more effectively alleviated spontaneous locomotor deficits and depressive-like behaviors and markedly prolonged median survival of transgenic mice, compared with monotreatment.65 Lithium use was also investigated in clinical trials of HD. Two recent clinical studies demonstrated lithium’s mood-stabilizing effects in HD patients of varying ages and duration of illness.184,185 The addition of lithium to tetrabenazine eliminated suicidal symptoms in all patients and improved depressive symptomts.185 Moreover, it prevented the progression of chorea in three of these patients.184 Despite these findings, clinical trials reported no benefits of lithium administration on improving choreiform movements.181 However, it is important to notice that these studies may be inconclusive due to the methodological limitations and small sample size.
Evidence of the neuroprotective effects of lithium from ALS models is inconsistent.186–190 In mouse models of ALS, treatment with either lithium alone or in combination with an antioxidant has been shown to improve motor function and slow down disease progression.189,190 The mainly hypothesized mechanism for such improvement was the stimulation of autophagy by lithium.186,191 Combined treatment of ALS mice with lithium and valproate produced a greater and more consistent effect in delaying the onset of disease symptoms, prolonging the lifespan, and decreasing the neurological deficit scores, in comparison with monotreatment with either drug.191 On the contrary, some preclinical studies reported no benefits of lithium administration in improving disease symptoms.187,188 In a small, 15-month pilot study on randomized ASL patients, a significant effect on disease progression and survival was reported in the lithium plus riluzole group (lithium plasma levels ranging from 0.4 to 0.8 mEq/L) compared with the riluzole only.186 A subsequent randomized, double-blind, placebo-controlled trial showed no benefits of lithium (serum levels maintained between 0.4 and 0.8 mEq/L) plus riluzole combination in slowing down the progression of ALS comparing with riluzole alone.192 More recently, a multicenter, double-blind, randomized, placebo-controlled trial (lithium carbonate in ALS trial) was designed to test the hypothesis that lithium may improve survival rates in ALS.193 Oral doses of lithium carbonate (mean serum levels ranging from 0.4 to 0.8 mmol/L) or placebo were continuously administered for 18 months. Lithium treatment was shown to be safe, but study results did not support any evidence of lithium benefit on survival compared to placebo.194,195
Results from preclinical studies also suggest that lithium may be useful for therapeutic intervention in autoimmune and inflammatory diseases, such as MS, the most common inflammatory demyelinating disease of the central nervous system. Lithium pretreatment at therapeutic doses in experimental autoimmune encephalomyelitis models not only markedly suppressed the clinical symptoms of experimental autoimmune encephalomyelitis but also greatly reduced demyelination, microglia activation, and leukocyte infiltration in the spinal cord.172
Finally, lithium efficacy was tested in a knock-in mouse model of SCA1 (Sca1[154Q/2Q] mice) that replicates many features of the human disease, a dominantly inherited neurodegenerative disorder characterized by progressive motor and cognitive dysfunction.173 In this study, Sca1(154Q/2Q) mice and their wild-type littermates were fed either regular chow or chow that contained 0.2% lithium carbonate. Dietary lithium carbonate supplementation resulted in the improvement of motor coordination, learning, and memory in Sca1(154Q/2Q) mice, both presymptomatically and after symptom onset. Neuropathologically, lithium treatment improved hippocampal dendritic arborization in mutant hippocampal pyramidal neurons.
Lithium in neurodevelopmental disorders
Parallel to research on neurodegenerative disorders, lithium potential neuroprotective functions have been explored in neurodevelopmental disorders, with the strongest evidence emerging in preclinical studies of FXS, the most commonly inherited form of mental retardation and autism. FXS is caused by suppressed expression of the fragile X mental retardation protein,196 an RNA-binding translational regulator important for establishing and maintaining functional neuronal networks.197,198 An altered synaptic plasticity, with pathologically enhanced metabotropic glutamate receptor (mGluR)-dependent LTD in the hippocampal CA1 neurons and deficits in NMDAR-dependent LTP at medial perforant path synapses in the DG, has been detected in mouse models of FXS (FX mice),199–201 suggesting enhanced mGluR signaling to be a major cause of this syndrome.202 According to this, the inhibition of mGluR5 has been shown to reverse several behavioral and morphological phenotypes, including cognitive deficits, in both developing and adult FX mice.203,204 Overall, these findings established a clear link between synaptic dysfunction and learning deficits in FXS.
Furthermore, following the discovery that the GSK-3 activity and the mGluR5 signaling were coordinately elevated in the hippocampus, striatum, and cerebral cortices of FX mice, with the inhibition of mGluR5 leading to the inhibition of GSK-3, research studies investigated this kinase as a therapeutic target for the treatment of FXS.205 According to this, GSK-3 has been identified as a bidirectional regulator of NMDAR-dependent synaptic plasticity, as LTP induction requiring the inhibition of GSK-3β, while GSK-3β activity is required for LTD induction.206,207
Lithium inhibition of GSK-3 was found to revert the altered phenotypes of FX mice, that is, to attenuate enhanced mGluR-mediated LTD, to ameliorate locomotor hyperactivity, audiogenic seizure hypersensitivity, increased spine density, excess protein synthesis, social behavior deficits, deficient passive avoidance learning and synaptic plasticity, and to normalize impaired hippocampus-dependent cognitive tasks.205,208–214 To the best of our knowledge, only one small open-label trial has been conducted in FXS patients,215 showing lithium treatment titrated to levels of 0.8–1.2 mEq/L to improve cognitive performance modifying the disrupted regulation of group 1 metabotropic glutamate receptor (mGluR and mGluR5)-dependent translation in dendrites.
Neuropathological alterations have also been described in autism spectrum disorders (ASDs). A preclinical study on neonatal rats isolated from their mother showed apparent autistic-like symptoms, such as social deficits, excessive repetitive self-grooming behavior, and increased anxiety- and depressive-like behaviors that were accompanied by impaired adult hippocampal neurogenesis, and enhanced basal inhibitory synaptic transmission in the hippocampal CA1 pyramidal neurons.216 Chronic lithium administration was effective in ameliorating these pathological changes, completely overcoming autistic-like behaviors, and restoring adult hippocampal neurogenesis and the balance between excitatory and inhibitory synaptic activities to physiological levels, supporting the use of lithium as a potential therapeutic agent against ASDs. Interestingly, two recent case studies described the effects of lithium treatment in two patients diagnosed with ASDs in childhood and presented regression with progressive loss of verbal and motor skills, catatonic features, aggressive and impulsive behaviors, and sleep disturbances after a stressful event during adolescence.217 They both presented a mutation/microdeletion of the SHANK3 gene on chromosome 22q13.3 that encodes a scaffold protein located at the glutamatergic synapses and involved in the regulation of the structural organization of dendritic spines and binding partner of neuroligins, accounting for approximately 2% of cases of intellectual disability in ASDs patients.218 After deletion of SHANK3 gene, the nervous system is thought to become more susceptible to developmental problems and psychiatric disorders, less able to recover after psychiatric and somatic events and more vulnerable to degeneration in the long term.219 Various psychotropic medications administered to the two patients (antipsychotics, benzodiazepines, antiepileptic drugs, antidepressants, and methylphenidate) failed to improve clinical symptoms and led to multiple side effects. In contrast, lithium therapy permitted reversibility of regression symptoms and catatonic features and the stabilization of behavioral disorders without significant side effects,217 suggesting that SHANK3 patients with ASDs may represent a more vulnerable subgroup with an atypical form of BD,219 in agreement with previous observations.220,221
Finally, lithium use has been explored in preclinical models of DS. DS is the leading genetic cause of intellectual disability and has been associated to impaired hippocampal neurogenesis. This latter plays a key role in establishing functional neuronal network and synaptic connectivity. The altered synaptic plasticity (eg, excitation/inhibition imbalance) discovered in mouse models of DS has been implicated in cognitive dysfunctions.143,222–224 Recent studies showed that 1-month administration of lithium in the Ts65Dn mouse, a model of DS, was able to restore adult neurogenesis in the DG and subventricular zone to physiological levels by stimulating the proliferation of neuronal precursor cells through the pharmacological activation of the Wnt/β-catenin pathway.143,153 The restoration of adult neurogenesis completely rescued the synaptic plasticity of newborn neurons in the DG and led to the full recovery of behavioral performance in three distinct hippocampal-dependent cognitive tasks (contextual fear conditioning, object location, and novel object recognition).143
Lithium in cerebral traumatic/ischemic injury
Emerging evidence from preclinical research suggests that lithium can mitigate neurological deficits incurred from traumatic or ischemic brain injury.40,70
The pathophysiology of TBI, after an initial mechanical injury that damages neurons, glia, and vascular structures, includes a cascade of secondary events that finally lead to neurodegeneration and loss of neurological functions: oxidative stress; excitotoxicity via excess glutamate release; increased NMDAR activation; neuroinflammation via proinflammatory cytokines, NO, or prostaglandins; mitochondrial disruption; failure of the blood–brain barrier (BBB) involving cerebral edema, hypoxia, and ischemia; and cellular death via necrosis and caspase-dependent (caspase-3) and/or caspase-independent apoptosis.70,225,226 Neuropsychiatric sequelae such as depression, anxiety, and PTSD and behavioral and cognitive deficits accompany secondary injuries.70 Patients afflicted by TBI show increased memory impairment, and increasing evidence suggests that TBI is a major risk factor for the development of AD.227–229
Given the neuroprotective effects of lithium, recent research has investigated its putative use in preclinical studies on TBI paradigms.70 Lithium pretreatment was found to attenuate interleukin-1β expression, brain edema, hippocampal neurodegeneration, and loss of hemispheric tissues, improve memory and spatial learning, and alleviate depressive behaviors in mouse models of mild TBI.230,231 Postinjury injections with therapeutic doses of lithium also reduced lesion volume and attenuated TBI-induced neuroinflammation by inhibiting microglia activation and cyclooxygenase-2 induction, while BBB integrity was maintained through the inhibition of matrix metallopeptidase-9 expression. TBI-induced hyperlocomotor activity, anxiety-like behaviors, and impairments in motor coordination were all normalized by lithium treatment. In addition, GSK-3β phosphorylation was found to be robustly increased after postinjury administration of lithium, with subsequent β-catenin accumulation, and reduced neuronal loss in the hippocampal CA3 region, as well as decreased hippocampal-dependent deficits in learning and memory.70,232,233
Similarly, GSK-3β has been strongly implicated in the pathogenesis of neuronal death caused by ischemic injury. While GSK-3β, in preclinical models of transient focal cerebral ischemia, was found to be activated inducing apoptotic cell death, its inactivation shortly after permanent focal or global cerebral ischemia was found to promote survival of vulnerable neurons, thus identifying GSK-3β inhibition as a major target for treatments.49
Pretreatment with lithium and valproate was found to attenuate the hypoxia-induced serine dephosphorylation of GSK-3α and GSK-3β in mouse cerebral cortex, hippocampus, and striatum.234 Consistently, in a model of neonatal rat hypoxic–ischemic brain injury, postinsult treatment with lithium therapeutic doses reduced the ischemia-induced dephosphorylation of GSK-3β and the extracellular signal-regulated kinase, the activation of calpain and caspase-3, the mitochondrial release of cytochrome c and apoptosis-inducing factor, as well as autophagy.128 Lithium inhibition of GSK-3β is also accompanied by the upregulation of Bcl-2, increase in the DNA-binding activity of heat shock factor-1 to the heat-shock element, thus superinducing Hsp70, downregulation of p53, and decreased expression of Bcl-2-associated X protein in rat models of stroke.235–238 These findings highlight the prominent role of GSK-3β inhibition in the antiapoptotic effects of lithium under ischemic conditions.
Besides the inhibition of GSK-3, lithium showed to provide neuroprotection in rat hippocampus following cerebral ischemia through the downregulation of NMDAR function, overstimulated as a consequence of the glutamate extracellular accumulation.122
Long-term lithium pretreatment at therapeutically relevant doses decreased brain infarct volume and apoptotic cell death and improved neurological deficits in a permanent rat model of brain focal ischemia.238,239 Moreover, it prevented ischemia-induced exploratory behavioral changes and memory impairments in an animal model of global cerebral ischemia.235 These benefits were associated with an increase in the number of viable cells and a decrease in apoptotic cells in the CA1 hippocampal area of ischemic brain. Infarct volume, neurological deficits, and the number of neurons showing DNA damage in the ischemic brain were also reduced by postinsult subcutaneous injection of lithium at 0.5 mEq/kg in a rat ischemia/reperfusion model.237
Lithium has been demonstrated to inhibit neuroinflammation resulting from ischemic brain injury by preventing inflammatory disorder of the hematoencephalic barrier and neurodegeneration.233 Long-term lithium treatment of neonatal rats subjected to hypoxic–ischemic brain injury significantly reduced total tissue loss, at least partly by inhibiting microglia activation and attenuating proinflammatory cytokines (ie, interleukin-1β) or chemokine levels.157 Overexpression of Hsp70, leading to the inactivation of the inflammatory transcription factor NF-κB,240 has been proposed as a possible mechanism by which lithium exerts its anti-inflammatory activity in experimental stroke models.
The stimulation of hippocampal neurogenesis has also been implicated in the neuroprotective effect of lithium against brain ischemia.157,158 In addition, lithium treatment showed to improve stroke recovery in rats by augmenting neurovascular remodeling and vascular neoformation.241 Specifically, it was found to increase the VEGF protein levels via a mechanism involving the PI3-kinase and GSK-3 signaling pathways in cultured rat brain endothelial cells.109 Since VEGF has been linked to angiogenesis, neurogenesis, and neuroprotection,242 this growth factor signaling mechanism may contribute to lithium’s reported ability to promote neurovascular remodeling after ischemic stroke.
Finally, studies on mesenchymal stem cells in rat models of brain ischemia showed that incubation with lithium chloride increased their migratory ability and homing to damaged tissues, reduced infarct volume, enhanced regenerative potential and angiogenesis in the infarcted penumbra regions, and improved functional recovery.243,244
Conclusion
Increasing evidence highlights BD, as well as other neuropsychiatric conditions, to be associated with morphological brain abnormalities, such as regional impairments in neuroplasticity, cellular resilience, and hippocampal neurogenesis, which highlight the role of neurodegeneration in BD. This new perspective has induced most recent research to focus on the possible neuroprotective effects of psychotropic medications, particularly those whose efficacy in the treatment of BD has been corroborated.9–11,13–15,20,245 Consistently, over the past few years, increasing promising results have emerged for lithium salts, a medication historically used to treat BD.
Since the GSK-3, the main target of lithium, plays a key role in the pathogenesis of many neuropsychiatric disorders, the therapeutic potentials of lithium have been investigated in various preclinical paradigms of different neurodegenerative diseases associated with cell atrophy/loss and impairment of cellular resilience, as well as, most recently, of neurodevelopmental disorders.49,117,246–248 There is now evidence that the neuroprotective benefits of this agent in the human brain are exerted through several complementary mechanisms leading to increased neuronal viability/function, enhanced angiogenesis and neurogenesis, BBB integrity maintenance, and anti-inflammation as well as disease-specific neuroprotective properties.10 Thus, the findings reported in the present review provide intriguing support for the neurotrophic/neuroprotective effects of chronic treatment with lithium, suggesting possible implications for its use in other neuropsychiatric conditions, including those neurodevelopmental disorders, such as ASD, recently suggested as the possible common vulnerability factor for most mental disorders.249,250 The encouraging results reported so far suggest the need for further studies exploring the new therapeutic potentials of this old drug in clinical samples.
Acknowledgment
We thank Giulia Gray for the English language text revision.
Disclosure
The authors report no conflicts of interest in this work.
References
Yatham LN, Kennedy SH, Parikh SV, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2013. Bipolar Disord. 2013;15(1):1–44. | ||
Yildiz A, Vieta E, Leucht S, Baldessarini RJ. Efficacy of antimanic treatments: meta-analysis of randomized, controlled trials. Neuropsychopharmacology. 2011;36(2):375–389. | ||
Baldessarini RJ, Tondo L, Davis P, Pompili M, Goodwin FK, Hennen J. Decreased risk of suicides and attempts during long-term lithium treatment: a meta-analytic review. Bipolar Disord. 2006;8(5 Pt 2):625–639. | ||
Cipriani A, Pretty H, Hawton K, Geddes JR. Lithium in the prevention of suicidal behavior and all-cause mortality in patients with mood disorders: a systematic review of randomized trials. Am J Psychiatry. 2005;162(10):1805–1819. | ||
Cipriani A, Hawton K, Stockton S, Geddes JR. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMC. 2013;346:f3646. | ||
Geddes JR, Burgess S, Hawton K, Jamison K, Goodwin GM. Long-term lithium therapy for bipolar disorder: systematic review and meta-analysis of randomized controlled trials. Am J Psychiatry. 2004;161(2):217–222. | ||
Grof P, Müller-Oerlinghausen B. A critical appraisal of lithium’s efficacy and effectiveness: the last 60 years. Bipolar Disord. 2009;11(Suppl 2):S10–S19. | ||
Nivoli AM, Colom F, Murru A, et al. New treatment guidelines for acute bipolar depression: a systematic review. J Affect Disord. 2011;129(1–3):14–26. | ||
Bauer M, Alda M, Priller J, Young LT; International Group for the Study of Lithium Treated Patients (IGSLI). Implications of the neuroprotective effects of lithium for the treatment of bipolar and neurodegenerative disorders. Pharmacopsychiatry. 2003;36(Suppl 3):S250–S254. | ||
Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65(1):105–142. | ||
Eriksson PS, Perfilieva E, Björk-Eriksson T, et al. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4(11):1313–1317. | ||
Duman RS. Depression: a case of neuronal life and death? Biol Psychiatry. 2004;56(3):140–145. | ||
Machado-Vieira R, Manji HK, Zarate CA Jr. The role of lithium in the treatment of bipolar disorder: convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord. 2009;11(Suppl 2):S92–S109. | ||
Machado-Vieira R, Soeiro-De-Souza MG, Richards EM, Teixeira AL, Zarate CA Jr. Multiple levels of impaired neural plasticity and cellular resilience in bipolar disorder: developing treatments using an integrated translational approach. World J Biol Psychiatry. 2014;15(2):84–95. | ||
Zarate CA Jr, Singh J, Manji HK. Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry. 2006;59(11):1006–1020. | ||
Chang K, Karchemskiy A, Barnea-Goraly N, Garrett A, Simeonova DI, Reiss A. Reduced amygdalar gray matter volume in familial pediatric bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2005;44(6):565–573. | ||
Drevets WC, Price JL, Simpson JR, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386(6627):824–827. | ||
Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK. Lithium-induced increase in human brain grey matter. Lancet. 2000;356(9237):1241–1242. | ||
Sassi RB, Brambilla P, Hatch JP, et al. Reduced left anterior cingulate volumes in untreated bipolar patients. Biol Psychiatry. 2004;56(7):467–475. | ||
Selvaraj S, Arnone D, Job D, et al. Grey matter differences in bipolar disorder: a meta-analysis of voxel-based morphometry studies. Bipolar Disord. 2012;14(2):135–145. | ||
Angelucci F, Brenè S, Mathé AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry. 2005;10(4):345–352. | ||
Monkul ES, Malhi GS, Soares JC. Anatomical MRI abnormalities in bipolar disorder: do they exist and do they progress? Aust N Z J Psychiatry. 2005;39(4):222–226. | ||
Bearden CE, Thompson PM, Dalwani M, et al. Greater cortical gray matter density in lithium-treated patients with bipolar disorder. Biol Psychiatry. 2007;62(1):7–16. | ||
Drevets WC. Neuroimaging and neuropathological studies of depression: implications for the cognitive emotional features of mood disorders. Cur Opin Neurobiol. 2001;11(2):240–249. | ||
Hajek T, Kopecek M, Höschl C, Alda M. Smaller hippocampal volumes in patients with bipolar disorder are masked by exposure to lithium: a meta-analysis. J Psychiatry Neurosci. 2012;37(5):333–343. | ||
Hajek T, Cullis J, Novak T, et al. Hippocampal volumes in bipolar disorders: opposing effects of illness burden and lithium treatment. Bipolar Disord. 2012;14(3):261–270. | ||
Lyoo IK, Dager SR, Kim JE, et al. Lithium-induced gray matter volume increase as a neural correlate of treatment response in bipolar disorder: a longitudinal brain imaging study. Neuropsychopharmacology. 2010;35(8):1743–1750. | ||
Moore GJ, Cortese BM, Glitz DA, et al. A longitudinal study of the effects of lithium treatment on prefrontal and subgenual prefrontal gray matter volume in treatment-responsive bipolar disorder patients. J Clin Psychiatry. 2009;70(5):699–705. | ||
Yucel K, McKinnon MC, Taylor VH, et al. Bilateral hippocampal volume increases after long-term lithium treatment in patients with bipolar disorder: a longitudinal MRI study. Psychopharmacology (Berl). 2007;195(3):357–367. | ||
Yucel K, Taylor VH, McKinnon MC, et al. Bilateral hippocampal volume increase in patients with bipolar disorder and short-term lithium treatment. Neuropsychopharmacology. 2008;33(2):361–367. | ||
Hajek T, Weiner MW. Neuroprotective effects of lithium in human brain? Food for thought. Curr Alzheimer Res. Epub 2016 Feb 18. | ||
Hajek T, Bauer M, Simhandl C, et al. Neuroprotective effect of lithium on hippocampal volumes in bipolar disorder independent of long-term treatment response. Psychol Med. 2014;44(3):507–517. | ||
Moore GJ, Bebchuk JM, Hasanat K. Lithium increases N-acetyl-aspartate in the human brain: in vivo evidence in support of bcl-2’s neurotrophic effects? Biol Psychiatry. 2000;48(1):1–8. | ||
Silverstone PH, Wu RH, O’Donnell T, Ulrich M, Asghar SJ, Hanstock CC. Chronic treatment with lithium, but not sodium valproate, increases cortical N-acetyl-aspartate concentrations in euthymic bipolar patients. Int Clin Psychopharmacol. 2003;8(2):73–79. | ||
Hajek T, Bauer M, Pfennig A, et al. Large positive effect of lithium on prefrontal cortex N-acetylaspartate in patients with bipolar disorder: 2-centre study. J Psychiatry Neurosci. 2012;37(3):185–192. | ||
Forlenza OV, Coutinho AM, Aprahamian I, et al. Long-term lithium treatment reduces glucose metabolism in the cerebellum and hippocampus of nondemented older adults: an [18F]FDG-PET study. ACS Chem Neurosci. 2014;5(6):484–489. | ||
Camins A, Verdaguer E, Junyent F, et al. Potential mechanisms involved in the prevention of neurodegenerative diseases by lithium. CNS Neurosci Ther. 2009;15(4):333–344. | ||
Chuang DM. Neuroprotective and neurotrophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases? Crit Rev Neurobiol. 2004;16(1–2):83–90. | ||
Plotnikov EY, Silachev DN, Zorova LD, et al. Lithium salts – simple but magic. Biochemistry (Mosc). 2014;79(8):740–749. | ||
Wada A, Yokoo H, Yanagita T, Kobayashi H. Lithium: potential therapeutics against acute brain injuries and chronic neurodegenerative diseases. J Pharmacol Sci. 2005;99(4):307–321. | ||
Forlenza OV, De-Paula VJ, Diniz BS. Neuroprotective effects of lithium: implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem Neurosci. 2014;5(6):443–450. | ||
Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010;31(1):24–31. | ||
Bosetti F, Rintala J, Seemann R, et al. Chronic lithium downregulates cyclooxygenase-2 activity and prostaglandin E(2) concentration in rat brain. Mol Psychiatry. 2002;7(8):845–850. | ||
Goldstein BI, Kemp DE, Soczynska JK, McIntyre RS. Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J Clin Psychiatry. 2009;70(8):1078–1090. | ||
Nahman S, Belmaker RH, Azab AN. Effects of lithium on lipopolysaccharide-induced inflammation in rat primary glia cells. Innate Immun. 2012;18(3):447–458. | ||
Rapaport MH, Manji HK. The effects of lithium on ex vivo cytokine production. Biol Psychiatry. 2001;50(3):217–224. | ||
Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamateinduced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci U S A. 1999;96(15):8745–8750. | ||
Chen RW, Chuang DM. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression: a prominent role in neuroprotection against excitotoxicity. J Biol Chem. 1999;274(10):6039–6042. | ||
Chuang DM, Wang Z, Chiu CT. GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front Mol Neurosci. 2011;4:15. | ||
Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog Neurobiol. 2001;65(4):391–426. | ||
Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang DM. Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology. 2002;43(7):1173–1179. | ||
Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem. 2002;80(4):589–597. | ||
Jope RS. Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Biochem Sci. 2003;24(9):441–443. | ||
Kopnisky KL, Chalecka-Franaszek E, Gonzalez-Zulueta M, Chuang DM. Chronic lithium treatment antagonizes glutamate-induced decrease of phosphorylated CREB in neurons via reducing protein phosphatase 1 and increasing MEK activities. Neuroscience. 2003;116(2):425–435. | ||
Manji HK, Moore GJ, Chen G. Lithium up-regulates the cytoprotective protein Bcl-2 in the CNS in vivo: a role for neurotrophic and neuroprotective effects in manic depressive illness. J Clin Psychiatry. 2000;61(Suppl 9):S82–S96. | ||
Patel S, Yenush L, Rodríguez PL, Serrano R, Blundell TL. Crystal structure of an enzyme displaying both inositol-polyphosphate-1-phosphatase and 3′-phosphoadenosine-5′-phosphate phosphatase activities: a novel target of lithium therapy. J Mol Bio. 2002;315(4):677–685. | ||
Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93(16):8455–8459. | ||
Mendes CT, Mury FB, de Sá Moreira E, et al. Lithium reduces Gsk3b mRNA levels: implications for Alzheimer Disease. Eur Arch Psychiatry Clin Neurosci. 2009;259(1):16–22. | ||
Liang MH, Chuang DM. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem. 2007;282(6):3904–3917. | ||
Quiroz JA, Machado-Vieira R, Zarate CA Jr, Manji HK. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010;62(1):50–60. | ||
Chin PC, Majdzadeh N, D’Mello SR. Inhibition of GSK3beta is a common event in neuroprotection by different survival factors. Brain Res Mol Brain Res. 2005;137(1–2):193–201. | ||
Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7(11):1421–1434. | ||
Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. | ||
Aghdam SY, Barger SW. Glycogen synthase kinase-3 in neurodegeneration and neuroprotection: lessons from lithium. Curr Alzheimer Res. 2007;4(1):21–31. | ||
Carmichael J, Sugars KL, Bao YP, Rubinsztein DC. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J Biol Chem. 2002;277(37):33791–33798. | ||
Diniz BS, Machado-Vieira R, Forlenza OV. Lithium and neuroprotection: translational evidence and implications for the treatment of neuropsychiatric disorders. Neuropsychiatr Dis Treat. 2013;9:493–500. | ||
Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3beta-catalyzed tau phosphorylation in Parkinson’s disease models. FASEB J. 2009;23(9):2820–2830. | ||
Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem. 2008;104(6):1433–1439. | ||
Koh SH, Yoo AR, Chang DI, Hwang SJ, Kim SH. Inhibition of GSK-3 reduces infarct volume and improves neurobehavioral functions. Biochem Biophys Res Commun. 2008;371(4):894–899. | ||
Leeds PR, Yu F, Wang Z, et al. A new avenue for lithium: intervention in traumatic brain injury. ACS Chem Neurosci. 2014;5(6):422–433. | ||
Li DW, Liu ZQ, Chen W, Yao M, Li GR. Association of glycogen synthase kinase-3β with Parkinson’s disease (review). Mol Med Rep. 2014;9(6):2043–2050. | ||
King MK, Pardo M, Cheng Y, Downey K, Jope RS, Beurel E. Glycogen synthase kinase-3 inhibitors: rescuers of cognitive impairments. Pharmacol Ther. 2014;141(1):1–12. | ||
Muyllaert D, Kremer A, Jaworski T, et al. Glycogen synthase kinase-3b, or a link between amyloid and tau pathology? Genes Brain Behav. 2008;7(Suppl 1):S7–S66. | ||
Phiel CJ, Wilson CA, Lee VMY, Klein PS. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature. 2003;423(6938):435–439. | ||
Lovestone S, Davis DR, Webster MT, et al. Lithium reduces tau phosphorylation: effects in living cells and in neurons at therapeutic concentrations. Biol Psychiatry. 1999;45(8):995–1003. | ||
Noble W, Planel E, Zehr C, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102(19):6990–6995. | ||
Wexler EM, Geschwind DH, Palmer TD. Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol Psychiatry. 2008;13(3):285–292. | ||
Bradley CA, Peineau S, Taghibiglou C, et al. A pivotal role of GSK-3 in synaptic plasticity. Front Mol Neurosci. 2012;5:13. | ||
Hooper C, Markevich V, Plattner F, et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci. 2007;25(1):81–86. | ||
Beaulieu JM, Zhang X, Rodriguiz RM, et al. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A. 2008;105(4):1333–1338. | ||
Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on betacatenin in mouse hippocampus. Biol Psychiatry. 2004;55(8):781–784. | ||
Kalinichev M, Dawson LA. Evidence for antimanic efficacy of glycogen synthase kinase-3 (GSK3) inhibitors in a strain-specific model of acute mania. Int J Neuropsychopharmacol. 2011;14(8):1051–1067. | ||
Rowe MK, Wiest C, Chuang DM. GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci Biobehav Rev. 2007;31(6):920–931. | ||
Beaulieu JM, Sotnikova TD, Yao WD, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101(14):5099–5104. | ||
Gould TD, Einat H, Bhat R, Manji HK. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacol. 2004;7(4):387–390. | ||
Prickaerts J, Moechars D, Cryns K, et al. Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci. 2006;26(35):9022–9029. | ||
Kaidanovich-Beilin O, Lipina TV, Takao K, et al. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol Brain. 2009;2:35. | ||
Gould TD, Einat H, O’Donnell KC, et al. Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology. 2007;32(10):2173–2183. | ||
Gould TD, Chen G, Manji HK. In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology. 2004;29(1):32–38. | ||
Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology. 2001;158(1):100–106. | ||
Chen G, Zeng WZ, Yuan PX, et al. The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J Neurochem. 1999;72(2):879–882. | ||
Huang X, Wu D-Y, Chen G, Manji H, Chen DF. Support of retinal ganglion cell survival and axon regeneration by lithium through a Bcl-2-dependent mechanism. Invest Ophthalmol Vis Sci. 2003;44(1):347–354. | ||
Goodwin FK, Jamison KR. Manic-Depressive Illness: Bipolar and Recurrent Unipolar Disorders. 2nd ed. New York: Oxford University Press; 2007. | ||
Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14(1):51–59. | ||
Dell’Osso L, Del Debbio A, Veltri A, et al. Associations between brain-derived neurotrophic factor plasma levels and severity of the illness, recurrence and symptoms in depressed patients. Neuropsychobiology. 2010;62(4):207–212. | ||
Machado-Vieira R, Dietrich MO, Leke R, et al. Decreased plasma brain derived neurotrophic factor levels in unmedicated bipolar patients during manic episode. Biol Psychiatry. 2007;61(2):142–144. | ||
Piccinni A, Marazziti D, Catena M, et al. Plasma and serum brain-derived neurotrophic factor (BDNF) in depressed patients during 1 year of antidepressant treatments. J Affect Disord. 2008;105(1–3):279–283. | ||
Piccinni A, Veltri A, Costanzo D, et al. Decreased plasma levels of brain-derived neurotrophic factor (BDNF) during mixed episodes of bipolar disorder. J Affect Disord. 2015;171:167–170. | ||
Post RM. Role of BDNF in bipolar and unipolar disorder: clinical and theoretical implications. J Psychiatr Res. 2007;41(12):979–990. | ||
Dell’Osso L, Carmassi C, Del Debbio A, et al. Brain-derived neurotrophic factor plasma levels in patients suffering from post-traumatic stress disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(5):899–902. | ||
Fernandes BS, Steiner J, Berk M, et al. Peripheral brain-derived neurotrophic factor in schizophrenia and the role of antipsychotics: meta-analysis and implications. Mol Psychiatry. 2015;20(9):1108–1119. | ||
Frey BN, Andreazza AC, Cereser KM, et al. Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci. 2006;79(3):281–286. | ||
Einat H, Yuan P, Gould TD, et al. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci. 2003;23(19):7311–7316. | ||
Jacobsen JP, Mørk A. The effect of escitalopram, desipramine, electroconvulsive seizures and lithium on brain-derived neurotrophic factor mRNA and protein expression in the rat brain and the correlation to 5-HT and 5-HIAA levels. Brain Res. 2004;1024(1–2):183–192. | ||
de Sousa RT, van de Bilt MT, Diniz BS, et al. Lithium increases plasma brain-derived neurotrophic factor in acute bipolar mania: a preliminary 4-week study. Neurosci Lett. 2011;494(1):54–56. | ||
Suwalska A, Sobieska M, Rybakowski JK. Serum brain-derived neurotrophic factor in euthymic bipolar patients on prophylactic lithium therapy. Neuropsychobiology. 2010;62(4):229–234. | ||
Angelucci F, Aloe L, Jiménez-Vasquez P, Mathé AA. Lithium treatment alters brain concentrations of nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor in a rat model of depression. Int J Neuropsychopharmacol. 2003;6(3):225–231. | ||
Frey BN, Andreazza AC, Rosa AR, et al. Lithium increases nerve growth factor levels in the rat hippocampus in an animal model of mania. Behav Pharmacol. 2006;17(4):311–318. | ||
Guo S, Arai K, Strins MF, Chuang DM, Lo EH. Lithium upregulates vascular endothelial growth factor in brain endothelial cells and astrocytes. Stroke. 2009;40(2):652–655. | ||
Hellweg R, Lang UE, Nagel M, Baumgartner A. Subchronic treatment with lithium increases nerve growth factor content in distinct brain regions of adult rats. Mol Psychiatry. 2002;7(6):604–608. | ||
Silva R, Martins L, Longatto-Filho A, et al. Lithium prevents stress-induced reduction of vascular endothelium growth factor levels. Neurosci Lett. 2007;429(1):33–38. | ||
Warner-Schmidt JL, Duman RS. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14–19. | ||
Jope RS. A bimodal model of the mechanism of action of lithium. Mol Psychiatry. 1999;4(1):21–25. | ||
Gould TD, Chen G, Manji HK. Mood stabilizer psychopharmacology. Clin Neurosci. 2002;2(3–4):193–212. | ||
Yuan PX, Huang LD, Jiang YM, Gutkind JS, Manji HK, Chen G. The mood stabilizer valproic acid activates mitogen-activated protein kinases and promotes neurite growth. J Biol Chem. 2001;276(34):31674–31683. | ||
Nonaka S, Hough CJ, Chuang DM. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc Natl Acad Sci U S A. 1998;95(5):2642–2647. | ||
Chuang DM, Priller J. Potential use of lithium in neurodegenerative disorders. In: Bauer M, Grof P, Muller-Oerlinghausen B, editors. Lithium in Neuropsychiatry: The Comprehensive Guide. Abingdon, Oxon: Informa UK Ltd; 2006:381–398. | ||
Chiu CT, Chuang DM. Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol Ther. 2010;128(2):281–304. | ||
Hashimoto R, Fujimaki K, Jeong MR, Christ L, Chuang DM. Lithium-induced inhibition of Src tyrosine kinase in rat cerebral cortical neurons: a role in neuroprotection against N-methyl-D-aspartate receptor-mediated excitotoxicity. FEBS Lett. 2003;538(1–3):145–148. | ||
Liu Y, Zhang G, Gao C, Hou X. NMDA receptor activation results in tyrosine phosphorylation of NMDA receptor subunit 2A(NR2A) and interaction of Pyk2 and Src with NR2A after transient cerebral ischemia and reperfusion. Brain Res. 2001;909(1–2):51–58. | ||
Hou XY, Zhang GY, Yan JZ, Chen M, Liu Y. Activation of NMDA receptors and L-type voltage-gated calcium channels mediates enhanced formation of Fyn-PSD95-NR2A complex after transient brain ischemia. Brain Res. 2002;955(1–2):123–132. | ||
Ma J, Zhang GY. Lithium reduced N-methyl-D-aspartate receptor subunit 2A tyrosine phosphorylation and its interactions with Src and Fyn mediated by PSD-95 in rat hippocampus following cerebral ischemia. Neurosci Lett. 2003;348(3):185–189. | ||
Berridge MJ, Downes CP, Hanley MR. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 1989;59(3):411–419. | ||
Manji HK, Bersudsky Y, Chen G, Belmaker RH, Potter WZ. Modulation of protein kinase C isozymes and substrates by lithium: the role of myo-inositol. Neuropsychopharmacology. 1996;15(4):370–381. | ||
Yildiz A, Guleryuz S, Ankerst DP, Ongür D, Renshaw PF. Protein kinase C inhibition in the treatment of mania: a double-blind, placebo-controlled trial of tamoxifen. Arch Gen Psychiatry. 2008;65(3):255–263. | ||
Sarkar S, Floto RA, Berger Z, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170(7):1101–1111. | ||
Sarkar S, Rubinsztein DC. Inositol and IP3 levels regulate autophagy: biology and therapeutic speculations. Autophagy. 2006;2(2):132–134. | ||
Li Q, Li H, Roughton K, et al. Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death Dis. 2010;1:e56. | ||
Chiu CT, Chuang DM. Neuroprotective action of lithium in disorders of the central nervous system. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2011;36(6):461–476. | ||
Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278(27):25009–25013. | ||
Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–595. | ||
Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6(4):304–312. | ||
Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3(6):331–338. | ||
Kang HJ, Noh JS, Bae YS, Gwag BJ. Calcium-dependent prevention of neuronal apoptosis by lithium ion: essential role of phosphoinositide 3-kinase and phospholipase Cgamma. Mol Pharmacol. 2003;64(2):228–234. | ||
Everall IP, Bell C, Mallory M, et al. Lithium ameliorates HIV-gp120-mediated neurotoxicity. Mol Cell Neurosci. 2002;21(3):493–501. | ||
Shalbuyeva N, Brustovetsky T, Brustovetsky N. Lithium desensitizes brain mitochondria to calcium, antagonizes permeability transition, and diminishes cytochrome C release. J Biol Chem. 2007;282(25):18057–18068. | ||
Bachmann RF, Wang Y, Yuan P, et al. Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage. Int J Neuropsychopharmacol. 2009;12(6):805–822. | ||
Bosche B, Schäfer M, Graf R, Härtel FV, Schäfer U, Noll T. Lithium prevents early cytosolic calcium increase and secondary injurious calcium overload in glycolytically inhibited endothelial cells. Biochem Biophys Res Commun. 2013;434(2):268–272. | ||
Ngok-Ngam P, Watcharasit P, Thiantanawat A, Satayavivad J. Pharmacological inhibition of GSK3 attenuates DNA damage-induced apoptosis via reduction of p53 mitochondrial translocation and Bax oligomerization in neuroblastoma SH-SY5Y cells. Cell Mol Biol Lett. 2013;18(1):58–74. | ||
de Sousa RT, Zarate CA Jr, Zanetti MV, et al. Oxidative stress in early stage bipolar disorder and the association with response to lithium. J Psychiatr Res. 2014;50:36–41. | ||
Khairova R, Pawar R, Salvadore G, et al. Effects of lithium on oxidative stress parameters in healthy subjects. Mol Med Rep. 2012;5(3):680–682. | ||
Ghasemi M, Dehpour AR. The NMDA receptor/nitric oxide pathway: a target for the therapeutic and toxic effects of lithium. Trends Pharmacol Sci. 2011;32(7):420–434. | ||
Contestabile A, Greco B, Ghezzi D, Tucci V, Benfenati F, Gasparini L. Lithium rescues synaptic plasticity and memory in Down syndrome mice. J Clin Invest. 2013;123(1):348–361. | ||
Ge S, Yang CH, Hsu KS, Ming GL, Song H. A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron. 2007;54(4):559–566. | ||
Schmidt-Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature. 2004;429(6988):184–187. | ||
Mongiat LA, Espósito MS, Lombardi G, Schinder AF. Reliable activation of immature neurons in the adult hippocampus. PLoS One. 2009;4(4):e5320. | ||
Marín-Burgin A, Mongiat LA, Pardi MB, Schinder AF. Unique processing during a period of high excitation/inhibition balance in adult-born neurons. Science. 2012;335(6073):1238–1242. | ||
Crews L, Adame A, Patrick C, et al. Increased BMP6 levels in the brains of Alzheimer’s disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J Neurosci. 2010;30(37):12252–12262. | ||
Garza JC, Guo M, Zhang W, Lu XY. Leptin restores adult hippocampal neurogenesis in a chronic unpredictable stress model of depression and reverses glucocorticoid-induced inhibition of GSK-3β/β-catenin signaling. Mol Psychiatry. 2012;17(8):790–808. | ||
Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 2011;476(7361):458–461. | ||
Mao Y, Ge X, Frank CL, et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell. 2009;136(6):1017–1031. | ||
Coras R, Siebzehnrubl FA, Pauli E, et al. Low proliferation and differentiation capacities of adult hippocampal stem cells correlate with memory dysfunction in humans. Brain. 2010;133(11):3359–3372. | ||
Bianchi P, Ciani E, Contestabile A, Guidi S, Bartesaghi R. Lithium restores neurogenesis in the subventricular zone of the Ts65Dn mouse, a model for Down syndrome. Brain Pathol. 2010;20(1):106–118. | ||
Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK. Enhancement of hippocampal neurogenesis by lithium. J Neurochem. 2000;75(4):1729–1734. | ||
Hashimoto R, Senatorov V, Kanai H, Leeds P, Chuang D-M. Lithium stimulates progenitor proliferation in cultured brain neurons. Neuroscience. 2003;117(1):55–61. | ||
Kim JS, Chang MY, Yu IT, et al. Lithium selectively increases neuronal differentiation of hippocampal neural progenitor cells both in vitro and in vivo. J Neurochem. 2004;89(2):324–336. | ||
Li H, Li Q, Du X, et al. Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. J Cereb Blood Flow Metab. 2011;31(10):2106–2115. | ||
Yan XB, Hou HL, Wu LM, Liu J, Zhou JN. Lithium regulates hippocampal neurogenesis by ERK pathway and facilitates recovery of spatial learning and memory in rats after transient global cerebral ischemia. Neuropharmacology. 2007;53(4):487–495. | ||
Pfennig A, Alda M, Young T, et al. Prophylactic lithium treatment and cognitive performance in patients with a long history of bipolar illness: no simple answers in complex disease-treatment interplay. Int J Bipolar Disord. 2014;2:1. | ||
Wingo AP, Harvey PD, Baldessarini RJ. Neurocognitive impairment in bipolar disorder patients: functional implications. Bipolar Disord. 2009;11(2):113–125. | ||
Young AH. More good news about the magic ion: lithium may prevent dementia. Br J Psychiatry. 2011;198(5):336–337. | ||
Nunes PV, Forlenza OV, Gattaz WF. Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br J Psychiatry. 2007;190:359–360. | ||
Kessing LV, Sondergard L, Forman JL, Andersen PK. Lithium treatment and risk of dementia. Arch Gen Psychiatry. 2008;65(11):1331–1335. | ||
Kessing LV, Forman JL, Andersen PK. Does lithium protect against dementia? Bipolar Disord. 2010;12(1):87–94. | ||
Macdonald A, Briggs K, Poppe M, Higgins A, Velayudhan L, Lovestone S. A feasibility and tolerability study of lithium in Alzheimer’s disease. Int J Geriatr Psychiatry. 2008;23(7):704–711. | ||
Hampel H, Ewers M, Bürger K, et al. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70(6):922–931. | ||
Leyhe T, Eschweiler GW, Stransky E, et al. Increase of BDNF serum concentration in lithium treated patients with early Alzheimer’s disease. J Alzheimers Dis. 2009;16(3):649–656. | ||
Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198(5):351–356. | ||
Nunes MA, Viel TA, Buck HS. Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr Alzheimer Res. 2013;10(1):104–107. | ||
Mauer S, Vergne D, Ghaemi SN. Standard and trace-dose lithium: a systematic review of dementia prevention and other behavioral benefits. Aust N Z J Psychiatry. 2014;48(9):809–818. | ||
Vo TM, Perry P, Ellerby M, Bohnert K. Is lithium a neuroprotective agent? Ann Clin Psychiatry. 2015;27(1):49–54. | ||
De Sarno P, Axtell RC, Raman C, Roth KA, Alessi DR, Jope RS. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2008;181(1):338–345. | ||
Watase K, Gatchel JR, Sun Y, et al. Lithium therapy improves neurological function and hippocampal dendritic arborization in a spinocerebellar ataxia type 1 mouse model. PLoS Med. 2007;4(5):e182. | ||
Forlenza OV, Aprahamian I, de Paula VJ, Hajek T. Lithium, a therapy for AD: current evidence from clinical trials of neurodegenerative disorders. Curr Alzheimer Res. Epub 2016 Feb 18. | ||
King TD, Bijur GN, Jope RS. Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3beta and attenuated by lithium. Brain Res. 2001;919(1):106–114. | ||
Arraf Z, Amit T, Youdim MB, Farah R. Lithium and oxidative stress lessons from the MPTP model of Parkinson’s disease. Neurosci Lett. 2012;516(1):57–61. | ||
Kim YH, Rane A, Lussier S, Andersen JK. Lithium protects against oxidative stress-mediated cell death in α-synuclein-overexpressing in vitro and in vivo models of Parkinson’s disease. J Neurosci Res. 2011;89(10):1666–1675. | ||
Youdim MB, Arraf Z. Prevention of MPTP (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) dopaminergic neurotoxicity in mice by chronic lithium: involvements of Bcl-2 and Bax. Neuropharmacology. 2004;46(8):1130–1140. | ||
Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18(10):1162–1164. | ||
Yong Y, Ding H, Fan Z, Luo J, Ke ZJ. Lithium fails to protect dopaminergic neurons in the 6-OHDA model of Parkinson’s disease. Neurochem Res. 2011;36(3):367–374. | ||
Scheuing L, Chiu CT, Liao HM, Linares GR, Chuang DM. Preclinical and clinical investigations of mood stabilizers for Huntington’s disease: what have we learned? Int J Biol Sci. 2014;10(9):1024–1038. | ||
Wei H, Qin ZH, Senatorov VV, et al. Lithium suppresses excitotoxicity-induced striatal lesions in a rat model of Huntington’s disease. Neuroscience. 2001;106(3):603–612. | ||
Senatorov VV, Ren M, Kanai H, Wei H, Chuang DM. Short-term lithium treatment promotes neuronal survival and proliferation in rat striatum infused with quinolinic acid, an excitotoxic model of Huntington’s disease. Mol Psychiatry. 2004;9(4):371–385. | ||
Danivas V, Moily NS, Thimmaiah R, et al. Off label use of lithium in the treatment of Huntington’s disease: a case series. Indian J Psychiatry. 2013;55(1):81–83. | ||
Raja M, Soleti F, Bentivoglio AR. Lithium treatment in patients with Huntington disease and suicidal behavior. J Clin Psychopharmacol. 2013;33(6):819–821. | ||
Fornai F, Longone P, Cafaro L, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105(6):2052–2057. | ||
Gill A, Kidd J, Vieira F, Thompson K, Perrin S. No benefit from chronic lithium dosing in a sibling-matched, gender balanced, investigator-blinded trial using a standard mouse model of familial ALS. PLoS One. 2009;4(8):e6489. | ||
Pizzasegola C, Caron I, Daleno C, et al. Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph Lateral Scler. 2009;10(4):221–228. | ||
Ferrucci M, Spalloni A, Bartalucci A, et al. A systematic study of brainstem motor nuclei in a mouse model of ALS, the effects of lithium. Neurobiol Dis. 2010;37(2):370–383. | ||
Shin JH, Cho SI, Lim HR, et al. Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model of amyotrophic lateral sclerosis. Mol Pharmacol. 2007;71(4):965–975. | ||
Feng HL, Leng Y, Ma CH, Zhang J, Ren M, Chuang DM. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience. 2008;155(3):567–572. | ||
Aggarwal SP, Zinman L, Simpson E, et al. Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(5):481–488. | ||
Al-Chalabi A, Shaw PJ, Young CA, et al; UKMND-LiCALS. Protocol for a double-blind randomised placebo-controlled trial of lithium carbonate in patients with amyotrophic lateral sclerosis (LiCALS) [Eudract number: 2008-006891-31]. BMC Neurol. 2011;11:111. | ||
Morrison KE, Dhariwal S, Hornabrook R, et al; for UKMND-LiCALS Study Group. Lithium in patients with amyotrophic lateral sclerosis (LiCALS): a phase 3 multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2013;12(4):339–345. | ||
Chiò A, Mora G. The final chapter of the ALS lithium saga. Lancet Neurol. 2013;12(4):324–325. | ||
Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J Clin Invest. 2012;122(12):4314–4322. | ||
Bhakar AL, Dolen G, Bear MF. The pathophysiology of fragile X (and what it teaches about synapses). Annu Rev Neurosci. 2012;35:417–433. | ||
Gatto CL, Broadie K. The fragile X mental retardation protein in circadian rhythmicity and memory consolidation. Mol Neurobiol. 2009;39(2):107–129. | ||
Eadie BD, Cushman J, Kannangara TS, Fanselow MS, Christie BR. NMDA receptor hypofunction in the dentate gyrus and impaired context discrimination in adult Fmr1 knockout mice. Hippocampus. 2012;22(2):241–254. | ||
Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95(5):3291–3295. | ||
Yun SH, Trommer BL. Fragile X mice: reduced long-term potentiation and N-methyl-D-aspartate receptor-mediated neurotransmission in dentate gyrus. J Neurosci Res. 2011;89(2):176–182. | ||
Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–377. | ||
Dolen G, Bear MF. Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol. 2008;586(Pt 6):1503–1508. | ||
Michalon A, Sidorov M, Ballard TM, et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74(1):49–56. | ||
Min WW, Yuskaitis CJ, Yan Q, et al. Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology. 2009;56(2):463–472. | ||
Mines Ma, Jope RS. Glycogen synthase kinase-3: a promising therapeutic target for fragile x syndrome. Front Mol Neurosci. 2011;4:35. | ||
Peineau S, Taghibiglou C, Bradley C, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53(5):703–717. | ||
Choi CH, Schoenfeld BP, Bell AJ, et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380:106–119. | ||
Franklin AV, King MK, Palomo V, Martinez A, McMahon LL, Jope RS. Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol Psychiatry. 2014;75(3):198–206. | ||
King MK, Jope RS. Lithium treatment alleviates impaired cognition in a mouse model of fragile X syndrome. Genes Brain Behav. 2013;12(7):723–731. | ||
Liu ZH, Chuang DM, Smith CB. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol. 2011;14(5):618–630. | ||
Liu ZH, Huang T, Smith CB. Lithium reverses increased rates of cerebral protein synthesis in a mouse model of fragile X syndrome. Neurobiol Dis. 2012;45(3):1145–1152. | ||
Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One. 2010;5(3):e9706. | ||
Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem Pharmacol. 2010;79(4):632–646. | ||
Berry-Kravis E, Sumis A, Hervey C, et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. 2008;29(4):293–302. | ||
Wu X, Bai Y, Tan T, et al. Lithium ameliorates autistic-like behaviors induced by neonatal isolation in rats. Front Behav Neurosci. 2014;8:234. | ||
Serret S, Thümmler S, Dor E, Vesperini S, Santos A, Askenazy F. Lithium as a rescue therapy for regression and catatonia features in two SHANK3 patients with autism spectrum disorder: case reports. BMC Psychiatry. 2015;15:107. | ||
Durand CM, Betancur C, Boeckers TM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39(1):25–27. | ||
Denayer A, Van Esch H, de Ravel T, et al. Neuropsychopathology in 7 patients with the 22q13 deletion syndrome: presence of bipolar disorder and progressive loss of skills. Mol Syndromol. 2012;3(1):14–20. | ||
Verhoeven WM, Egger JI, Willemsen MH, de Leijer GJ, Kleefstra T. Phelan-McDermid syndrome in two adult brothers: atypical bipolar disorder as its psychopathological phenotype? Neuropsychiatr Dis Treat. 2012;8:175–179. | ||
Verhoeven WM, Egger JI, Cohen-Snuijf R, Kant SG, de Leeuw N. Phelan-McDermid syndrome: clinical report of a 70-year-old woman. Am J Med Genet A. 2013;161A(1):158–161. | ||
Fernandez F, Morishita W, Zuniga E, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci. 2007;10(4):411–413. | ||
Chakrabarti L, Galdzicki Z, Haydar TF. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci. 2007;27(43):11483–11495. | ||
Chakrabarti L, Best TK, Cramer NP, et al. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat Neurosci. 2010;13(8):927–934. | ||
Algattas H, Huang JH. Traumatic brain injury pathophysiology and treatments: early, intermediate, and late phases post-injury. Int J Mol Sci. 2014;15(1):309–341. | ||
Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76(5):886–899. | ||
Olsson A, Csajbok L, Ost M, et al. Marked increase of beta-amyloid (1–42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J Neurol. 2004;251(7):870–876. | ||
Sivanandam TM, Thakur MK. Traumatic brain injury: a risk factor for Alzheimer’s disease. Neurosci Biobehav Rev. 2012;36(5):1376–1381. | ||
Uryu K, Chen XH, Martinez D, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208(2):185–192. | ||
Shapira M, Licht A, Milman A, Pick CG, Shohami E, Eldar-Finkelman H. Role of glycogen synthase kinase-3beta in early depressive behavior induced by mild traumatic brain injury. Mol Cell Neurosci. 2007;34(4):571–577. | ||
Zhu ZF, Wang QG, Han BJ, William CP. Neuroprotective effect and cognitive outcome of chronic lithium on traumatic brain injury in mice. Brain Res Bull. 2010;83(5):272–277. | ||
Dash PK, Johnson D, Clark J, et al. Involvement of the glycogen synthase kinase-3 signaling pathway in TBI pathology and neurocognitive outcome. PLoS One. 2011;6(9):e24648. | ||
Yu F, Wang Z, Tchantchou F, Chiu CT, Zhang Y, Chuang DM. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J Neurotrauma. 2012;29(2):362–374. | ||
Roh MS, Eom TY, Zmijewska AA, De Sarno P, Roth KA, Jope RS. Hypoxia activates glycogen synthase kinase-3 in mouse brain in vivo: protection by mood stabilizers and imipramine. Biol Psychiatry. 2005;57(3):278–286. | ||
Bian Q, Shi T, Chuang DM, Qian Y. Lithium reduces ischemia-induced hippocampal CA1 damage and behavioral deficits in gerbils. Brain Res. 2007;1184:270–276. | ||
Bijur GN, Jope RS. Opposing actions of phosphatidylinositol 3-kinase and glycogen synthase kinase-3 beta in the regulation of HSF-1 activity. J Neurochem. 2000;75(6):2401–2408. | ||
Ren M, Senatorov VV, Chen R-W, Chuang D-M. Post-insult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc Natl Acad Sci U S A. 2003;100(10):6210–6215. | ||
Xu JH, Culman J, Blume A, Brecht S, Gohlke P. Chronic treatment with a low dose of lithium protects the brain against ischemic injury by reducing apoptotic death. Stroke. 2003;34(5):1287–1292. | ||
Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. Neuroreport. 1998;9(9):2081–2084. | ||
Zheng Z, Kim JY, Ma H, Lee JE, Yenari MA. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J Cereb Blood Flow Metab. 2008;28(1):53–63. | ||
Kim YR, van Meer MP, Tejima E, et al. Functional MRI of delayed chronic lithium treatment in rat focal cerebral ischemia. Stroke. 2008;39(2):439–447. | ||
Fan Y, Yang GY. Therapeutic angiogenesis for brain ischemia: a brief review. J Neuroimmune Pharmacol. 2007;2(3):284–289. | ||
Tsai LK, Leng Y, Wang Z, Leeds P, Chuang DM. The mood stabilizers valproic acid and lithium enhance mesenchymal stem cell migration via distinct mechanisms. Neuropsychopharmacology. 2010;35(11):2225–2237. | ||
Tsai LK, Wang Z, Munasinghe J, Leng Y, Leeds P, Chuang DM. Mesenchymal stem cells primed with valproate and lithium robustly migrate to infarcted regions and facilitate recovery in a stroke model. Stroke. 2011;42(10):2932–2939. | ||
Gray JD, McEwen BS. Lithium’s role in neural plasticity and its implications for mood disorders. Acta Psychiatr Scand. 2013;128(5):347–361. | ||
Bachmann RF, Schloesser RJ, Gould TD, Manji HK. Mood stabilizers target cellular plasticity and resilience cascades: implications for the development of novel therapeutics. Mol Neurobiol. 2005;32(2):173–202. | ||
Chuang DM, Manji HK. In search of the Holy Grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked? Biol Psychiatry. 2007;62(1):4–6. | ||
Manji HK, Moore GJ, Chen G. Clinical and preclinical evidence for the neurotrophic effects of mood stabilizers: implications for the pathophysiology and treatment of manic-depressive illness. Biol Psychiatry. 2000;48(8):740–754. | ||
Dell’Osso L, Dalle Luche R, Maj M. Adult autism spectrum as a transnosographic dimension. CNS Spectr. 2016;21(2):131–133. | ||
Insel TR. Rethinking schizophrenia. Nature. 2010;468(7321):187–193. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.