")
Back to Journals » International Journal of Nanomedicine » Volume 16
A Multiepitope Peptide, rOmp22, Encapsulated in Chitosan-PLGA Nanoparticles as a Candidate Vaccine Against Acinetobacter baumannii Infection
Authors Du X , Xue J, Jiang M, Lin S, Huang Y, Deng K, Shu L, Xu H, Li Z, Yao J, Chen S, Shen Z, Feng G
Received 14 December 2020
Accepted for publication 13 February 2021
Published 4 March 2021 Volume 2021:16 Pages 1819—1836
DOI https://doi.org/10.2147/IJN.S296527
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ebrahim Mostafavi
Xingran Du,1,* Jianpeng Xue,2,* Mingzi Jiang,3 Shaoqing Lin,4 Yuzhen Huang,4 Kaili Deng,5 Lei Shu,5 Hanmei Xu,2 Zeqing Li,2 Jing Yao,4 Sixia Chen,4 Ziyan Shen,4 Ganzhu Feng4
1Department of Infectious Disease, The Second Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China; 2State Key Laboratory of Natural Medicines, The Engineering Research Center of Synthetic Polypeptide Discovery and Evaluation of Jiangsu Province, China Pharmaceutical University, Nanjing, Jiangsu, People’s Republic of China; 3Department of Respiratory and Critical Care Medicine, The First People’s Hospital of Kunshan, Suzhou, Jiangsu, People’s Republic of China; 4Department of Respiratory and Critical Care Medicine, The Second Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China; 5Department of Respiratory Medicine, Sir Run Run Hospital, Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ganzhu Feng
Department of Respiratory and Critical Care Medicine, The Second Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, People’s Republic of China
Tel +86-025-58509837
Fax +86-025-58509994
Email [email protected]
Background: The development of vaccines is a promising and cost-effective strategy to prevent emerging multidrug-resistant (MDR) Acinetobacter baumannii (A. baumannii) infections. The purpose of this study was to prepare a multiepitope peptide nanovaccine and evaluate its immunogenicity and protective effect in BALB/c mice.
Methods: The B-cell and T-cell epitopes of Omp22 from A. baumannii were predicted using bioinformatics methods and identified by immunological experiments. The optimal epitopes were conjugated in series by 6-aminocaproic acid and chemically synthesized multiepitope polypeptide rOmp22. Then, rOmp22 was encapsulated by chitosan (CS) and poly (lactic-co-glycolic) acid (PLGA) to prepare CS-PLGA-rOmp22 nanoparticles (NPs). The immunogenicity and immunoprotective efficacy of the vaccine were evaluated in BALB/c mice.
Results: CS-PLGA-rOmp22 NPs were small (mean size of 272.83 nm) with apparently spherical structures, positively charged (4.39 mV) and nontoxic to A549 cells. A high encapsulation efficiency (54.94%) and a continuous slow release pattern were achieved. Compared with nonencapsulated rOmp22, CS-PLGA-rOmp22 immunized BALB/c mice induced higher levels of rOmp22-specific IgG in serum and IFN-γ in splenocyte supernatant. Additionally, lung injury and bacterial burdens in the lung and blood were suppressed, and potent protection (57.14%-83.3%) against acute lethal intratracheal A. baumannii challenge was observed in BALB/c mice vaccinated with CS-PLGA-rOmp22.
Conclusion: CS-PLGA-rOmp22 NPs elicited specific IgG antibodies, Th1 cellular immunity and protection against acute lethal intratracheal A. baumannii challenge. Our results indicate that this nanovaccine is a desirable candidate for preventing A. baumannii infection.
Keywords: A. baumannii, epitope vaccine, polymeric nanoparticles, PLGA, chitosan
Introduction
Acinetobacter baumannii (A. baumannii) has recently emerged as one of the most important health problems due to its propensity to acquire multidrug, extensive drug and even pandrug resistance phenotypes at rates not previously seen.1–3 The main antibiotic resistance mechanisms include alterations in target sites, failure of degradation-specific enzymes, perfusion defects and modifications of multidrug effusion pumps.1,4 In addition, A. baumannii has a number of potential virulence factors, such as a siderophore-mediated iron-acquisition system and biofilm formation, which could possibly affect clinical outcomes.5 The global emergence of multi-drug resistant (MDR) and pan-drug resistant (PDR) A. baumannii has resulted in significantly increased mortality rates with limited or no options for therapeutic interventions.6,7
Vaccination strategies are emerging as a viable option to prevent or treat MDR or PDR infections, but there is still no licensed vaccine against A. baumannii. Conventional vaccines developed from live attenuated or inactivated whole cells could induce strong humoral and cellular immunity, however, the clinical applications of such vaccines have been limited due to their complex compositions and potential safety concerns.8–10 In recent decades, research on vaccines against A. baumannii has primarily focused on various forms of recombinant antigens, including biofilm-associated protein Bap,11 auto-transporter (Ata),12 outer membrane protein A (OmpA),13 outer membrane protein assembly factor (BamA),14 poly-N-acetyl-β-(1-6)-glucosamine (PNAG),15 and outer membrane protein 22 (Omp22).16 Animal studies showed that some single recombinant protein based vaccines provided only weak protection against A. baumannii infection or poor cross-protection against certain strains.17 In addition, the formulation of such vaccines often leads to reactogenic and/or allergenic responses that are often not desired.18 Thus, identifying an antigen that has high immunogenicity and avoids the virulence of structural proteins is the key to preparing an A. baumannii vaccine.
Recently, the design of epitope-driven or peptide-based vaccines has become more attractive, because they are comparatively easier to produce and construct, lack any infectious potential and offer chemical stability.18–20 There are many multiepitope vaccine design studies involving various bacteria, such as Klebsiella pneumoniae,21 Shigella sonnei19 and meningitis-inducing bacteria (Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae Type b).22 Ren et al23 first designed a multiepitope assembly peptide (MEP) of A. baumannii and evaluated its immunogenicity and protective immunity in BALB/c mice. The results of that study indicated that rMEP is a promising vaccine candidate for the control of infections caused by A. baumannii. However, to be optimally effective, peptide-based vaccines need to be administered with adjuvants.24 Many currently available adjuvants are toxic, not biodegradable and invariably invoke adverse reactions, including allergic responses and excessive inflammation.25 A nontoxic, biodegradable, biocompatible vaccine delivery system is urgently needed.
The nanotechnology-based approach is considered to be one of the most advantageous methods for the development of peptide-based vaccines.26 Poly(lactic-co-glycolic) acid (PLGA) is a synthetic copolymer that has been approved by the United States Food and Drug Administration (FDA) and European Medicine Agency (EMA) for various medical and pharmaceutical applications in humans.27 PLGA NPs have been shown to be efficient for antigen delivery because of their effectiveness in enhancing immune responses, controlled release, high encapsulation efficiency and tissue biodistribution, along with being biodegradable, nontoxic and small in size.28–30 While PLGA NPs display many advantages in antigen delivery, in comparison with cationic biopolymers, they can suffer poor encapsulation efficiency and instability when loading negatively charged molecules, such as protein or peptide antigens. Chitosan (CS) is a nontoxic and nonimmunogenic naturally occurring linear amino polysaccharide (poly 1.4-beta-glucosamine), with the ability to enhance the penetration of large molecules across mucosal surfaces.31 Such anionic PLGA NPs can be subjected to chitosan-surface coating, thus resulting in cationic chitosan-coated PLGA (CS-PLGA) NPs, which hold promise as innovative formulations for targeted delivery.29 Previous studies have demonstrated that CS-PLGA NPs are particularly effective for antigen delivery to antigen presenting cells (APCs), such as inducing antigen presentation to lymphocytes or modulating APC function.29
In this study, we chose A. baumannii outer membrane protein 22 (Omp22), a highly conserved and highly immunogenic protein, as the candidate antigen. A previous study found that immunization with recombinant Omp22 efficiently elicited high titers of specific IgG, increased the survival rates of mice, and suppressed the bacterial burdens in the organs and peripheral blood.16 However, Omp22 is not only a key protein involved in the A. baumannii metabolic process, but also has toxicity.16 Therefore, we used bioinformatics techniques and immunological methods to predict and identify optimal T-cell and B-cell epitopes on the A. baumannii Omp22 protein. Subsequently, the identified dominant epitopes were connected in series by 6-aminocaproic acid and chemically synthesized to generate the multiepitope peptide rOmp22. Then, rOmp22 was encapsulated by CS-PLGA to prepare a multiepitope peptide nanovaccine (CS-PLGA-rOmp22). The physical-structural characterization, immunogenicity and protective efficacy of the vaccine were evaluated comprehensively in vitro and in vivo. This novel nanovaccine can retain the immunogenicity of Omp22 and avoid its harmful effects on the host, and it should become a high-priority strategy against A. baumannii infection.
Materials and Methods
Materials
PLGA (lactide: glycolide=50:50; MW=30,000–60,000), polyvinyl alcohol (PVA; MW=85,000–124 000, 99% hydrolyzed), HRP-labelled goat anti-mouse IgG, Freund’s complete adjuvant and Freund’s incomplete adjuvant were purchased from Sigma-Aldrich (St Louis, MO). The human lung adenocarcinoma epithelial cell line A549 was obtained from American Type Culture Collection (ATCC). RPMI-1640 medium, fetal bovine serum (FBS) and antibiotic-antimycotic were all purchased from Invitrogen (Carlsbad, CA). The Cell Counting Kit-8 (CCK-8) Assay kit was purchased from KeyGEN BioTECH (Nanjing, China). Fc blocking antibody, mouse I-Ab APC, PerCP-Cy5.5 anti-mouse CD11c, PE anti-mouse F4/80, FITC anti-mouse CD11b, PE anti-mouse CD3 and APC anti-mouse CD19 were all purchased from BD-Biosciences (San Diego, CA). DNA ligase, DNA polymerase (Taq enzyme), restriction enzymes BamHI and XhoI were purchased from American Thermo Company.
A. baumannii ATCC19606 strain was obtained from ATCC. Three clinical A. baumannii strains were collected from the Second Affiliated Hospital of Nanjing Medical University. All clinical A. baumannii strains were confirmed to be multidrug resistant (MDR) strains by drug sensitivity experiments (Table S1 Supporting information) according to Clinical and Laboratory Standards Institute (CLSI) M100. The E. coli BL21 (DE3) and the plasmid pET28a (+) used in the study were purchased from Novagen Company (Beijing, China) and kept in our laboratory. For all experiments, unless otherwise stated, bacteria were grown on Luria-Bertani (LB: 10 g/L tryptone, 5 g/L yeast extract, and 10 g/L sodium chloride) agar plates or in LB broth at 37°C.
Animals
All animal experiments were performed using 6- to 8-week-old female BALB/c mice purchased from Shanghai Super-B&K Laboratory Animal Corp. Ltd. (Shanghai, China). The mice were raised under specific pathogen-free (SPF) conditions at an ambient temperature of 25°C and provided sterile food and water ad libitum. All animal experiments were performed in accordance with the Guide of the National Institutes of Health for the Care and Use of Laboratory Animals (NIH Publications No. 8023) and approved by the Ethics Committee of Animal Care and Welfare, Nanjing Medical University (Approval No. IACUC-1904044). All efforts were made to minimize animal suffering.
Expression and Purification of Omp22
The gene encoding Omp22 was obtained through PCR amplification using the genomic DNA of the A. baumannii ATCC 17978 strain as the template. The primers were Omp22-F (5′ CAA GGA TCC CTG GGC GGC GTT GAA TAT G 3′) and Omp22-R (5′ CAC AAG CTT TTA CTG TTT CGC GTA AAT G 3′). The PCR products were digested using the BamHI and XhoI enzymes and ligated into the plasmid pET28a. The recombinant plasmid pET28a-omp22 was transfected into E. coli BL21 (DE3). The recombinant protein Omp22 was obtained through induced expression and purification. After elution with elution buffer (1 M imidazole in binding buffer), the collected fractions were analysed with SDS-PAGE.
Prediction and Identification of B-Cell Epitopes and T-Cell Epitopes
The physicochemical properties of the A. baumannii Omp22 protein were analysed and all possible dominant B-cell and T-cell epitopes were predicted using an immunoinformatics approach. The B-cell epitopes were predicted using the bioinformatics software OptimumAntigen Design Tool (GenScript, China). B-cell epitopes were predicted according to their secondary structure, surface accessibility, hydrophilicity, flexibility, and antigenic index. Four candidate B-cell epitopes with a length of 14 amino acids and no overlap with each other were selected after comprehensive analysis. When predicting T-cell epitopes, the Immune Epitope Database Analysis Resource (IEDB) (https://tools.iedb.org/mhci/) was used to analyse and predict the A and E subregions of MHC-II genes in mice. Four nonoverlapping candidate T-cell epitopes were stored based on their scores.32,33 The predicted B-cell and T-cell epitope peptides were chemically synthesized by GenScript Biotechnology Co. Ltd (China).
Twelve female BALB/c mice (6 to 8 weeks) were randomly divided into two groups: the Omp22 immunized-group and the phosphate-buffered saline (PBS) control group. Mice were subcutaneously injected with 100 μg of recombinant Omp22 (1 μg/μL in PBS) or an equal volume of PBS three times at two-week intervals. In the Omp22 vaccine group, Freund’s adjuvant was added to enhance the immune effect. One week after the last immunization, the serum of each mouse was collected to detect B-cell epitope specific antigens by indirect ELISA. Splenocytes were isolated from vaccinated mice and adjusted to a concentration of 1×106 cells/mL, and 200 μL of the cell suspension was added to each well of a 96-well plate and stimulated with 20 μg/mL candidate T-cell epitopes. After incubation for 72 h, supernatants were collected and the levels of gamma interferon (IFN-γ) were measured using mouse IFN-γ ELISA kits. Based on the above analysis, three B-cell epitopes (amino acid sequences: NIPLSQARAQSVKNY; YATLDKVAQTL and SVQLIMP) and two T-cell epitopes (amino acid sequences: VPSSRIDAQGY and TFDTNKSNIKP) from Omp22 were selected to design the multiepitope protein.
Design and Synthesis of rOmp22
Three optimal B-cell epitopes and two T-cell epitopes were connected in series by 6-aminocaproic acid. The multiepitope peptide rOmp22 of 59 aa, with a molecular weight of 6536.4 Da was chemically synthesized and identified by liquid chromatography and mass spectrometry. The synthesis and identification of rOmp22 was completed by GenScript Co. Ltd, China. The secondary structure and tertiary structure of the peptide were predicted by the PSIPRED protein sequence analysis workbench (http://bioinf.cs.ucl.ac.uk/psipred) and Gromacs 2020 package, respectively.
Preparation of Nanoparticles
CS-PLGA NPs were prepared using a modified water/oil/water double emulsion evaporation technique.34 Briefly, 2 mg of rOmp22 dissolved in PBS containing 0.003% sodium alginate was the inner water phase, and 80 mg of PLGA (lactide: glycolide=50:50; MW=30,000–60,000) (Sigma-Aldrich) was dissolved in 2 mL of dichloromethane (DCM) as the organic phase. A 2% (w/V) polyvinyl alcohol (PVA; MW=85,000–124,000, 99% hydrolysed; Sigma-Aldrich) solution including 0.2% chitosan (MW=50,000; Sigma-Aldrich) was prepared as the external aqueous phase. The emulsion was stirred at 1500 rpm at room temperature overnight. The nanoparticles were obtained by ultracentrifugation (30,000 g for 30 min), washed three times with deionized water to remove excess polyvinyl alcohol and rOmp22, and lyophilized to obtain CS-PLGA-rOmp22. A volume of PBS equivalent to that used for rOmp22 was similarly encapsulated in CS-PLGA to obtain CS-PLGA-PBS to serve as a negative control. All lyophilized nanoparticles were stored at −80°C until use.
Encapsulation Efficiency and Peptide Loading Level
The encapsulation efficiency of rOmp22 in CS-PLGA was measured using high performance liquid chromatography (HPLC) (Agilent, USA) by quantitating rOmp22 in the supernatant after ultracentrifugation. The rOmp22 encapsulation efficiency (EE) and the peptide loading capacity (LC) were calculated using the following formulas:
EE = (A-B)/A × 100
LC = (A-B)/C × 100
where A is the total amount of rOmp22, B is the free amount of rOmp22, and C is the CS-PLGA-rOmp22 weight. These measurements were performed three times.
Particle Size and Zeta Potential
The particle size, polydispersity index (PDI) and zeta potential were measured by particle size analyser (Anton Paar, Graz, Austria). CS-PLGA-rOmp22 or CS-PLGA-PBS was suspended in filtered distilled water, sonicated, and placed in a cuvette to measure size and zeta potential. Each sample was measured three times and reported as the mean of triplicates for size (diameter in nanometers) and zeta potential (millivolts). These experiments were conducted at least three times.
Transmission Electron Microscopy (TEM)
The morphology of CS-PLGA-rOmp22 and CS-PLGA-PBS was observed using high-resolution TEM (Hitachi, HT7700 Exalens). One drop of the complex was deposited on a copper grid (carbon-coated copper grid, 200 mesh). After adding phosphotungstic acid, the grids were dried for 10 min prior to TEM analysis.
In vitro Peptide Release
The release of the rOmp22 peptide from CS-PLGA was determined following the method of Abd EI Hady et al.35 Briefly, CS-PLGA-rOmp22 NPs were suspended in normal saline (NS) in Eppendorf tubes. The suspensions were incubated at 37°C and at various time intervals (30 min, 1, 2, 4, 6, 12, 24, 48 and 72 h), supernatants were collected by centrifugation. The released peptide in the supernatants was measured using HPLC and replaced with fresh buffer. HPLC analysis was performed with a mobile phase of phosphate buffer-acetonitrile and gradient elution (Table S1 Supporting information). The temperature was set at 35°C, the flow rate was 1 mL/min, the injection volume was 20 μL and the detection wavelength was 220 nm. The particles were resuspended by brief sonication. Background was obtained using the supernatants from blank particles.
Cytotoxicity Studies
The cytotoxicity of rOmp22, CS-PLGA-PBS and CS-PLGA-rOmp22 to human lung adenocarcinoma epithelial cell line A549 (ATCC) was detected by CCK-8 assay. In the cell viability assay, A549 cells were seeded at 1×105 cells/well in 96-well plates in 100 μL of medium and incubated overnight at 37°C in a humidified atmosphere with 5% CO2 to allow cell attachment. Then the cells were incubated with different concentrations of rOmp22 (1.25–80 μg/mL), CS-PLGA-PBS (12.5–800 μg/mL) or CS-PLGA-rOmp22 (12.5–800 μg/mL) for 6, 24 and 48 h. Then, 10 μL of CCK-8 solution was added to the culture medium and incubated for an additional 2 h. Optical density (OD) values were measured at 450 nm wavelength using a microplate reader (Bio-Rad iMark).
Mouse Immunization
BALB/c mice were randomly divided into four groups: the CS-PLGA-rOmp22-treated group, CS-PLGA-PBS-treated group, rOmp22-immunized group and adjuvant-treated group. Mice in the CS-PLGA-rOmp22 group were immunized subcutaneously with 40 μg/200 μL of encapsulated rOmp22 in NS, and those in the CS-PLGA-PBS group were immunized with an equivalent weight of CS-PLGA-PBS NPs. Each mouse in the rOmp22 group was immunized with 40 μg of nonencapsulated rOmp22 in 100 μL of NS formulated with an equal volume of Freund’s complete adjuvant (Sigma-Aldrich), and boosted with the same dose of rOmp22 in 100 μL of NS formulated with Freund’s incomplete adjuvant on days 14 and 28. In the adjuvant-treated group, each mouse was injected subcutaneously with 100 μL of NS formulated with an equal volume of Freund’s complete adjuvant on day 0 or Freund’s incomplete adjuvant on days 14 and 28. Six mice were randomly selected from each group for immunological assays. The other animals were used for the challenge test two weeks after the third immunization.
Antibody Titer Measurement with ELISA
The multiepitope peptide rOmp22 was first diluted to an optimal concentration (10 μg/mL) to coat a 96-well plate. The resulting solution was then added to each well (100 μL per well) and incubated for 12–18 h at 4°C. After washing five times with PBS plus 0.05% Tween 20 (PBST), 200 μL of 2% bovine serum albumin (BSA) was added to each well and incubated for 2 h at 37°C to block the unoccupied sites. After washing, serial dilutions of the pooled serum in each group ranging from 1:200 to 1:51,200 were added to the wells in triplicate and incubated at 37°C for 2 h. The plates were washed 5 times as described above. Then, 100 μL per well of horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (diluted 1:10,000) was added and the plates were incubated for 1 h. The plates were then washed 5 times and incubated with 100 μL per well of 3,3ʹ,5,5ʹ-tetramethylbenzidine solution (TMB) as the substrate until the desired absorbance was reached. The reaction was stopped by the addition of 100 μL of 2 M H2SO4. The optical density of the samples was measured at 450 nm using an ELISA plate reader. The endpoint titer was defined as the highest dilution for which the OD at 450 nm was at least 0.1 above that of the background well.
Splenocyte Stimulation and Cytokine Secretion
Splenocytes were isolated from vaccinated mice as described previously seven days after the last immunization.10 Splenocytes were adjusted to a concentration of 1×106 cells/mL, and 200 μL of the cell suspension was added to each well of a 96-well plate and either stimulated with rOmp22 (20 μg/mL) or left unstimulated. After incubation for 72 h, supernatants were collected and the levels of gamma interferon (IFN-γ) and interleukin 4 (IL-4) were detected using mouse IFN-γ and IL-4 ELISA kits.
Flow Cytometry Analysis
Single-spleen cell suspensions and draining lymph node cell suspensions were obtained from mice one week after the last immunization. Cells (1×106/mL) were blocked with Fc blocking antibody (BD Bioscience) in fluorescence-activated cell sorting (FACS) buffer (phosphate buffered saline, 1.0% fetal bovine serum) for 15 min at 4°C. The cells were washed and stained with fluorochrome-conjugated antibodies against lymphocyte surface receptors, APC-Mouse I-Ab, PerCP-Cy5.5-CD11c, PE-F4/80, FITC-CD11b, PE-CD3 and APC-CD19 (BD Biosciences) for 30 min at 4°C. The cells were then washed and fixed with 2% paraformaldehyde solution for 20 min at 4°C. Data were acquired on a BD FACS Canto II flow cytometer (BD Bioscience) with at least 1 × 105 events for each sample and analysed using FlowJo software (Tree Star Inc, Ashland, OR, USA).
Establishment of Pneumonia Models
A. baumannii strain ATCC19606 and 3 clinical A. baumannii strains (CS-MDR-AB, CRAB and PDR-AB) were grown in LB broth to the late-logarithmic phase at 37°C/150 rpm. Cells were harvested by centrifugation at 4000 g for 10 min, washed and resuspended in PBS and mixed with porcine mucin (Sigma-Aldrich) to a final concentration of 5% mucin. The desired CFU/mL was obtained by appropriate dilutions and the final concentration was quantified by plating serial dilutions onto LB agar plates. The mice were anaesthetized via an intraperitoneal (i. p.) injection of pentobarbital sodium in a supine position, and their trachea were exposed surgically. Lethal doses of ATCC19606 (2×108 CFU) and three clinical A. baumannii strains, CS-MDR-AB (1×109 CFU), CRAB (5×108 CFU) or PDR-AB (5×108 CFU), in a total volume of 100 μL were intratracheally administered to mice to induce acute pneumonia. The incised area was sealed with sterile surgical sutures.
The mice were monitored for 7 days, and the body weight, clinical score and survival number of each group were recorded every day. The overall clinical sign for each mouse was scored on a sliding scale of 0 to −5. Individual clinical scores were assigned as 0 (normal, active, healthy), −1 (slightly sick, slightly ruffled fur, otherwise normal), −2 (sick, ruffled fur, slow movement, hunching), −3 (very sick, ruffled fur, very slow movement, hunched, eyes shut), −4 (moribund), or −5 (dead).
Bacterial Load Assessment in the Blood and Lung
Blood samples were collected from six mice in each group at 24 h post-challenge. To determine the bacterial loads in the blood, samples were serially diluted and plated on blood agar plates. After blood collection, the mice were killed, and the lungs were removed aseptically, weighed, and homogenized. Serial dilutions of tissue homogenates were plated onto blood agar plates. Bacterial CFUs were enumerated after 24 hours of incubation at 37°C.
Histopathological Examination
Lungs were removed under aseptic conditions and fixed in 4% formalin. Histopathological examination of sections after embedding in paraffin was performed under a microscope after staining with haematoxylin-eosin (HE). Lung injury was estimated by the percentage of the lesion area among the total lung area using an ImagePro macro.
Statistical Analyses
Statistical analyses were performed using the Statistical Package of Social Sciences (SPSS, version 23.0; SPSS Inc., Chicago, IL) and Graphpad Prism (version 6.0; Graphpad software Inc., La Jolla, CA). All data were expressed as the mean ± SD. One-way analysis of variance (ANOVA) was used for multiple comparisons, followed by Bonferroni’s post hoc test. Survival data were compared using the Log rank test. A P value of < 0.05 was considered significant.
Results
Heterologous Expression and Purification of Recombinant Omp22
The recombinant Omp22 protein was synthesized and purified for further study. The Omp22-encoding gene was amplified by PCR, cloned into the plasmid pET-28a (+), and transformed into Escherichia coli (E. coli) BL21 (DE3). The positive clones were confirmed by restriction (Figure 1A) and sequencing (Figure S1 Supporting information). The recombinant protein Omp22 was expressed in E. coli BL21 (DE3) and purified with Ni-IDA resin. The purified protein was analysed by SDS-PAGE, and a specific band was seen at 22 kDa, which was consistent with the expected size (Figure 1B).
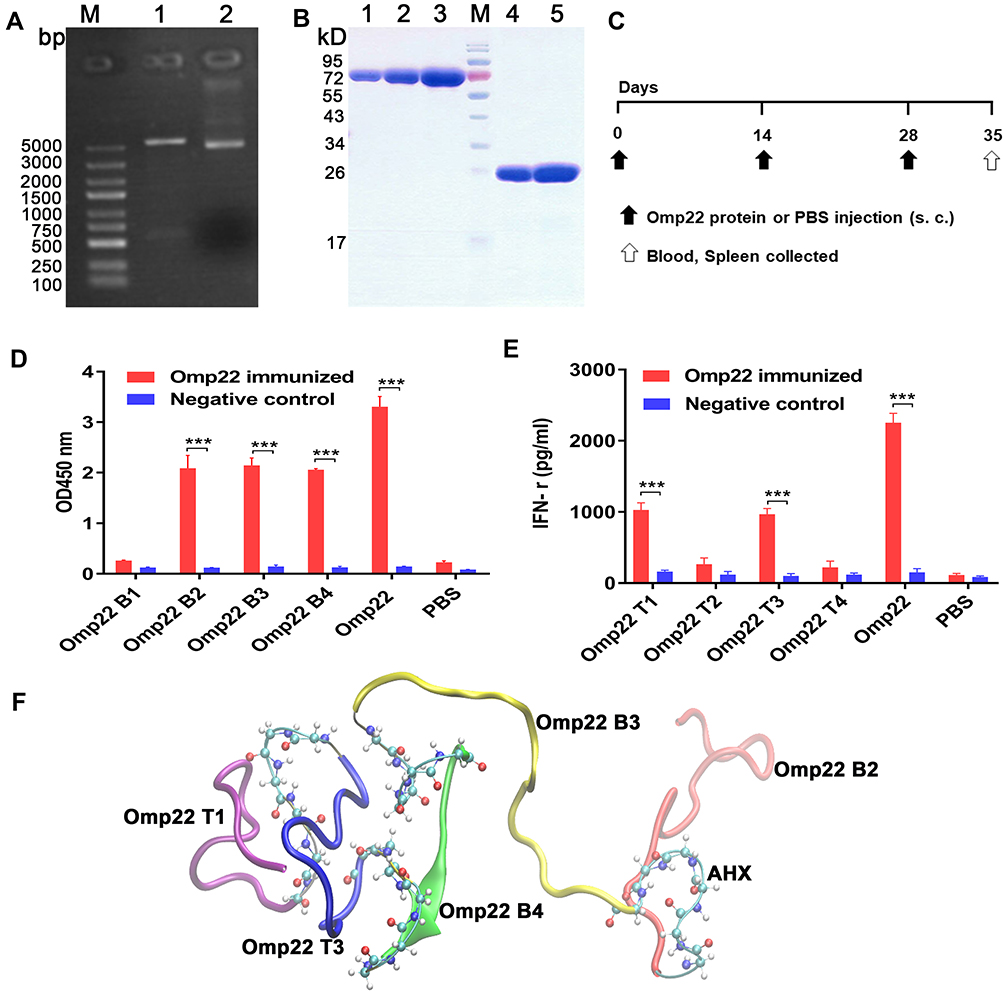 |
Figure 1 Epitope prediction and design of the multiepitope peptide rOmp22. In all studies, ***P < 0.001. Abbreviation: AHX, 6-aminocaproic acid. Notes: (A) Recombinant plasmid pET-28a-omp22.LaneM is the marker, lane 1 is the pET-28a-omp22 digested with BamHI and Hind III, and lane 2 is the undigested pET-28a-omp22. (B) SDS-PAGE of purified recombinant Omp22 protein. LaneM is the protein marker, lanes 1, 2 and 3 are Bovine Serum Albumin (BSA) calibrators at 1μg, 2μg and 4μg, respectively; lanes 4 and 5 are His-omp22 protein 2μg and 4μg, respectively. (C) Immunization scheme of mice. (D) Indirect ELISA test of candidate B-cell epitopes and Omp22-immunized mouse serum. (E) IFN-γ levels in the splenocytes supernatant. (F) Tertiary structure of the multiepitope peptide rOmp22, three optimal B-cell epitopes and two optimal T-cell epitopes were conjugated in series by 6-aminocaproic acid. |
Epitope Prediction and Design of the Multiepitope Peptide rOmp22
According to bioinformatics analysis, such as the secondary structure of the epitope, surface accessibility, hydrophilicity, flexibility and antigen index, four candidate B-cell epitope peptides (named Omp22 B1-Omp22 B4) and four candidate T-cell epitope peptides (named Omp22 T1-Omp22 T4) (Table 1) were predicted and chemically synthesized.
 |
Table 1 Locations and Amino Acid Sequences of Candidate Antigen Epitopes of the Omp22 Protein |
BALB/c mice were subcutaneously injected with 100 μg of recombinant Omp22 (1 μg/μL in PBS) or an equal volume of PBS three times with a two-week interval. One week after the last immunization, the blood and spleen were collected for further immunological study. The schedule of vaccination is shown in Figure 1C. Three B-cell epitopes (amino acid sequences: NIPLSQARAQSVKNY, YATLDKVAQTL and SVQLIMP) were recognized in serum from mice immunized with the whole-length Omp22 protein (Figure 1D). Splenocytes from Omp22 protein-immunized mice could be stimulated by two synthetic T-cell epitopes (amino acid sequences: VPSSRIDAQGY and TFDTNKSNIKP) (Figure 1E).
The identified optimal T-cell epitopes and B-cell epitopes were conjugated in series by 6-aminocaproic acid to design and generate a novel multiepitope peptide (MEP), rOmp22 (Figure 1F). The amino acid sequence and secondary structure of MEP rOmp22 are shown in Figure S2A and S2B (Supporting information). MEP rOmp22 with 59 amino acids and a molecular weight of 6535.48 Da was chemically synthesized and identified by liquid chromatography (Figure S3A Supporting information) and mass spectrometry (Figure S3B Supporting information).
Physical-Structural Characterization of Nanoparticles
The characteristics of CS-PLGA-PBS and CS-PLGA-rOmp22 nanoparticles were summarized in Table 2. Transmission electron microscopy (TEM) techniques were employed to assess the morphology and size of the nanoparticles. By TEM analysis, CS-PLGA-PBS (Figure 2A) and CS-PLGA-rOmp22 (Figure 2B) NPs appeared to be apparently smooth with evenly double spherical structures. Particle size analysis showed that the diameters of CS-PLGA-PBS (Figure 2C) and CS-PLGA-rOmp22 NPs (Figure 2D) were 295.78±21.28 nm and 272.83±15.57 nm, respectively, with nano-range sizes (100–300 nm) and moderate uniformity (PDI of approximately 0.228). As shown in Figure 2E and F, the absolute values of the zeta potential for CS-PLGA-PBS and CS-PLGA-rOmp22 were approximately 3.70 and 4.39 mV, respectively. The stability test of the CS-PLGA-rOmp22 NPs in saline solution showed that there was no nanoparticle aggregation until 72 h (Table S2).
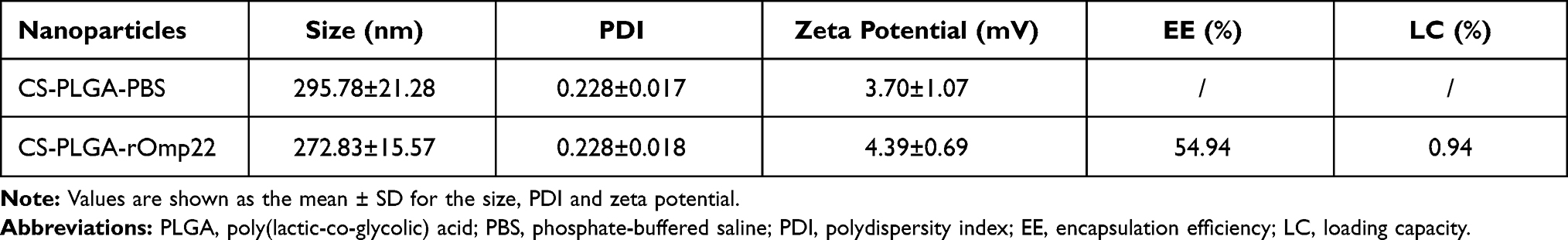 |
Table 2 Physical-Structural Characterization of CS-PLGA Nanoparticles |
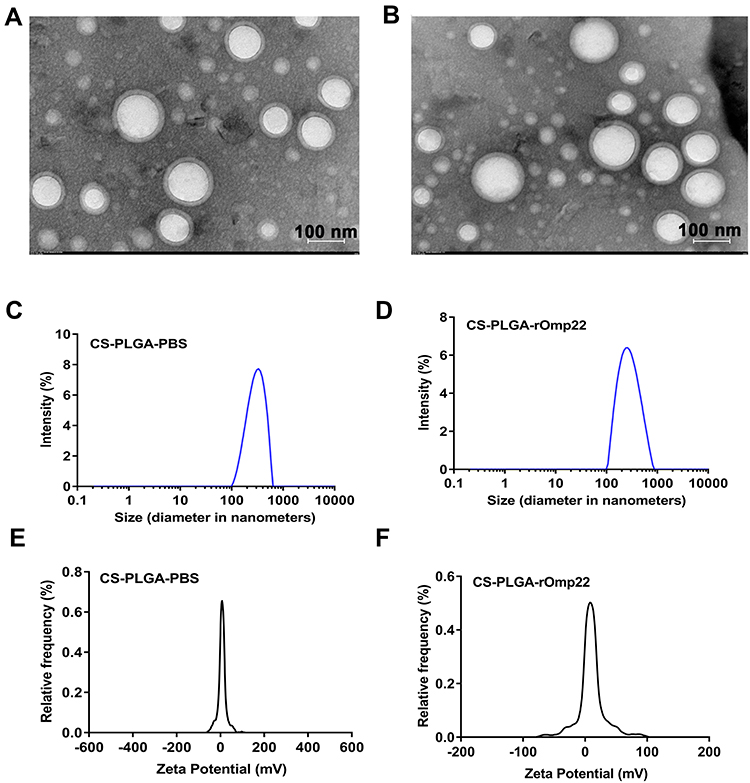 |
Figure 2 Physical-structural characterization of nanoparticles. TEM of CS-PLGA-PBS (A) and CS-PLGA-rOmp22 (B). Size distribution of CS-PLGA-PBS (C) and CS-PLGA-rOmp22 (D). Zeta potential analyses of CS-PLGA-PBS (E) and CS-PLGA-rOmp22 (F). |
Encapsulation Efficiency and Release Studies of CS-PLGA-rOmp22 Nanoparticles
We calculated the encapsulation efficiency (EE) by HPLC. The results showed that the EE was approximately 55% and the loading capacity was approximately 0.94% (Table 2). The release of the rOmp22 peptide from CS-PLGA-rOmp22 NPs was measured by a sustained slow release over a 72 h period. The release of the rOmp22 peptide from CS-PLGA-rOmp22 was 26.2%, 40.6%, 51.5% and 60% of the total encapsulated peptide at 12 h, 24 h, 48 h and 72 h, respectively (Figure 3).
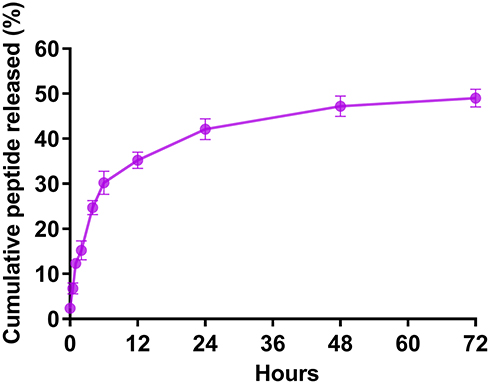 |
Figure 3 In vitro release of rOmp22 from CS-PLGA-rOmp22 NPs. |
Cytotoxic Effect of rOmp22 and CS-PLGA Nanoparticles on A549 Cells
Toxicity is of major concern when using nanoparticles, even biodegradable polymers, in biomedical applications. We tested the toxicity of rOmp22 and nanoparticles against the human lung adenocarcinoma epithelial cell line A549. Both dose- and time-dependent toxicity studies were conducted in A549 cells exposed to rOmp22 at concentrations ranging from 1.25 μg/mL to 80 μg/mL and CS-PLGA nanoparticles at concentrations ranging from 12.5 μg/mL to 800 μg/mL over a 6-, 24- and 48-h time periods. At all examined time points, no concentration of rOmp22 (Figure 4A) and nanoparticles (Figure 4B and C) affected the viability of A549 cells, which demonstrated that they were nontoxic to A549 cells, and substantiated their safety and biocompatibility for studies in vivo.
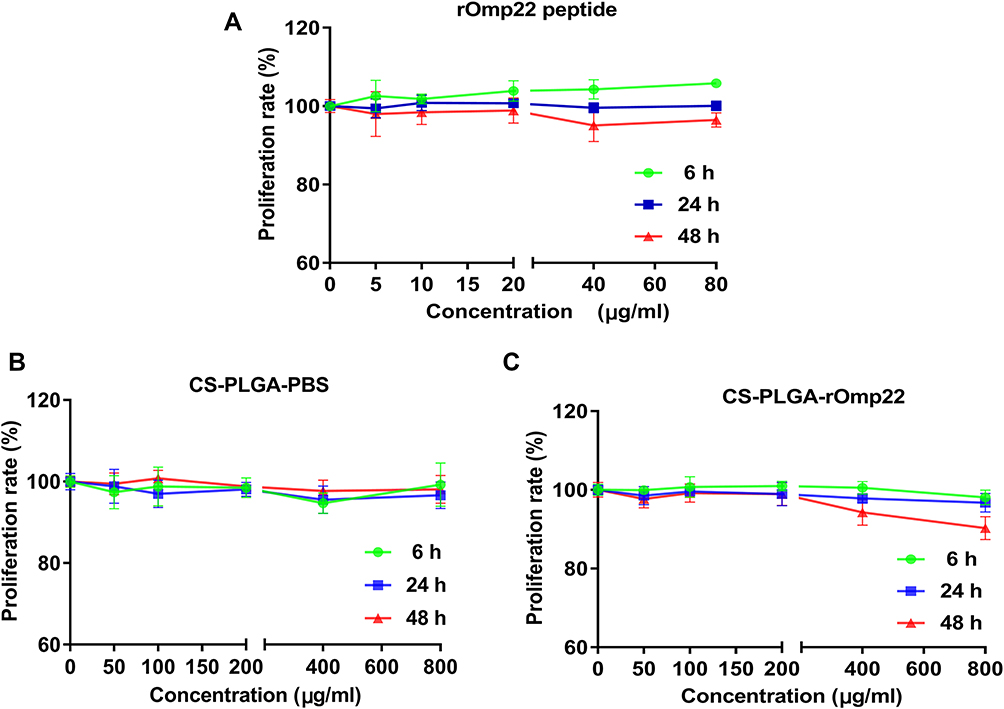 |
Figure 4 The cytotoxicity of rOmp22 (A), CS-PLGA-PBS (B) and CS-PLGA-rOmp22 NPs (C) to A549 cells. The cytotoxicity of rOmp22 and CS-PLGA NPs to A549 cells was detected by Cell Counting Kit-8 (CCK-8) assay. Experiments were performed in triplicate. |
Generation of Antibody-Mediated Immune Responses
To measure the antibody-mediated immune responses at different vaccination regimens, BALB/c mice were subcutaneously injected with 40 μg of rOmp22 or CS-PLGA-rOmp22 nanovaccine containing an equal amount of rOmp22. Mice injected with CS-PLGA-PBS or adjuvant were as control groups. Serum samples were collected from immunized mice after the third immunization (Figure 5A). We investigated the capacity of encapsulated rOmp22 to potentiate antibody immune responses in mice by quantifying rOmp22-specific antibodies using ELISA. As shown in Figure 5B, immunization with CS-PLGA-rOmp22 elicited significantly (P < 0.01) higher IgG than that of rOmp22 immunized mice. No antigen-specific antibody was detected in the serum from the CS-PLGA-PBS control group or the adjuvant-treated group.
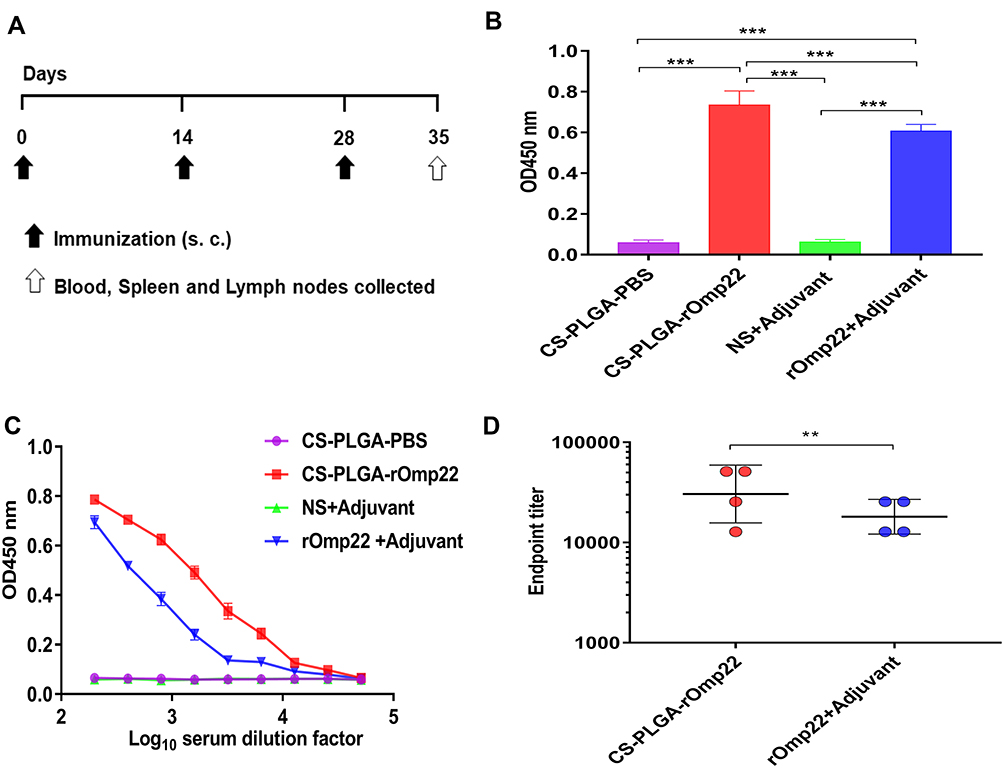 |
Figure 5 Humoral immune responses in BALB/c mice one week after the last immunization. Notes: (A) Immunization and specimen collection process. (B) rOmp22-specific IgG at 1:200 serum dilutions from mice in different groups. (C) rOmp22-specific IgG in serial dilutions from mice one week after the last immunization. (D) The endpoint titer was higher in the CS-PLGA-rOmp22 group than in the nonencapsulated rOmp22 group. |
To further define the robustness of the rOmp22 specific antibody, we next performed serial twofold dilutions of sera to determine rOmp22 specific antibody titers. As shown in Figure 5C and Figure 5D, sera from the CS-PLGA-rOmp22 mice had higher antibody titers and endpoint titers than those from nonencapsulated rOmp22 immunized mice and control mice.
Splenocyte Stimulation and Measurements of the Cytokines IL-4 and IFN-γ
Splenocytes were isolated from six mice in each group at day 7 after the third immunization and cultured with rOmp22 stimulation. IL-4 and IFN-γ secretion in the culture supernatants was detected by ELISA. The splenocytes from the CS-PLGA-rOmp22 immunized mice produced higher levels of IFN-γ than those from the rOmp22-immunized group (P <0.01) (Figure 6A), whereas IL-4 secretion showed no difference between the CS-PLGA-rOmp22 group and the non-encapsulated rOmp22 group (Figure 6B).
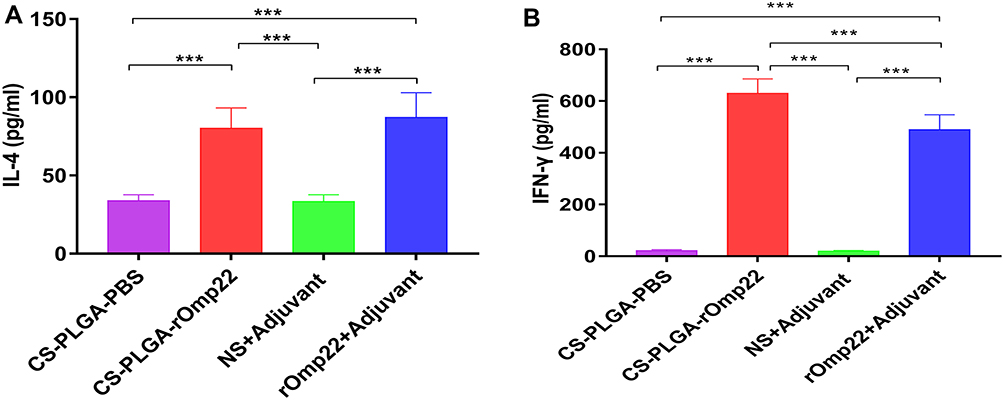 |
Figure 6 Cytokine levels, IL-4 (A) and IFN-γ (B) concentrations released from splenocyte supernatants of the adjuvant control and immunized groups. In all studies, ***P < 0.001. |
Flow Cytometry Analysis
Seven days after the last immunization, cells were harvested from the spleen and the draining lymph node. Flow cytometry analysis showed that the nanovaccine CS-PLGA-rOmp22 induced twofold CD19+ B cells and fewer CD3+ T cells in the lymph node after immunization than nonencapsulated rOmp22 (Figure S4, Figure 7A and C). There was no difference in CD19+ cell levels in the spleen among mice in different groups (Figure 7B). The frequencies of CD3+ cells were somewhat increased in the spleen of the CS-PLGA-rOmp22-immunized group (Figure 7D). No significant differences were observed in the frequencies of CD11c+ dendritic cells (DCs) and CD11b+ F4/80+ macrophages (MACs) in the spleen and draining lymph nodes (Figure 7E–H). Compared to the unencapsulated rOmp22 peptide, the CS-PLGA nanovaccine increased the expression of major histocompatibility complex class II (MHCII) markers on CD11c+ DCs by twofold in the draining lymph node (Figure 7I), while no significant difference was observed in the spleen across any group (Figure 7J). The expression of MHC II markers on CD11b+ F4/80+ MACs in the spleen and lymph nodes increased in the CS-PLGA-rOmp22 administered mice, although there was no significant difference compared with the nonencapsulated rOmp22 group (Figure 7K and L).
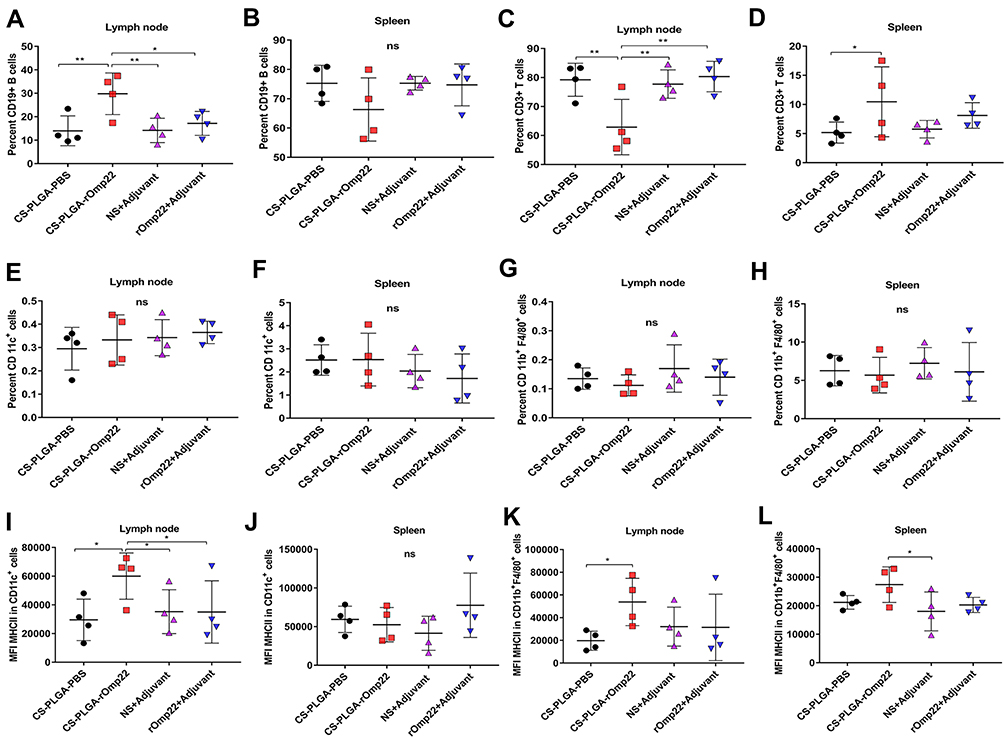 |
Figure 7 Flow cytometry analysis of the draining lymph nodes and splenocytes. Abbreviation: MFI, mean fluorescence intensity. Notes: (A–D)The scatterplot presents the percentage of CD19+B cells and CD3+T cells in the lymph node and spleen 7 days after the last immunization. (E–H) The scatterplot presents the percentage of CD11c+ dendritic cells or CD11b+F4/80+ cells in the lymph node and spleen. (I–L) Scatterplots present the major histocompatibility complex class II (MHC II) expression on CD11c+ dendritic cells or CD11b+F4/80+ cells in the lymph node and spleen. Statistics were performed using one-way ANOVA with Tukey’s post hoc correction (n=4). In all studies, *P < 0.05, **P < 0.01 . |
Immunization Reduced Bacterial Loads
Antibodies and cell-mediated immunity are predictors of immune responses. To determine whether the CS-PLGA-rOmp22 vaccine conferred protection, we assessed its effectiveness using an acute pneumonia model in BALB/c mice. Two weeks after the last boost immunization, all groups were challenged with lethal doses of A. baumannii ATCC19606 (2×108 CFU/mouse) and three clinical A. baumannii strains, CS-MDR-AB (1×109 CFU/mouse), CRAB (5×108 CFU/mouse) and PDR-AB (5×108 CFU/mouse) via the trachea. The drug sensitive test results of the four A. baumannii strains are shown in Table S3. Six mice were randomly selected from each group for the detection of bacterial load in the blood and lung tissue (Figure 8A).
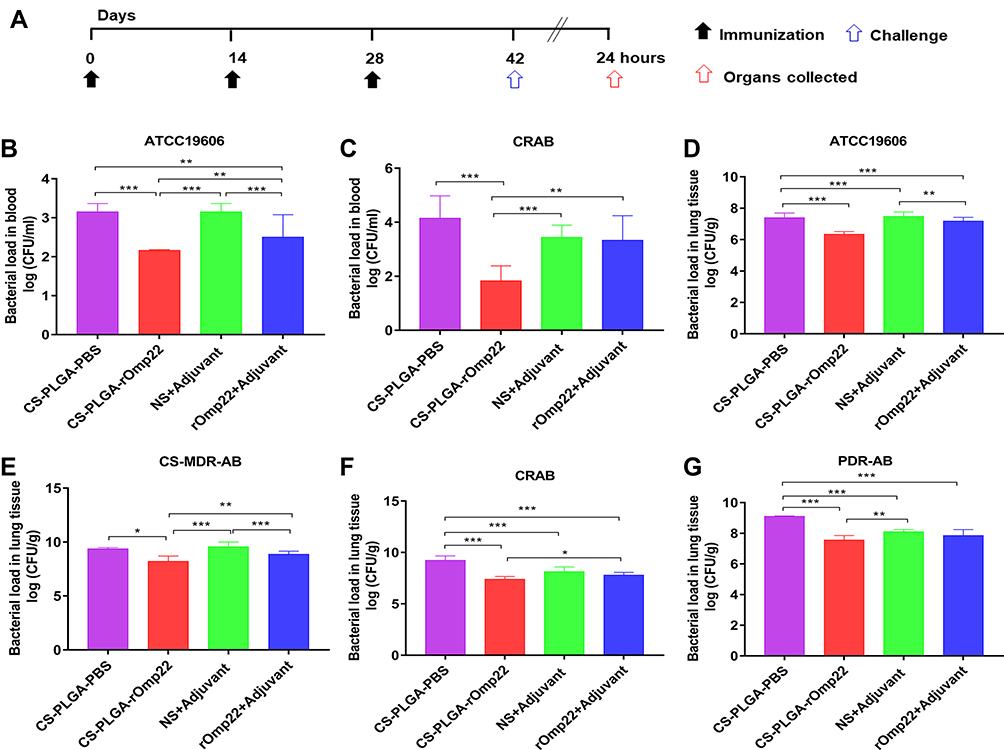 |
Figure 8 Bacterial loads in the blood and lung tissues of BALB/c mice. Notes: (A) Immunization and A. baumannii challenge process. Two weeks after the last immunization, BALB/c mice were intratracheally infected with lethal doses of A. baumannii ATCC19606 (2×108 CFU/mouse) and three clinical A. baumannii strains, CS-MDR-AB (1×109 CFU/mouse), CRAB (5×108 CFU/mouse) and PDR-AB (5×108 CFU/mouse). (B and C) Bacterial loads in the blood of mice at 24 hours post-challenge. (D–G) Bacterial loads in lungs. Lungs were removed at 24h after intratracheal challenge with A. baumannii. Bars indicate the mean ± SD (n=6). *P < 0.05, **P < 0.01, ***P < 0.001. |
Blood samples collected from six mice in each group at 24 h post-challenge were serially diluted and plated on LB agar plates followed by incubation at 37°C overnight. The number of CFUs was counted, and the log10 CFU/mL was calculated and compared. Mice immunized with CS-PLGA-rOmp22 had lower bacterial loads in the blood than mice from the other three groups (Figure 8B and C).
After blood collection, six mice in each group were sacrificed, and the lungs were collected. The right lung lobes were prepared for CFU assessment. As shown in Figure 8D–G, the lungs from the CS-PLGA-rOmp22 groups showed a significantly lower bacterial burden than those from the two control groups. Furthermore, the bacterial burden was significantly reduced in the CS-PLGA-rOmp22 vaccinated group compared with the nonencapsulated rOmp22 group. These results indicated that immunization with CS-PLGA-rOmp22 could partially reduce the colonization of A. baumannii in the lungs of mice.
Pathological Changes in Mouse Lung Tissue
The left lung tissue was aseptically collected 24 hours post-challenge, fixed in 4% buffered formalin, stained with haematoxylin-eosin and observed under a microscope. As shown in Figure 9A, the lung slices of the CS-PLGA-rOmp22 group showed less extensive lung lesions, alveolar oedema, lymphocyte infiltration and structural damage caused by the inflammatory response than those of the rOmp22 vaccine group, CS-PLGA-PBS group and adjuvant control group. Moreover, in the CS-PLGA-rOmp22 vaccine groups, there were more normal structures and clearer alveoli than in the other three groups. Figure 9B shows lung slices of unimmunized and uninfected normal BALB/c mice raised in the same environment and of the same age as other mice. The severity score results showed that the severity of lung injury in the CS-PLGA-rOmp22 groups was significantly lower than that in the other groups (Figure 9C–F), suggesting that mice immunized with the CS-PLGA-rOmp22 vaccine showed a decreased inflammatory response in the lungs.
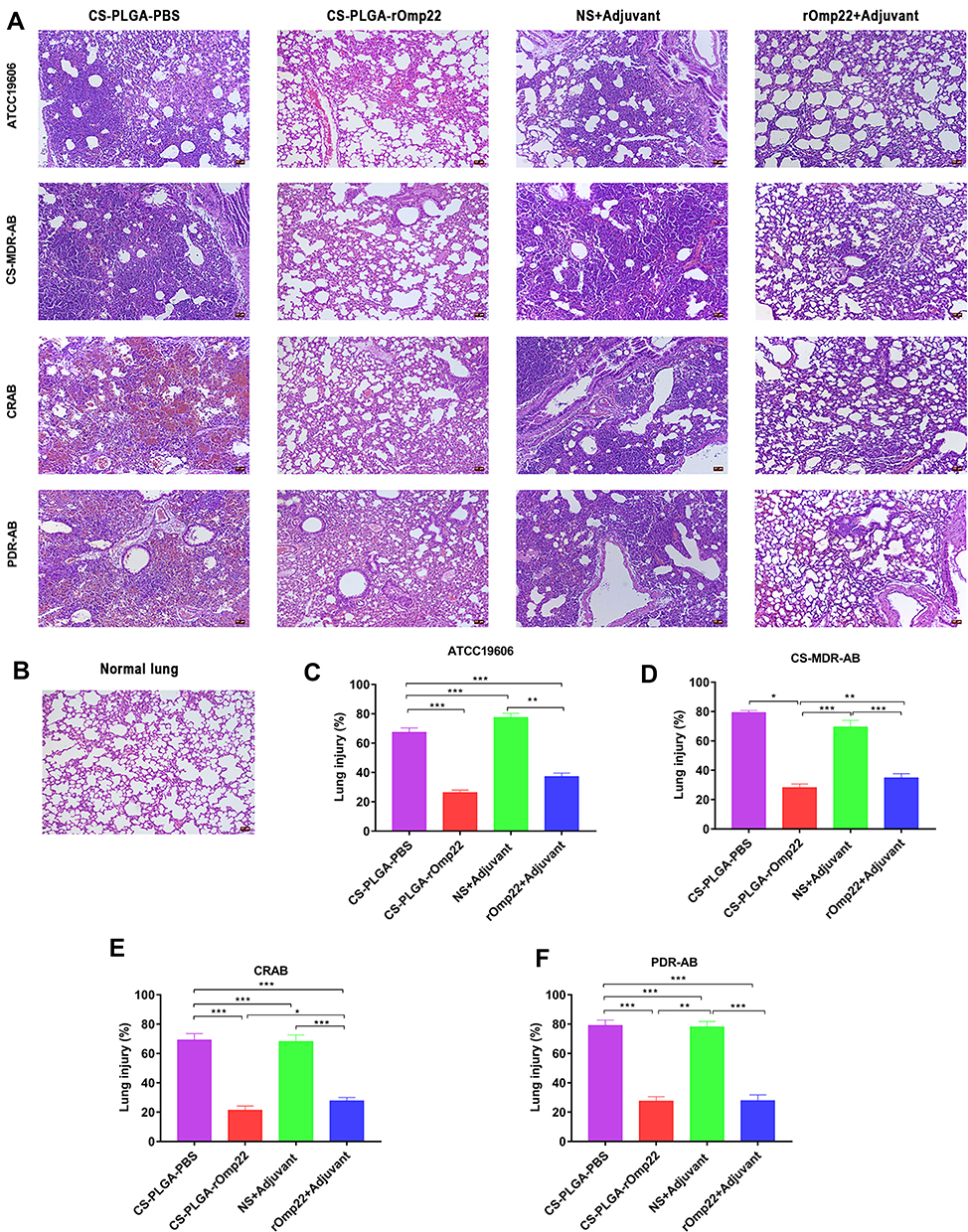 |
Figure 9 Lung histopathology. Notes: Lung specimens were taken from six mice challenged with A. baumannii in each group at 24 h post-challenge. Lung tissue sections were stained with haematoxylin-eosin and observed under a microscope (200×). (A) Lung tissue from different immunized groups subjected to A. baumannii ATCC 19606 and three clinical A. baumannii strains (scale bar, 50 μm). (B) Lung tissue from unimmunized uninfected normal BALB/c mouse showing normal histological characteristics (Scale bar, 50 μm). (C–F) Semiquantitative analysis of the inflammatory area in the lung tissue (n=6). Histograms show the mean percentage of lesion area within the total lung. Data are presented as the means± SD (n=6). *P < 0.05, **P < 0.01, ***P < 0.001. |
Survival Rate, Body Weight Changes and Clinical Scores of Mice Post-Challenge
Six mice challenged with A. baumannii were randomly selected for recording survival, body weight changes and clinical scores every day for seven days. As shown in Figure 10A–D, all mice in the CS-PLGA-PBS- and adjuvant-treated groups died 72 h post-challenge. After the lethal dose of ATCC19606, CS-MDR-AB, CRAB and PDR-AB strains challenged, the 7-day survival rates of mice immunized with CS-PLGA-rOmp22 were 83.33%, 71.43%, 66.67% and 57.14%, respectively, which were significantly higher than those immunized with rOmp22 (60%, 60%, 50% and 42.86%, respectively).
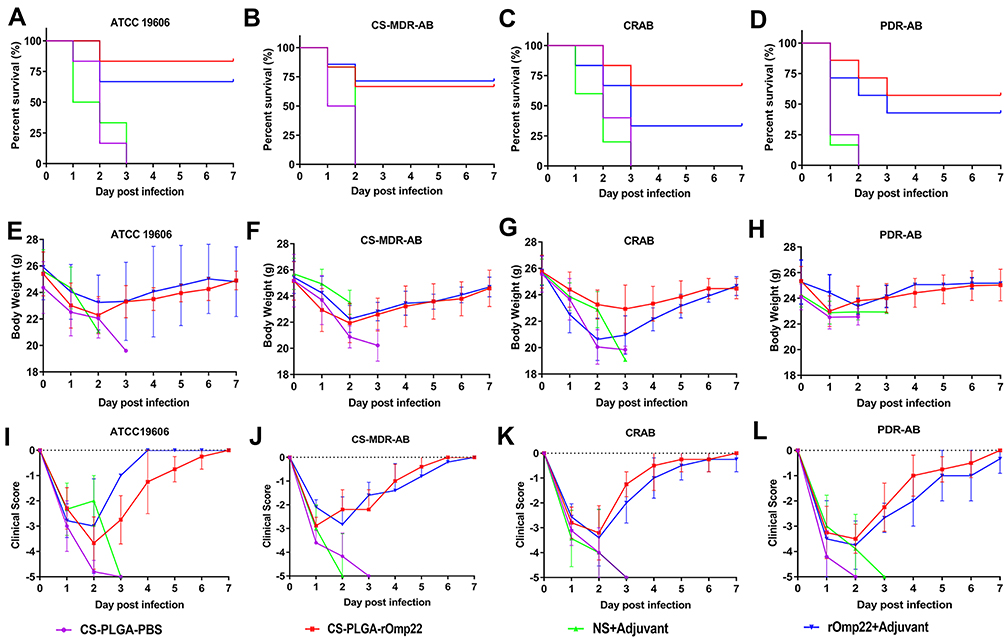 |
Figure 10 Survival rates, body weight changes and clinical scores each day after A. baumannii challenge. Notes: (A–D) Survival rates. Data are shown as the percentage survival at each time point. (E–H) Body weight of mice in each group post challenge. (I–L) Clinical scores of mice post-challenge. |
After A. baumannii challenge, the body weight (Figure 10E–H) and clinical symptom scores (Figure 10I–L) of each group decreased to their lowest levels two to three days post infection. Then, the symptoms of mice in the CS-PLGA-rOmp22 group and rOmp22 vaccinated group gradually improved. The body weight of the mice returned to that before challenge, and the symptoms disappeared seven days post-challenge. These results demonstrated that mice immunized with the CS-PLGA-rOmp22 nanovaccine were better protected from lethal doses of A. baumannii infection than mice vaccinated with nonencapsulated rOmp22.
Discussion
Multidrug-resistant A. baumannii is a rapidly emerging pathogen causing infections with high mortality rates due to inadequate medical treatment.1,2,6 New ways to prevent and treat such infections are of critical medical need. Despite decades of effort in the development of A. baumannii vaccines, there is still no effective vaccine against this pathogen.
Recently, there has been increasing interest in the development of vaccines that use only minimal components from pathogens. Such vaccines are based on recombinant proteins or even minimal fragments carrying immunological information from this protein, called peptide epitopes.26,36 Epitopes or antigenic determinants are the minimal immunogenic part of any particular antigen and are capable of inducing specific immune responses.26 In this research, we chose A. baumannii Omp22, a highly conserved and highly immunogenic protein, as the candidate antigen. Bioinformatics techniques and immunological methods were used to predict and identify optimal T-cell and B-cell epitopes on the Omp22 protein. Then, the identified dominant epitopes were connected in series by 6-aminocaproic acid and chemically synthesized to generate a novel multiepitope peptide rOmp22, which preserved the antigenic epitopes and avoided toxic structural fragments of Omp22.
Peptide-based subunit vaccines hold great potential as safer and more efficient alternatives to traditional vaccination strategies. However, nontoxic and effective delivery systems are needed to protect their respective immunogens from rapid degradation and to potentiate immunological responses.37 A novel type of composite microsphere, CS-PLGA, was demonstrated to possess the advantages of improving the stability of encapsulated proteins and increasing the subsequent release. In this view, we encapsulated the multiepitope peptide rOmp22 in CS-PLGA NPs with a high encapsulation efficiency rate and slow release pattern. To the best of our knowledge, this study was the first to undertake the encapsulation of the multi-epitope peptide rOmp22 in CS-PLGA NPs, physical-structural characterization studies, and elucidation of its immunogenicity and protection efficacy. Additionally, there was no report of CS-PLGA NP delivery system used in the design of bacterial epitope vaccines was retrieved prior to this study.
The morphological characteristics of nanoparticles can affect the release of biomaterials from nanoparticles. A leaky or porous structure enhances the release of biomaterials by diffusion, whereas a smooth surface reduces burst release.38 The physical and structural characteristics of CS-PLGA-rOmp22 revealed that it had a homogeneous morphology with a smooth spherical shape. CS-PLGA-rOmp22 was also uniform in particle size distribution and fairly dispersed without aggregation. The zeta potential value is one of the most important particle characterization parameters because it can affect both particle stability and particle adhesion.39 As shown in Figure 2E and F, CS-PLGA NPs exhibited a positive charge, which could adsorb more peptides, increase encapsulation efficiency and improve stability.
The release pattern of a peptide in nanoparticles is important for the development of a nanovaccine, since it can influence the immune response and the immunization regimen, such as the peptide concentration and the frequency of immunization. In the present study, the CS-PLGA-rOmp22 release profiles were biphasic, characterized by a burst of peptide followed by a sustained release. The burst release on the first day could induce a strong immune response. The sustained slow release of the peptide was an attractive property for a vaccine candidate as this might reduce the number of immunizations as well as enhance the presentation of the peptide to APCs. Cell toxicity studies showed that CS-PLGA NPs were not toxic to cells at concentrations as high as 800 μg/mL, thus indicating the safety of this delivery system as reported by several researchers.29,30
The present study showed that subcutaneous administration of the CS-PLGA-rOmp22 nanovaccine induced systemic antibody responses. As shown in Figure 5B–D, high titers of antigen-specific IgG antibody were detected in the serum of mice immunized with CS-PLGA-rOmp22 after boosting twice. In addition, CS-PLGA-rOmp22 vaccination induced much higher levels of antigen-specific IFN-γ and IL-4 secretion in the spleen cell culture supernatants in the CS-PLGA-rOmp22 and non-encapsulated rOmp22 groups compared to those in control mice. The CS-PLGA-rOmp22 vaccination induced much higher levels of antigen-specific IFN-γ production from splenocytes than the rOmp22-immunized group (P <0.01), whereas IL-4 secretion showed no difference between the CS-PLGA-rOmp22 group and the nonencapsulated rOmp22 group (Figure 6A and B). These results indicated that humoural and cellular immune responses, especially Th1-type immune responses, were induced and created full protection.
The challenge experiments proved that the mice immunized with CS-PLGA-rOmp22 acquired potent protection against infection with A. baumannii ATCC 19606 and three clinical A. baumannii strains. The bacterial load in the blood of the mice inoculated with CS-PLGA-rOmp22 after infection was much lower than that of the nonencapsulated rOmp22 group and the control groups. Almost no pathological change was observed in the lung tissue of the mice immunized with CS-PLGA-rOmp22. These results indicated that the high titer of antigen-specific antibody contributed to potent protection in the mice immunized with CS-PLGA-rOmp22.
Conclusion
In summary, this work reported a novel multiepitope peptide nanovaccine against A. baumannii. CS-PLGA-rOmp22 NPs elicited specific IgG antibodies, Th1 cellular immunity and protection against acute lethal intratracheal A. baumannii challenge in BALB/c mice. Our results indicate that this nanovaccine is a desirable candidate to prevent A. baumannii infection.
Ethics Approval
The animal experimental procedures were approved by the Ethics Committee of Animal Care and Welfare, Nanjing Medical University (Nanjing, China) (Approval Number: IACUC-1904044).
Acknowledgments
This research was supported by the National Natural Science Foundation of China (NO.81870009), Beijing Medical and Health Foundation (YWJKJJHKYJJ-B17543) and Science and Technology Development Foundation of Nanjing Medical University (NMUB2019053 and NMUB2018036).
We appreciate suggestions from Wen Qiu of Nanjing Medical University. We thank Yanfeng Zhao and Shuidi Zhao for providing the clinical strains of A. baumannii and assistance in the microbiology experiments. We further thank Ping Zhou for assistance with the flow cytometry analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vazquez-Lopez R, Solano-Galvez SG, Juarez Vignon-Whaley JJ, et al. Acinetobacter baumannii resistance: a real challenge for clinicians. Antibiotics. 2020;9(4):205. doi:10.3390/antibiotics9040205
2. Chuang YC, Cheng A, Sun HY, et al. Microbiological and clinical characteristics of Acinetobacter baumannii bacteremia: implications of sequence type for prognosis. J Infect. 2019;78(2):106–112. doi:10.1016/j.jinf.2018.10.001
3. Asif M, Alvi IA, Rehman SU. Insight into Acinetobacter baumannii: pathogenesis, global resistance, mechanisms of resistance, treatment options, and alternative modalities. Infect Drug Resist. 2018;11:1249–1260. doi:10.2147/IDR.S166750
4. De Oliveira DMP, Forde BM, Kidd TJ, et al. Antimicrobial Resistance in ESKAPE Pathogens. Clin Microbiol Rev. 2020;33(3):1–49.
5. Harding CM, Hennon SW, Feldman MF. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat Rev Microbiol. 2018;16(2):91–102. doi:10.1038/nrmicro.2017.148
6. Mulani MS, Kamble EE, Kumkar SN, Tawre MS, Pardesi KR. Emerging Strategies to Combat ESKAPE pathogens in the era of antimicrobial resistance: a review. Front Microbiol. 2019;10:539.
7. Garcia-Quintanilla M, Pulido MR, Lopez-Rojas R, Pachon J, McConnell MJ. Emerging therapies for multidrug resistant Acinetobacter baumannii. Trends Microbiol. 2013;21(3):157–163.
8. McConnell MJ, Rumbo C, Bou G, Pachon J. Outer membrane vesicles as an acellular vaccine against Acinetobacter baumannii. Vaccine. 2011;29(34):5705–5710.
9. McConnell MJ, Pachon J. Active and passive immunization against Acinetobacter baumannii using an inactivated whole cell vaccine. Vaccine. 2010;29(1):1–5.
10. McConnell MJ, Dominguez-Herrera J, Smani Y, et al. Vaccination with outer membrane complexes elicits rapid protective immunity to multidrug-resistant Acinetobacter baumannii. Infect Immun. 2011;79(1):518–526. doi:10.1128/IAI.00741-10
11. Fattahian Y, Rasooli I, Mousavi Gargari SL, et al. Protection against Acinetobacter baumannii infection via its functional deprivation of biofilm associated protein (Bap). Microb Pathog. 2011;51(6):402–406. doi:10.1016/j.micpath.2011.09.004
12. Bentancor LV, Routray A, Bozkurt-Guzel C, et al. Evaluation of the trimeric autotransporter Ata as a vaccine candidate against Acinetobacter baumannii infections. Infect Immun. 2012;80(10):3381–3388. doi:10.1128/IAI.06096-11
13. Zhang X, Yang T, Cao J, et al. Mucosal immunization with purified OmpA elicited protective immunity against infections caused by multidrug-resistant Acinetobacter baumannii. Microb Pathog. 2016;96:20–25. doi:10.1016/j.micpath.2016.04.019
14. Singh R, Capalash N, Sharma P. Immunoprotective potential of BamA, the outer membrane protein assembly factor, against MDR Acinetobacter baumannii. Sci Rep. 2017;7(1):12411. doi:10.1038/s41598-017-12789-3
15. Bentancor LV, O’Malley JM, Bozkurt-Guzel C, Pier GB, Maira-Litran T. Poly-N-acetyl-beta-(1-6)-glucosamine is a target for protective immunity against Acinetobacter baumannii infections. Infect Immun. 2012;80(2):651–656. doi:10.1128/IAI.05653-11
16. Huang W, Yao Y, Wang S, et al. Immunization with a 22-kDa outer membrane protein elicits protective immunity to multidrug-resistant Acinetobacter baumannii. Sci Rep. 2016;6:20724. doi:10.1038/srep20724
17. Chen W. Current advances and challenges in the development of Acinetobacter vaccines. Hum Vaccin Immunother. 2015;11(10):2495–2500. doi:10.1080/21645515.2015.1052354
18. Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK. Peptide Vaccine: progress and Challenges. Vaccines. 2014;2(3):515–536. doi:10.3390/vaccines2030515
19. Baseer S, Ahmad S, Ranaghan KE, Azam SS. Towards a peptide-based vaccine against Shigella sonnei: a subtractive reverse vaccinology based approach. Biologicals. 2017;50:87–99. doi:10.1016/j.biologicals.2017.08.004
20. Abdulla F, Adhikari UK, Uddin MK, Exploring T. B-cell epitopes and designing multi-epitope subunit vaccine targeting integration step of HIV-1 lifecycle using immunoinformatics approach. Microb Pathog. 2019;137:103791. doi:10.1016/j.micpath.2019.103791
21. Babu L, Uppalapati SR, Sripathy MH, Reddy PN. Evaluation of recombinant multi-epitope outer membrane protein-based Klebsiella pneumoniae subunit vaccine in mouse model. Front Microbiol. 2017;8:1805. doi:10.3389/fmicb.2017.01805
22. Zahroh H, Ma’rup A, Tambunan US, Parikesit AA. Immunoinformatics approach in designing epitope-based vaccine against meningitis-inducing bacteria (Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type b). Drug Target Insights. 2016;10:19–29. doi:10.4137/DTI.S38458
23. Ren S, Guan L, Dong Y, et al. Design and evaluation of a multi-epitope assembly peptide vaccine against Acinetobacter baumannii infection in mice. Swiss Med Wkly. 2019;149:w20052. doi:10.4414/smw.2019.20052
24. Malonis RJ, Lai JR, Vergnolle O. Peptide-based vaccines: current progress and future challenges. Chem Rev. 2020;120(6):3210–3229. doi:10.1021/acs.chemrev.9b00472
25. Skwarczynski M, Zhao G, Boer JC, et al. Poly(amino acids) as a potent self-adjuvanting delivery system for peptide-based nanovaccines. Sci Adv. 2020;6(5):eaax2285. doi:10.1126/sciadv.aax2285
26. Parvizpour S, Pourseif MM, Razmara J, Rafi MA, Omidi Y. Epitope-based vaccine design: a comprehensive overview of bioinformatics approaches. Drug Discov Today. 2020;25(6):1034–1042. doi:10.1016/j.drudis.2020.03.006
27. Amjadi I, Rabiee M, Hosseini MS, Mozafari M. Synthesis and characterization of doxorubicin-loaded poly(lactide-co-glycolide) nanoparticles as a sustained-release anticancer drug delivery system. Appl Biochem Biotechnol. 2012;168(6):1434–1447. doi:10.1007/s12010-012-9868-4
28. Park K, Skidmore S, Hadar J, et al. Injectable, long-acting PLGA formulations: analyzing PLGA and understanding microparticle formation. J Control Release. 2019;304:125–134. doi:10.1016/j.jconrel.2019.05.003
29. Duran V, Yasar H, Becker J, et al. Preferential uptake of chitosan-coated PLGA nanoparticles by primary human antigen presenting cells. Nanomedicine. 2019;21:102073. doi:10.1016/j.nano.2019.102073
30. Arafa MG, Girgis GNS, El-Dahan MS. Chitosan-Coated PLGA nanoparticles for enhanced ocular anti-inflammatory efficacy of atorvastatin calcium. Int J Nanomed. 2020;15:1335–1347. doi:10.2147/IJN.S237314
31. Wang Y, Li P, Kong L. Chitosan-modified PLGA nanoparticles with versatile surface for improved drug delivery. AAPS PharmSciTech. 2013;14(2):585–592. doi:10.1208/s12249-013-9943-3
32. Vita R, Overton JA, Greenbaum JA, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43(Database issue):D405–D412. doi:10.1093/nar/gku938
33. Vita R, Mahajan S, Overton JA, et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019;47(D1):D339–D343. doi:10.1093/nar/gky1006
34. Fairley SJ, Singh SR, Yilma AN, et al. Chlamydia trachomatis recombinant MOMP encapsulated in PLGA nanoparticles triggers primarily T helper 1 cellular and antibody immune responses in mice: a desirable candidate nanovaccine. Int J Nanomed. 2013;8:2085–2099. doi:10.2147/IJN.S44155
35. Abd Hady WE, Mohamed EA, Soliman OAE, El-Sabbagh HM. In vitro-in vivo evaluation of chitosan-PLGA nanoparticles for potentiated gastric retention and anti-ulcer activity of diosmin. Int J Nanomed. 2019;14:7191–7213. doi:10.2147/IJN.S213836
36. Skwarczynski M, Toth I. Recent advances in peptide-based subunit nanovaccines. Nanomedicine. 2014;9(17):2657–2669. doi:10.2217/nnm.14.187
37. Azim KF, Hasan M, Hossain MN, et al. Immunoinformatics approaches for designing a novel multi epitope peptide vaccine against human norovirus (Norwalk virus). Infect Genet Evol. 2019;74:103936. doi:10.1016/j.meegid.2019.103936
38. Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3:1377–1397. doi:10.3390/polym3031377
39. Liang X, Duan J, Li X, et al. Improved vaccine-induced immune responses via a ROS-triggered nanoparticle-based antigen delivery system. Nanoscale. 2018;10(20):9489–9503. doi:10.1039/C8NR00355F
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.