Back to Journals » Journal of Pain Research » Volume 14
A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study of the Efficacy, Tolerability, and Safety of Celecoxib Oral Solution (ELYXYB) in Acute Treatment of Episodic Migraine with or without Aura
Authors Lipton RB ![]() , Munjal S
, Munjal S ![]() , Tepper SJ, Iaconangelo C, Serrano D
, Tepper SJ, Iaconangelo C, Serrano D
Received 1 June 2021
Accepted for publication 9 August 2021
Published 19 August 2021 Volume 2021:14 Pages 2529—2542
DOI https://doi.org/10.2147/JPR.S322292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Alexandre F DaSilva
Richard B Lipton,1 Sagar Munjal,2 Stewart J Tepper,3 Charlie Iaconangelo,4 Daniel Serrano4
1The Saul R. Korey Department of Neurology, Department of Psychiatry and Behavioral Sciences, Department of Epidemiology & Population Health, Albert Einstein College of Medicine and Montefiore Headache Center, New York, NY, USA; 2Operations & Medical Affairs Proprietary Products, Dr. Reddy’s Laboratories Inc., Princeton, NJ, USA; 3Geisel School of Medicine, Dartmouth College, Lebanon, NH, USA; 4Pharmerit - An Open Health Company, Bethesda, MD, USA
Correspondence: Sagar Munjal Email [email protected]
Background: Safe, effective, oral therapies are needed for acute treatment of migraine. This clinical trial assessed the efficacy, tolerability, and safety of celecoxib oral solution (ELYXYB) in a single migraine attack associated with moderate-to-severe pain.
Methods: This was a phase III, randomized (1:1), double-blind, placebo-controlled trial, conducted at 41 US centers from December 2016 to October 2017. Adults with episodic migraine (with or without aura) for ≥ 1 year were treated with a single 4.8 mL dose of 120-mg celecoxib oral solution or placebo. Co-primary endpoints were the proportion of patients who were pain-free and free from the most bothersome migraine symptom (MBS) at 2 hours post-dose. The MBS was identified at screening from among nausea, photophobia, or phonophobia.
Results: Six hundred thirty-one patients were randomized (celecoxib oral solution, n=316; placebo, n=315; mean age 41 years, range 18– 75; 84.3% female). One study site met prespecified outlier criteria (defined as a treatment effect estimate that was at least twice as large as all other sites) and was excluded from efficacy analyses. This site had a mean 2-hour pain freedom placebo response rate of 75% vs a combined mean of 23.5% for all other sites. In subsequent analysis, 2-hour post-dose pain freedom response rates were significantly higher in the celecoxib oral solution group vs placebo (32.8%, [27.2%, 38.8%]) vs 23.5%, [18.5%, 29.2%]; P=0.020). For 2-hour post-dose MBS freedom, response rates were significantly higher in the celecoxib oral solution group vs placebo (58.1% [51.4%, 64.5%] vs 43.9% [37.2%, 50.7%]; P=0.003). A total of 10.7% (31/289) of patients treated with celecoxib oral solution and 9.9% (28/283) of placebo-treated patients reported a treatment-emergent adverse event (TEAE). Study drug-related TEAEs were reported by 7.3% (21/289) and 7.4% (21/283) of celecoxib oral solution and placebo patients, respectively; the most common were nausea (celecoxib oral solution: 1.4% [4/289] vs placebo: 1.8% [5/283]) and dysgeusia (celecoxib oral solution: 1.7% [5/289] vs placebo: 1.1% [3/283]). No serious TEAEs, deaths, or drug-related TEAEs leading to withdrawal were reported.
Conclusion: Celecoxib oral solution is a safe, effective COX-2-selective nonsteroidal anti-inflammatory drug for the treatment of acute migraine. In this analysis, celecoxib oral solution was significantly more effective than placebo and was also associated with a low rate of gastric TEAEs. Celecoxib oral solution may provide a convenient, alternate option to currently available treatments.
Trial Registration: ClinicalTrials.gov Identifier: NCT03009019; registered January 4, 2017; retrospectively registered, https://clinicaltrials.gov/ct2/show/NCT03009019.
Keywords: cyclooxygenase 2 inhibitors, migraine disorders, non-steroidal anti-inflammatory agents, clinical trial, phase III
Plain Language Summary
- Patients with migraine lack safe, effective, and rapid-acting oral therapies for acute treatment.
- Celecoxib oral solution (ELYXYB) is a new liquid formulation of celecoxib, a COX-2 selective nonsteroidal anti-inflammatory drug (NSAID) with a more rapid onset of action than available celecoxib capsule formulations.
- Selective COX-2 NSAIDs have a reduced incidence of gastrointestinal adverse events compared to non-specific NSAIDs.
- This phase 3 clinical trial aimed to assess the efficacy, tolerability, and safety of celecoxib oral solution in a single, moderate-to-severe migraine attack among 631 patients with migraine.
- Two hours after treating the migraine, patients who used celecoxib had statistically significant higher rates of pain freedom (32.8%; P=0.020) and freedom from their most bothersome migraine symptom (58.1%; P=0.003) compared to patients treated with placebo (23.5% and 43.9%).
- Adverse event rates were similar for both groups, and gastric adverse events were infrequent.
- Celecoxib oral solution may provide an additional, convenient option for patients requiring migraine treatment.
Introduction
Migraine is a common neurologic disorder associated with disabling headache requiring acute treatment1 that is estimated to affect between 17.1% and 20.7% of females and between 5.6% and 9.7% of males in the United States.2,3 Beyond headache pain, the most prevalent symptoms of migraine include light sensitivity (photophobia), sound sensitivity (phonophobia), and nausea.1,4–6 Other common symptoms include hyperalgesia (an increased sensitivity to painful stimuli) and cutaneous allodynia (dermal pain provoked by non-painful stimuli).7
Key clinical research and expert guidance recommend the early acute treatment of migraine via targeted medications administered shortly after the headache begins.8,9 The primary goal of treatment is to safely and quickly achieve freedom from migraine pain and symptoms, without recurrence. However, as migraine severity and characteristics vary, patients typically require individualized approaches to achieve success.8 Triptans are the mainstay of acute migraine treatment and are available in several formulations.10 Oral triptans have a relatively slow rate of absorption (median time to peak plasma concentration [Tmax]: ~1.5 hours), and a corresponding lag in onset of action, which can result in delayed pain relief.11,12 Liquid triptan nasal sprays are more rapid-acting than oral triptans, but conventional intranasal spray pumps deposit only a limited amount of drug (2.5%) to the upper posterior nasal cavity, and a substantial amount of the dose is swallowed. Once swallowed, the triptan effectively becomes an orally administered medication subject to the previously noted disadvantages.13 Sumatriptan subcutaneous injections have a more rapid onset of action than oral triptans, but also have lower tolerability and patient acceptance.14,15 Other drugs, such as opioids, have been shown effective in treating migraine headache.1,8 Opioids are not recommended as regular1 or first-line therapy,16 in part because they increase patient risk for the development of chronic migraine17 along with potential addiction potential. Opioids also have high adverse event (AE) rates18–20 that affect up to 92% of patients who take these drugs for acute pain,21 and that lead to discontinuation in approximately 1 in 5 patients with episodic migraine.22
Some patients with migraine respond well to simple pain relievers, as measured by 2-hour post-dose pain relief.1 For example, nonsteroidal anti-inflammatory drugs (NSAIDs) have analgesic, antipyretic, and anti-inflammatory effects.23 Several NSAIDs, including diclofenac, ibuprofen, and naproxen, are established as effective in migraine and are recommended treatment options for acute attacks.1 These non-selective NSAIDs inhibit the biosynthesis of prostaglandins (which play a key role in migraine pathogenesis)24 through two isoenzymes, cyclooxygenase (COX)-1 and COX-2. However, up to 60% of patients experience gastrointestinal (GI) AEs with non-selective NSAIDs; these problems include GI bleeding and ulcers, which are related to COX-1 inhibition.23,25 However, the analgesic efficacy of NSAIDs is associated with COX-2 inhibition.23 Furthermore, since migraine is a chronic condition10 requiring episodic treatment,8,9 it is important to recognize that the cumulative consumption of non-selective NSAIDs can lead to serious AEs, including GI, cardiovascular, and renal events.26 Predicated on this, the use of COX-2-selective versus non-selective NSAIDs may provide a therapeutic advantage in the acute management of migraine.23
Celecoxib is a COX-2 selective NSAID; it has an analgesic profile similar to other NSAIDs, alongside a more favorable GI profile.27,28 A systematic review and meta-analysis of celecoxib capsules in patients with osteoarthritis and rheumatoid arthritis found a significantly reduced incidence of GI AEs with celecoxib capsules compared to non-specific NSAIDs such as naproxen (relative risk [RR]: 0.61 [95% CI: 0.55–0.67]). Incidence was also reduced non-significantly when celecoxib was compared to the COX-2-specific NSAID rofecoxib (RR: 0.72 [95% CI: 0.49–1.06]).27 In 2007, Loo et al evaluated 400-mg celecoxib capsules versus 550-mg naproxen sodium in an open-label, randomized, controlled study (N=60) for acute treatment of patients with migraine. Celecoxib capsules showed similar pain reduction as naproxen, based on Visual Analogue Scale results, as well as significantly reduced gastric pain incidence.29 However, celecoxib capsules are slow to provide pain relief, with a median Tmax of 2.5 hours.30 Because rapid pain relief is an important treatment goal,30,31 and due to GI issues with oral NSAIDs,30 celecoxib capsules have not been approved for the acute treatment of migraine in the U.S.32
ELYXYB (celecoxib oral solution, 25 mg/mL, previously known as DFN-15) is a new liquid formulation of celecoxib, approved in May 2020 for the acute treatment of migraine with or without aura in adults.33 It has a shorter demonstrated Tmax (median 0.7 hours) than available celecoxib capsule formulations,30 and has been evaluated and found effective versus placebo in a prior phase 3 randomized, double-blind, placebo-controlled trial of patients with episodic migraine (N=622). In this trial, celecoxib oral solution was significantly superior at 2 hours post-dose to placebo for the co-primary endpoints of pain freedom (35.6% vs 21.7%; P<0.001) and freedom from the most bothersome symptom (MBS) (57.8% vs 44.8%; P=0.007).32 The current trial was the second of two phase 3 placebo-controlled evaluations required by the US Food and Drug Administration (FDA) to substantiate the safety and effectiveness of celecoxib oral solution.
Materials and Methods
Study Design
This was a randomized, controlled, double-blind study conducted at 41 US centers from December 2016 to October 2017. Study sites were selected based on predefined criteria that included previous migraine trial experience and a history of successful patient enrollment. Patients were randomized 1:1 to receive the following: celecoxib oral solution (120 mg/4.8 mL celecoxib, liquid formulation with celecoxib 25 mg/mL) or matching placebo (0 mg/4.8 mL). The randomization scheme was generated by the biostatistics group of INC Research (now Syneos Health, Morrisville, NC). An interactive web response system assigned the appropriate study kit to each patient. All randomization data were kept strictly confidential and accessible only to authorized personnel until unblinding at study end. Using the assigned treatment, a single migraine attack, associated with moderate-to-severe pain, was treated as soon as possible (ie, within ≤1 hour). Treatment was only to be used for a new attack, not a recurrence, and ≥48 hours of freedom from pain and symptoms due to a previous migraine attack was required. The total duration of participation was up to 10 weeks from baseline.
Patients were provided with an electronic diary (eDiary) to record all pre-dose and post-dose efficacy data. This included migraine date and time, last food intake and fat content, pre-dose pain and functioning levels, and MBS data. Patients also recorded the following information: time to meaningful pain relief and pain freedom; post-dose pain levels and the Patient Perception of Migraine Questionnaire-Revised (PPMQ-R); and treatment satisfaction, all measured at predefined time points. Following treatment, patients had the option to take rescue medication after the 2-hour post-dose timepoint. Rescue medication selection was determined by the investigator and participant, and was exclusive of study-prohibited medications.
Study Patients
Patients were required to be previously diagnosed with episodic migraine, with or without aura, as defined by International Classification of Headache Disorders, 3rd edition (ICHD-3), and to have had the diagnosis for ≥1 year. Additional inclusion criteria were as follows: male or female, aged 18 to 75 years; migraine onset prior to 50 years of age; history of episodic migraine (ICHD-3) with 2–8 monthly attacks (with or without aura), ≤14 headache days per month, and ≥48 hours of headache-free time between attacks; reported usual migraine pain (without treatment) of 2 (moderate) or 3 (severe) on headache pain severity scale (range 0–3); able to differentiate between migraine and a tension-type or cluster headache; if female and of childbearing potential, a negative pregnancy test was required at all study visits; female patients were required not to be lactating, not to be planning to become pregnant, and to practice reliable birth control or abstinence throughout the study. Participants had to read, speak, and understand English proficiently, to provide written, informed consent, and to be willing and, in the opinion of the investigator, able to comply with study procedures and scheduling.
Exclusion criteria were as follows: prior exposure to celecoxib oral solution; intolerance to any celecoxib or sulfonamide formulation, or significant AEs or contraindications related to other NSAIDs (eg, due to gastrointestinal bleed, ulcer, or history of acute renal failure); treatment with an investigational drug or device within 30 days of randomization, or participation in a central nervous system clinical trial within 2 months of randomization. Patients were also excluded if they had experienced medication overuse in the 90 days before screening, defined as opioids or combination medications, including barbiturates, used ≥10 days; NSAIDs or other simple medications (such as over the counter medications) >14 days per month; and triptans or ergots ≥10 days per month. Alcohol or substance use disorder identified within 12 months of screening, a positive urine drug screen for recreational drugs or alcohol, or prescription drug use not explained by disclosed concomitant medication use also excluded patients from participation.
Patients with a history of the following drug treatments were also excluded: onobotulinum toxin A for migraine in the 4 months before screening (cosmetic use was acceptable); unstable dosage of migraine prophylactic medication use in the 30 days before and/or throughout screening; use of mini-prophylaxis for menstrual migraine; history of cluster headaches, or only a “probable migraine” diagnosis (per ICHD-3); history of cerebrovascular events, including but not limited to stroke or transient ischemic attack; chronic use of warfarin sodium or an equivalent drug; current antipsychotic use, or use within 30 days of randomization; treatment with cytochrome P450 (CYP)2C9 inducers, or with CYP2D6 substrates with a narrow therapeutic window (ie, thioridazine), within 7 days of randomization. Patients with the following medical conditions were excluded: concurrent seizure disorder or history of migralepsy (seizure following migraine); history of ischemic coronary artery disease or congenital heart disease; insulin-requiring diabetes mellitus or diabetes with A1C >7.9%; positive screening test for human immunodeficiency virus, hepatitis B surface antigen, or hepatitis C virus antibody; history of cancer within the past 5 years, except adequately treated basal cell or squamous cell skin carcinoma, or in situ cervical cancer; any medical condition or procedure that, in the judgment of the investigator, would have confounded the study objectives. Last, patients with the following vital signs or laboratory values were excluded: uncontrolled hypertension or screening systolic/diastolic blood pressure (SBP/DBP) >140/90 mm hg; Fridericia’s corrected QT interval >450 msec, serum creatinine >1.5x upper limit of normal (ULN), and/or serum total bilirubin >1.5x ULN; serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), or alkaline phosphate >2.5x ULN; or, any clinical laboratory or electrocardiogram (ECG) abnormality that could endanger the participant or interfere with study conduct.
Patients could be withdrawn subsequent to randomization if they became pregnant, violated any protocol requirements affecting study outcomes, developed significant illness, needed to undergo major acute surgery, or if the investigator deemed it unsafe for the participant to continue in the study. Patients could also be excluded if they took rescue medication before data collection at the 2-hour post-dose timepoint.
Study Procedures
During the screening visit, all patients received a full physical exam, 12-lead electroencephalogram, and had their vital signs and blood pressure checked. In addition, a migraine history assessment was used to determine the participant’s MBS (nausea, photophobia, or phonophobia) associated with migraine. Patients attended two subsequent study visits: at baseline (randomization) and following treatment. At study randomization, patients were provided with single-dose bottles with a deliverable volume of 4.8 mL and were instructed to drink the entire contents during the treated migraine attack. Patients returned to the study site within 2 to 7 days of treatment for assessments that included physical examination, vital signs, clinical laboratory tests, and ECG. At this visit, patient eDiary entries were also evaluated to ensure proper recording, and patients were required to return all unused drug.
This study was performed in compliance with International Council for Harmonisation Good Clinical Practices, as well as the ethical principles of the Declaration of Helsinki and the US Food and Drug Administration (FDA) Code of Federal Regulations. Before study initiation, the protocol, participant information, informed consent forms, and other relevant documentation were reviewed and approved by the Copernicus Group Institutional Review Board (Durham, North Carolina) and the Committee for the Protection of Human Subjects (Hanover, New Hampshire; one study site only). Prior to study initiation, all patients provided written consent to participate; as part of this process, patients were informed about the nature and purpose of the study, participation/termination conditions, and treatment risks and benefits. At each study site, the principal investigator was responsible for recruiting patients and for ensuring that no patients underwent study-related examinations or activities before informed consent was obtained.
Efficacy Assessments
The co-primary endpoints comparing celecoxib oral solution to placebo were the proportion of patients who were pain-free at 2 hours post-dose (defined as a reduction from pre-dose moderate [Grade 2] or severe [Grade 3] pain to none [Grade 0]) and the proportion of patients reporting freedom from their screening MBS (among nausea, photophobia, or phonophobia). The selection of co-primary endpoints was based on the most current guidance from the FDA for clinical trials in migraine.34
In addition, a secondary endpoint was participant treatment satisfaction as measured by total, total raw, subscale, and global scores on the PPMQ-R at 24 hours post-dose. The PPMQ-R is a validated instrument of 30 questions used to assess satisfaction with migraine medication. PPMQ-R subscales measure efficacy (11 questions), function (4 questions), ease of use (2 questions), and side effects (10 questions); and also includes 3 global questions measuring medication effectiveness, side effects, and overall satisfaction.35 Since patients did not pay for the study drug, the 2 PPMQ-R questions in the drug cost subscale were not included. Items were scored on a 7-point scale from 1 (very satisfied) to 7 (very dissatisfied), except items related to tolerability, which were scored on a 5-point scale from 1 (not at all) to 5 (extremely).
Other secondary endpoints included the proportion of patients free from nausea, photophobia, and phonophobia at 15, 30, and 45 minutes and 1, 1.5, 2, 4, and 24 hours post-dose; the proportion of patients with pain relief at 15, 30, and 45 minutes and 1, 1.5, 2, 4, and 24 hours post-dose; and the proportion of patients with sustained pain relief. Sustained pain relief was defined as pain-free at 2 hours post-dose, with no use of rescue medication and no recurrence of headache pain within 2 to 24 hours post-dose.
Safety and Tolerability Assessments
Safety assessments included the following: AEs from screening until study completion or discontinuation; compliance with study drug use; rescue medication use; concomitant medication review; physical examinations and suicidality check; pregnancy tests, as applicable; vital signs (sitting SBP/DBP, pulse rate, and body temperature); laboratory examinations (hematology, chemistry, and urinalysis); and 12-lead ECGs.
Statistical Analysis
Descriptive statistical methods only were used to summarize study data, including baseline patient characteristics, with sample size and hypothesis testing performed for the co-primary and secondary efficacy endpoints. For the first and second co-primary endpoint, respectively, a sample size of 600 patients was estimated to provide 88% power and 480 patients provided 94% power to detect the assumed difference between placebo and celecoxib oral solution (11.6% and 16.5% for the first and second co-primary endpoints, respectively), and anticipating a 15% dropout rate. These data points were based on phase 2 data for oral celecoxib, as well as historical literature.36 Comparisons between the celecoxib oral solution and placebo groups for all efficacy endpoints were conducted using the full analysis set (FAS). The FAS included all randomized patients who took 1 dose of study drug and had ≥1 post-baseline assessment for symptoms or pain. Missing efficacy data were imputed using last observation carried forward (LOCF), with results provided for LOCF and observed case (OC) data. The safety set included all patients who received ≥1 dose of study drug (recorded in their eDiary). Unless otherwise specified, all statistical testing and confidence intervals (CIs) were 2-sided and performed using a significance level of 0.05.
For both co-primary endpoints, a closed sequential procedure using Fisher’s exact test was used to test significance. If the first co-primary endpoint was statistically significant, the second co-primary endpoint was tested for statistical significance. To be included in the analysis of the second co-primary endpoint, patients must have experienced their screening MBS symptom pre-dose (but it did not have to be designated as their pre-dose MBS). Patients who did not report MBS at screening, did not have their screening MBS present at pre-dose, who took rescue medication prior to data collection at the 2-hour post-dose timepoint, and/or who had a Grade 1 (mild) pre-dose pain levels were excluded from analysis. The number of patients with response and with non-missing assessments, and the proportion of responders in the celecoxib oral solution and placebo groups, were calculated (including corresponding 95% CIs, odds ratios, and P-values). Exploratory subgroup analyses on the co-primary endpoints were conducted in the FAS using the following baseline categories: age (18–34 years, 35–49 years, 50–64 year and ≥65 years); sex (male and female); and ethnicity (Hispanic and non-Hispanic). For each subgroup, the odds ratio for response and corresponding CI for each co-primary endpoint was calculated using the treatment group, the subgroup variable, and the treatment-by-subgroup interaction term as covariates in the logistic regression model.
The secondary efficacy endpoint of PPMQ-R treatment satisfaction at 24 hours post-dose was compared between the celecoxib oral solution and placebo groups using Wilcoxon rank-sum testing. PPMQ-R scores at 24 hours post-dose were also compared to baseline scores (which assessed the patients’ usual migraine medication pre-study) for celecoxib oral solution patients only. The proportion of responders for the additional secondary efficacy endpoints was summarized by treatment group, and corresponding P-values from Fisher’s exact test were computed for between-group comparisons.
Last, based on a prespecified post-hoc analysis plan, case diagnostics were used to evaluate the strength of any site-specific influence on estimates. A logistic generalized linear mixed-effects model (GLMM) was used to detect potential site outliers, as follows: a GLMM was fit with treatment arm as the fixed-effect predictor and site as the random-effect predictor; next, outlier detection statistics were computed, including Cook’s distance and the covariance ratio.37 The Cook’s distance statistic evaluated how much the treatment effect estimate changed when each site was removed from the analysis sample. The covariance ratio statistic evaluated change in the treatment effect CI when each site was removed from the analysis sample. A site was considered an outlier if it led to a Cook’s distance or covariance ratio value that was at least twice as large as all other sites. This was more rigorous than the typical cut-off value of 4/n for Cook’s distance,38 where n would be the number of sites (40), which would have yielded a liberal cut-off value of 0.10 for defining an outlier. These conservative criteria were applied to ensure that any site identified as an outlier would be aberrant and influential. If any outlier sites were identified, they were to be removed from the analysis sample. Finally, the primary endpoints were re-computed by conducting the pre-specified analysis (Fisher’s exact test with LOCF imputation) using the modified analysis sample.
All statistical analyses were conducted using the SAS® software package (version 9.3) or R statistical software (version 3.4.4).
Results
Patients
A total of 926 patients were screened; 631 (68.1%) met entry criteria and were subsequently randomized (Figure 1). Patient demographics and baseline characteristics are summarized in Table 1; mean patient age was 41 years (range 18 to 75), and the majority were female (487 [84.3%]), non-Hispanic (493 [85.3%]), and white (426 [73.7%]). A total of 544 of 631 patients (86.2%) completed the treatment period, with 81 (12.8%) discontinuing. The primary reasons for discontinuation are detailed in Figure 1. Additionally, 6 randomized patients were recorded as not having taken any drug (celecoxib oral solution or placebo) and were excluded from the efficacy and safety analyses. The FAS comprised 567 of 631 (89.9%) randomized patients, including 287 in the celecoxib oral solution arm and 280 in the placebo arm. The safety set included 572 of 631 (90.6%) randomized patients, including 289 in the celecoxib oral solution arm and 283 in the placebo arm.
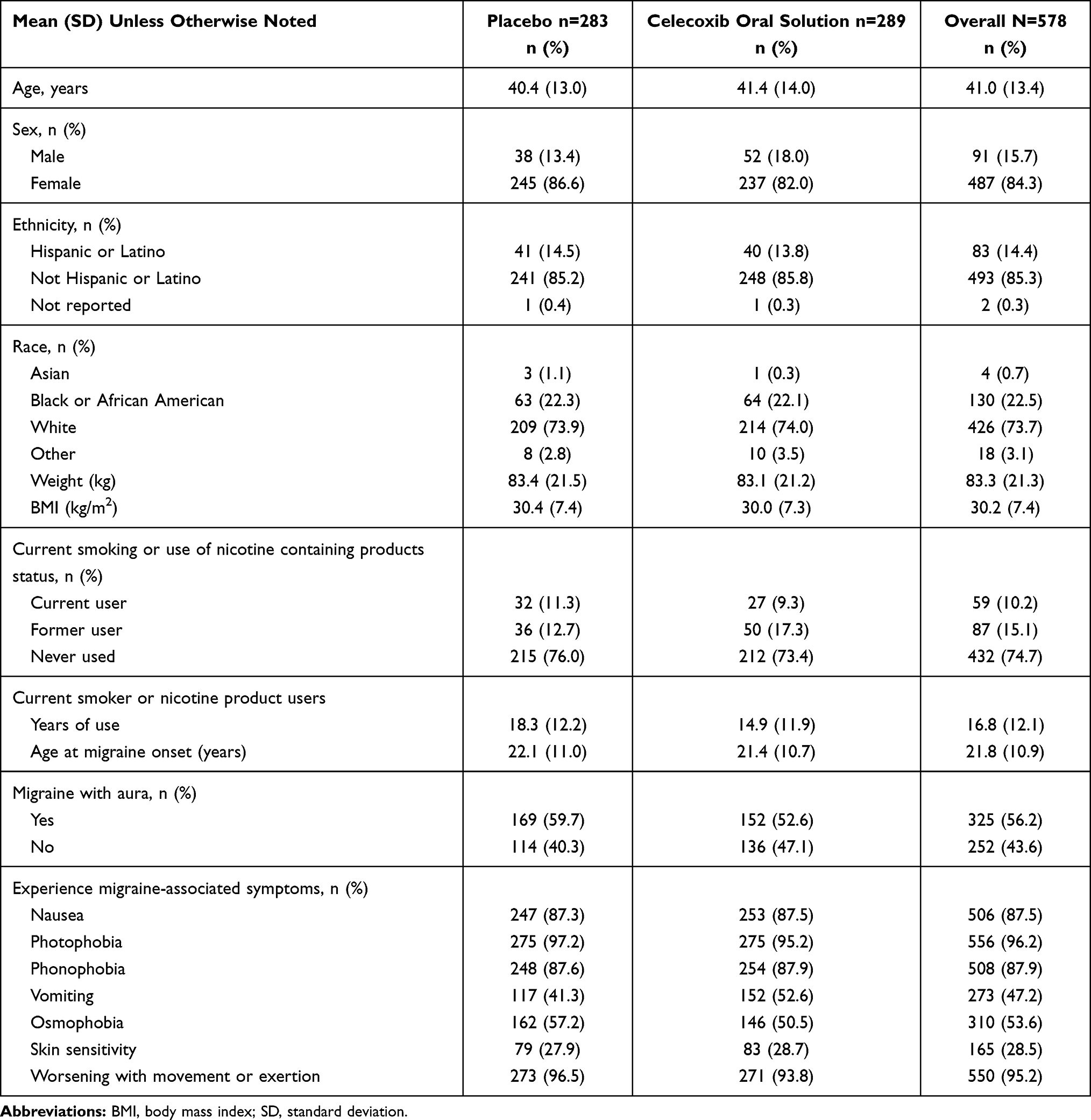 |
Table 1 Demographics and Baseline Characteristics (Safety Set) |
 |
Figure 1 Participant disposition. |
Primary Full Analysis Set Findings
Response rates for the co-primary endpoint of headache pain freedom at 2 hours post-dose (LOCF, 95% CI) were 32.9% (27.4%, 38.7%) in the celecoxib oral solution group and 25.8% (20.7%, 31.5%) in the placebo group (P=0.075). For the co-primary endpoint of 2-hour MBS freedom, response rates (LOCF, 95% CI) in the celecoxib oral solution group were 58.9% (52.4%, 65.2%) and 45.0% (38.5%, 51.7%) in the placebo group (P=0.003) (Supplemental Table 1). The pre-specified post-hoc analyses examined confounding effects that may have contributed to lack of separation in pain freedom, including site outlier analyses, and results are reported next.
Identification of Outlier Site
Based on the procedure outlined above, one study site met outlier criteria, with a mean 2-hour pain freedom placebo response rate of 75% (Cook’s distance, 0.8). At all other sites, the mean 2-hour pain freedom placebo response rate was 23.5% (Supplemental Figures 1). This site, which included 12 patients treated with placebo and 15 patients treated with celecoxib oral solution, was therefore excluded from efficacy analyses in the results presented below. Supplemental Figures 2 and 3 in the Appendix to this document provide all additional efficacy analyses with the outlier site included.
Co-Primary Efficacy Endpoints with Outlier Site Removed
For the co-primary endpoint of headache pain freedom at 2 hours post-dose, response rates (LOCF, 95% CI) in the celecoxib oral solution group were higher (32.8% [27.2%, 38.8%]) than placebo (23.5% [18.5%, 29.2%]), with statistically significant differences (using Fisher’s exact test) between treatment groups (P=0.020) in favor of celecoxib oral solution (Table 2). For the co-primary endpoint of 2-hour MBS freedom, response rates (LOCF, 95% CI) in the celecoxib oral solution group were higher (58.1% [51.4%, 64.5%]) than placebo (43.9% [37.2%, 50.7%]), with statistically significant differences between treatment groups (P=0.003) in favor of celecoxib oral solution (Table 2). The treatment effect of celecoxib oral solution was statistically significant for both co-primary endpoints (Table 2).
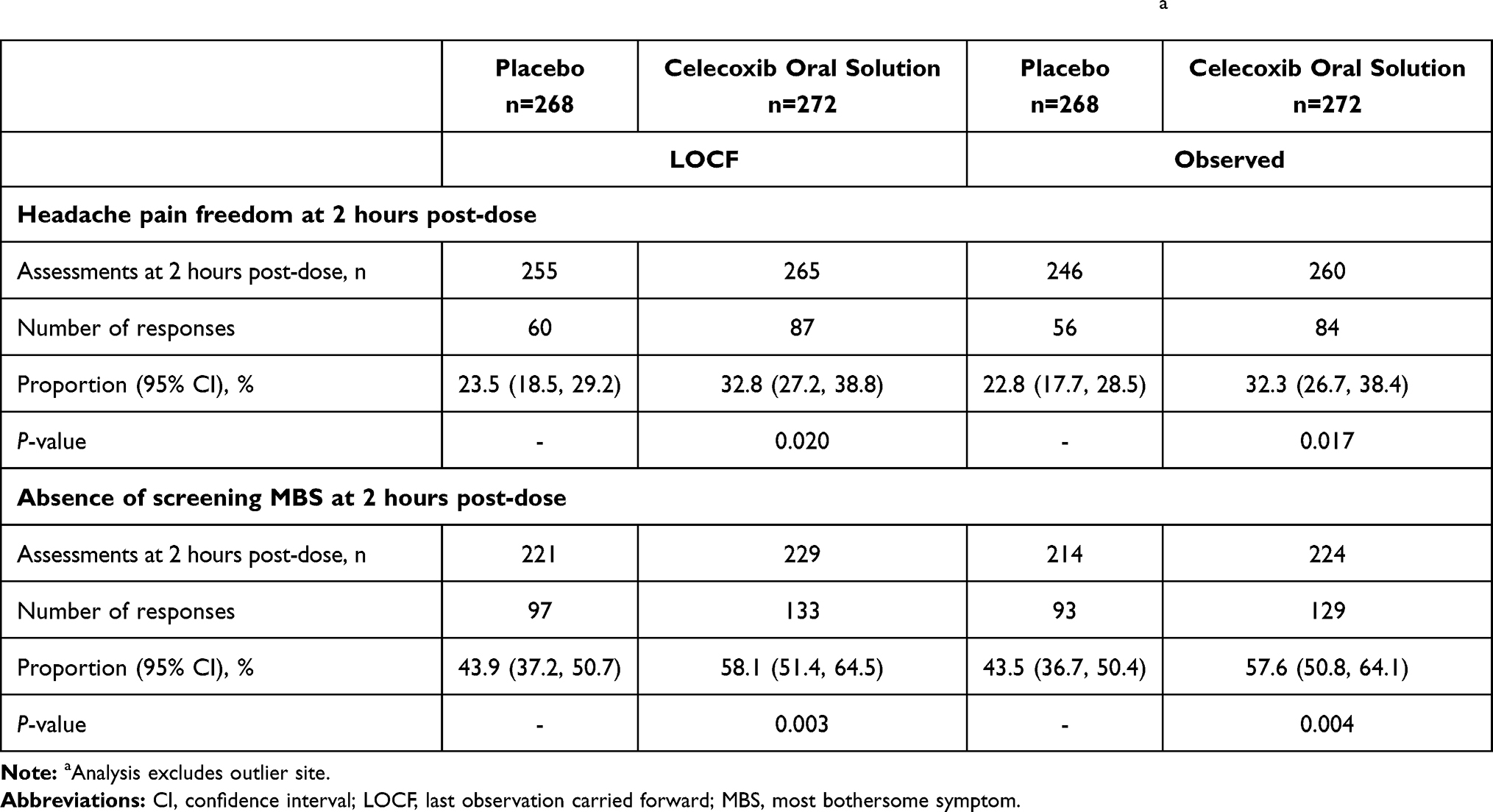 |
Table 2 Co-Primary Efficacy Analysis: Headache Pain Freedom and MBS Freedom 2 Hours Post-Dosea |
Secondary Endpoints with Outlier Site Removed
The change from baseline in treatment satisfaction, as measured by the PPMQ-R at 24 hours post-dose, was higher for celecoxib oral solution compared with placebo in total score (P=0.006), total raw score (P=0.003), the subscales of efficacy (P=0.003) and function (P=0.007), the global item of medication effectiveness (P=0.002), and overall satisfaction (P=0.010). For celecoxib oral solution patients only, the comparison between 24-hours post-dose PPMQ-R and baseline PPMQ-R (patients’ pre-study usual migraine medication) showed significantly greater satisfaction compared with baseline in total score (P=0.005), total raw score (P=0.018), and the subscales of function (P<0.001), ease of use (P=0.014), tolerability (P<0.001), and the global item of side effects (P=0.001).
In terms of freedom from migraine symptoms (nausea, photophobia, and phonophobia), freedom from photophobia response was significantly greater for celecoxib oral solution patients at 1.5 (P=0.013), 2 (P=0.001), and 4 hours post-dose (P=0.005). Freedom from phonophobia response was significantly greater for patients receiving celecoxib oral solution at 1.5 hours (P=0.030) and 2 hours post-dose (P=0.019). Freedom from nausea was significantly greater for patients receiving celecoxib oral solution at 4 hours (P=0.046) (Figure 2).
 |
Figure 2 Freedom from nausea, photophobia, and phonophobia at prespecified timepoints post-dose using LOCF, FAS†. †Analysis excludes outlier site. *Denotes statistical significance. Abbreviations: FAS, full analysis set; LOCF, last observation carried forward; Pbo, placebo. |
For headache pain relief at 2 hours post-dose, 180/265 (67.9%) celecoxib oral solution patients experienced headache pain relief compared with 141/255 (55.3%) placebo patients (P=0.004) (Figure 3). For headache pain relief at timepoints other than the 2-hour post-dose, the percentage of celecoxib oral solution responders was greater than the placebo group at all timepoints beyond 15 minutes post-dose, with statistical significance seen at 1 hour (P=0.021), 1.5 hours (P=0.014), and 4 hours post-dose (P=0.012). Finally, the percentage of celecoxib oral solution patients with sustained headache pain relief from 2 to 24 hours post-dose was higher (58.3%) than the placebo group (42.8%; P=0.002).
 |
Figure 3 Proportion of patients with headache pain relief by post-dose timepoint using LOCF, FAS†. †Analysis excludes outlier site. Abbreviations: LOCF, last observation carried forward. |
Subgroup analyses for age, sex, and ethnicity did not reveal any impact to reported efficacy results.
Safety and Tolerability with Outlier Site Included
Thirty-one of 289 (10.7%) celecoxib oral solution patients and 28 of 283 (9.9%) placebo patients reported a treatment-emergent AE (TEAE). Study-drug-related TEAEs were reported by 21/289 (7.3%) celecoxib oral solution patients and 21/283 (7.4%) placebo patients. The most commonly reported TEAEs were nausea (4 [1.4%] celecoxib oral solution patients and 5 [1.8%] placebo patients) and dysgeusia (5 [1.7%] celecoxib oral solution patients and 3 [1.1%] placebo patients). No study-drug-related TEAEs were reported by ≥2.0% of patients, and no deaths occurred during the study. There were no treatment-emergent serious AEs (SAEs); 4 non-treatment-emergent SAEs were reported in 3 patients. Three patients (all placebo) discontinued the study due to a TEAE, while 3 patients (2 placebo, 1 celecoxib oral solution) discontinued the study after experiencing AEs unrelated to the study drug.
Overall, there were no clinically meaningful changes from baseline for any clinical laboratory variables, vital statistics, or ECG readings, and no new safety signals were identified.
Discussion
In this randomized, placebo-controlled trial of patients with moderate-to-severe episodic migraine, celecoxib oral solution achieved statistically significant results for the co-primary efficacy endpoints of freedom from headache pain and MBS at 2 hours (32.8%, vs 23.5%; P=0.020, and 58.1% vs 43.9%; P=0.003, respectively). These results were obtained after excluding an outlier site with disproportionate placebo response from the analysis. Prior to this exclusion, 2-hour pain freedom response rates were essentially unchanged for celecoxib oral solution (32.9%), but rose to 25.8% for placebo, and no statistically significant difference was observed between treatment groups (P=0.075). Removal of the outlier site did not change the statistical significance of the MBS analysis: with the outlier site, 58.9% of patients receiving celecoxib oral solution reported MBS freedom, compared to 45.0% of placebo patients (P=0.003).
Additionally, response for celecoxib oral solution was significantly higher than placebo for many secondary endpoints, including treatment satisfaction, freedom from the migraine symptoms of photophobia and phonophobia, and early and sustained headache pain relief. Finally, no safety signals were identified, and celecoxib oral solution was well tolerated, with 31/289 (10.7%) patients reporting a TEAE, and 21 (7.3%) experiencing a TEAE considered related to the study drug. Notably, the incidence of GI-related TEAEs was low; for example, nausea occurred in 4 (1.4%) patients taking celecoxib oral solution versus 5 (1.8%) placebo patients. There were no deaths or treatment-emergent SAEs, although 4 non-treatment-emergent SAEs were reported. Three patients (0.5%, all placebo) discontinued due to a TEAE, and 3 patients (2 placebo, 1 celecoxib oral solution) had non-treatment-emergent AEs, leading to study drug discontinuation.
These findings support results from the first phase 3 randomized, placebo-controlled efficacy and safety trial of celecoxib oral solution. Among patients treated with celecoxib oral solution, 35.6% experienced freedom from migraine pain at 2 hours (P<0.001 vs placebo), with a low incidence of GI TEAEs (nausea occurred in 9 [3.2%] celecoxib oral solution and 5 [1.8%] placebo patients).32 The current study similarly found that approximately one-third of patients treated with celecoxib oral solution reported pain freedom at 2 hours, alongside a very low incidence of gastric TEAEs that were similar for active treatment and placebo.
A high placebo response was observed in this trial for the co-primary efficacy outcomes (23.5% headache pain freedom and 43.9% absence of screening MBS). A similarly high placebo response was observed in the first phase 3 trial of celecoxib oral solution (21.7% headache pain freedom and 44.8% absence of screening MBS),32 and historically, placebo response has been consistently high in migraine trials due to a large number of factors.39 For example, a review of the placebo effect in trials of oral triptans has noted a consistent high placebo response, with 2-hour headache pain freedom and pain relief rates in placebo patients as high as 17% and 50%, respectively.40 In the current study, despite a high placebo response, and after exclusion of the outlier site, the celecoxib oral solution treatment effect remained positive.
Also noteworthy to this study is that approximately one-half of patients receiving celecoxib oral solution reported headache pain relief at 1-hour post-dose. The potential for rapid pain relief with celecoxib oral solution is congruent with its pharmacology: celecoxib oral solution 120 mg reaches peak concentrations within 0.7 hours, giving it a more rapid onset of action than celecoxib capsules 400 mg capsules (Tmax 2.5 hours).30 Considered jointly, this information suggests that celecoxib oral solution may provide effective acute therapy for patients requiring rapid relief from migraine pain. This finding will need to be evaluated in greater depth using comparative research.
The current study results support existing research showing that NSAIDs are effective in migraine,1 and that COX-2-specific NSAIDs can address inflammation and pain while sparing patients from COX-1-mediated AEs.23 Additional research is needed to investigate the 48-hour migraine recurrence rate with celecoxib oral solution, and to evaluate the safety and tolerability associated with frequent use of the drug. Further studies are also needed to establish the role of celecoxib oral solution within existing treatment paradigms and in specific subgroups, such as patients with aura. Finally, oral celecoxib solution should be investigated alongside existing standard of care medications, such as triptans.
Strengths and Limitations
The strengths of this study include the large number of patients and the study design itself. Patients used eDiaries to immediately record real-time data regarding migraine attack severity; the impact of treatment on migraine pain, bothersome symptoms, and functional disability; treatment satisfaction; and rescue medication use. Therefore, investigators were able to rapidly assess the effects of celecoxib oral solution at multiple timepoints, and as early as 15 minutes.
Conclusions
Celecoxib oral solution (available in the US as ELYXYB) is a safe, effective COX-2-selective NSAID and the first celecoxib formulation approved for acute migraine relief. Treatment with celecoxib oral solution is associated with a low incidence of gastric adverse events, and may provide a convenient, alternate option to triptans, non-selective NSAIDs, and currently available celecoxib capsule formulations.
Abbreviations
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; CI, confidence interval; COX, cyclooxygenase; CYP, cytochrome P450; DBP, diastolic blood pressure; DFN-15, celecoxib oral solution, 25 mg/mL; ECG, electrocardiogram; eDiary, electronic diary; FAS, full analysis set; GI, gastrointestinal; ICHD-3, International Classification of Headache Disorders, 3rd edition; LOCF, last observation carried forward; MBS, most bothersome symptom; NSAIDs, nonsteroidal anti-inflammatory drugs; OC, observed cases; PPMQ-R, Patient Perception of Migraine Questionnaire-Revised; SAE, serious adverse event; SBP, systolic blood pressure; SD, standard deviation; TEAE, treatment-emergent adverse event; Tmax, time to maximal concentration; ULN, upper limit of normal; US, United States.
Data Sharing Statement
The datasets generated and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
The research protocol, patient information and informed consent form, and other relevant study documentation were reviewed and approved by the Institutional Review Boards for each study center.
Prior Presentations
The abstract of this paper was presented virtually at the American Headache Society Annual Meeting (June 15-30, 2020). The poster’s abstract, titled DFN-15 (celecoxib oral solution, 25 mg/mL) in the acute treatment of episodic migraine: efficacy results from two phase III randomized, double-blind, placebo-controlled studies was published in Special Issue: 62nd Annual Scientific Meeting American Headache Society®. Headache: The Journal of Head and Face Pain. 2020;60(1):1-156. https://doi.org/10.1111/head.13854.
Acknowledgments
The authors would also like to thank Preeti Singh, MD (Medical Affairs at Promius) for project management and manuscript review and Caitlin Rothermel, MPH (HLG Health Communications) for medical writing and editorial support.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Dr. Reddy’s Laboratories, Ltd (Basel, Switzerland) designed and executed the study and performed data analysis; Dr. Reddy’s also provided funding for independent third-party data collection, analysis of the data, and editorial assistance.
Disclosure
RBL has received research support from the NIH. He also receives support from the FDA, Migraine Research Foundation and the National Headache Foundation. He serves on the editorial board of Neurology, senior advisor to Headache, and associate editor to Cephalalgia. He has reviewed for the NIA and NINDS, holds stock options in eNeura Therapeutics and Biohaven Holdings; serves as consultant, advisory board member, or has received honoraria from American Academy of Neurology, Allergan, American Headache Society, AEON Biopharma, Amgen, Avanir, Biohaven, Biovision, Boston Scientific, CVS Health, Dr. Reddy’s (Promius), Electrocore, Eli Lilly, eNeura Therapeutics, Equinox, GlaxoSmithKline, Grifols, Invex, Lundbeck (Alder), Merck, Novartis, Pernix, Pfizer, Satsuma, Sun Pharma, Supernus, S&L Marx Foundation, Teva, Trigemina, Vector, Vedanta. He receives royalties from Oxford University Press (Wolff’s Headache 7th and 8th Edition), Wiley, and Informa. He reports stock options from CtrlM Health. SM is an employee of Dr. Reddy’s Laboratories and owns stock in the company. SJT received grants for research (no personal compensation) from Allergan/AbbVie, Amgen, Eli Lilly, Lundbeck, Neurolief, Novartis, Satsuma, and Zosano; served as a consultant and/or on advisory boards (honoraria) for Aeon, Allergan/AbbVie, Alphasights, Amgen, Atheneum, Axsome Therapeutics, Becker Pharmaceutical Consulting ClearView Healthcare Partners, CoolTech, CRG, Currax, DRG, Eli Lilly, ExpertConnect, FCB Health, GLG, Guidepoint Global, Health Science Communications, HMP Communications, Impel, InteractiveForums, Krog and Partners, Lundbeck, M3 Global Research, MJH Holdings, Neurolief, Novartis, Palion Medical, Pulmatrix, SAI MedPartners, Satsuma, Spherix Global Insights, Strategy Inc, System Analytic, Taylor and Francis, Teva, Theranica, Unity HA, XOC, and Zosano; received salary from Dartmouth-Hitchcock Medical Center, American Headache Society, Thomas Jefferson University; and received CMR honoraria from American Academy of Neurology, American Headache Society, Catamount Medical Education, Diamond Headache Clinic, Forefront Collaborative, Haymarket Medical Education, Peerview, Medical Education Speakers Network, Migraine Association of Ireland, North American Center for CME, The Ohio State University, Physicians’ Education Resource, PlatformQ Education, Primed, Texas Neurological Society, WebMD/Medscape, and Annenberg Center for Health Sciences. CI was a paid consultant of Dr. Reddy’s Laboratories. DS was a paid consultant of Dr. Reddy’s Laboratories. The authors report no other conflicts of interest related to this work.
References
1. Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: the American Headache Society evidence assessment of migraine pharmacotherapies. Headache. 2015;55(1):3–20.
2. Burch R, Rizzoli P, Loder E. The prevalence and impact of migraine and severe headache in the United States: figures and trends from government health studies. Headache. 2018;58(4):496–505.
3. Lipton RB, Bigal ME, Diamond M, et al. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68(5):343–349.
4. Bigal ME, Liberman JN, Lipton RB. Age-dependent prevalence and clinical features of migraine. Neurology. 2006;67(2):246–251.
5. Lipton RB, Buse DC, Saiers J, Fanning KM, Serrano D, Reed ML. Frequency and burden of headache-related nausea: results from the American Migraine Prevalence and Prevention (AMPP) study. Headache. 2013;53(1):93–103.
6. Munjal S, Singh P, Reed ML, et al. Most bothersome symptom in persons with migraine: results from the Migraine in America Symptoms and Treatment (MAST) study. Headache. 2020;60(2):416–429.
7. Lipton RB, Bigal ME, Ashina S, et al. Cutaneous allodynia in the migraine population. Ann Neurol. 2008;63(2):148–158.
8. American Headache Society. The American Headache Society position statement on integrating new migraine treatments into clinical practice. Headache. 2019;59(1):1–18.
9. Lipton RB, Stewart WF, Stone AM, Lainez MJ, Sawyer JP; Disability in Strategies of Care Study Group. Stratified care vs step care strategies for migraine: the Disability in Strategies of Care (DISC) study: a randomized trial. JAMA. 2000;284(20):2599–2605.
10. Bigal ME, Ferrari M, Silberstein SD, Lipton RB, Goadsby PJ. Migraine in the triptan era: lessons from epidemiology, pathophysiology, and clinical science. Headache. 2009;49(Suppl 1):S21–33.
11. Lacey LF, Hussey EK, Fowler PA. Single dose pharmacokinetics of sumatriptan in healthy volunteers. Eur J Clin Pharmacol. 1995;47(6):543–548.
12. Tfelt-Hansen P, De Vries P, Saxena PR. Triptans in migraine: a comparative review of pharmacology, pharmacokinetics and efficacy. Drugs. 2000;60(6):1259–1287.
13. Tepper SJ, Johnstone MR. Breath-powered sumatriptan dry nasal powder: an intranasal medication delivery system for acute treatment of migraine. Med Devices (Auckl). 2018;11:147–156.
14. Moore JC, Miner JR. Subcutaneous delivery of sumatriptan in the treatment of migraine and primary headache. Patient Prefer Adherence. 2012;6:27–37.
15. Tfelt-Hansen P. Efficacy and adverse events of subcutaneous, oral, and intranasal sumatriptan used for migraine treatment: a systematic review based on number needed to treat. Cephalalgia. 1998;18(8):532–538.
16. Loder E, Weizenbaum E, Frishberg B, Silberstein S. American Headache Society Choosing Wisely Task F. Choosing wisely in headache medicine: the American Headache Society’s list of five things physicians and patients should question. Headache. 2013;53(10):1651–1659.
17. Bigal ME, Serrano D, Buse D, Scher A, Stewart WF, Lipton RB. Acute migraine medications and evolution from episodic to chronic migraine: a longitudinal population-based study. Headache. 2008;48(8):1157–1168.
18. Buse DC, Pearlman SH, Reed ML, Serrano D, Ng-Mak DS, Lipton RB. Opioid use and dependence among persons with migraine: results of the AMPP study. Headache. 2012;52(1):18–36.
19. Levin M. Opioids in headache. Headache. 2014;54(1):12–21.
20. Paulozzi LJ, Zhang K, Jones CM, Mack KA. Risk of adverse health outcomes with increasing duration and regularity of opioid therapy. J Am Board Fam Med. 2014;27(3):329–338.
21. Gregorian RS
22. Holland S, Fanning KM, Serrano D, Buse DC, Reed ML, Lipton RB. Rates and reasons for discontinuation of triptans and opioids in episodic migraine: results from the American Migraine Prevalence and Prevention (AMPP) study. J Neurol Sci. 2013;326(1–2):10–17.
23. Gong L, Thorn CF, Bertagnolli MM, Grosser T, Altman RB, Klein TE. Celecoxib pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2012;22(4):310–318.
24. Antonova M, Wienecke T, Olesen J, Ashina M. Prostaglandins in migraine: update. Curr Opin Neurol. 2013;26(3):269–275.
25. Peura DA, Goldkind L. Balancing the gastrointestinal benefits and risks of nonselective NSAIDs. Arthritis Res Ther. 2005;7(Suppl 4):S7–13.
26. Solomon DH, Husni ME, Libby PA, et al. The risk of major NSAID toxicity with celecoxib, ibuprofen, or naproxen: a secondary analysis of the PRECISION trial. Am J Med. 2017;130(12):1415–1422.
27. Moore RA, Derry S, Makinson GT, McQuay HJ. Tolerability and adverse events in clinical trials of celecoxib in osteoarthritis and rheumatoid arthritis: systematic review and meta-analysis of information from company clinical trial reports. Arthritis Res Ther. 2005;7(3):R644–665.
28. Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N Engl J Med. 2016;375(26):2519–2529.
29. Loo CY, Tan HJ, Teh HS, Raymond AA. Randomised, open label, controlled trial of celecoxib in the treatment of acute migraine. Singapore Med J. 2007;48(9):834–839.
30. Pal A, Shenoy S, Gautam A, et al. Pharmacokinetics of DFN-15, a novel oral solution of celecoxib, versus celecoxib 400-mg capsules: a randomized crossover study in fasting healthy volunteers. Clin Drug Investig. 2017;37(10):937–946.
31. Lipton RB, Munjal S, Dodick DW, Tepper SJ, Serrano D, Iaconangelo C. Acute treatment of migraine with celecoxib oral solution: results of a randomized, placebo-controlled clinical trial. J Pain Res. 2021;14:549–560.
32. Lipton RB, Munjal S, Brand-Schieber E, Tepper SJ, Dodick DW. Efficacy, tolerability, and safety of DFN-15 (celecoxib oral solution, 25 mg/mL) in the acute treatment of episodic migraine: a randomized, double-blind, placebo-controlled study. Headache. 2020;60(1):58–70.
33. Food and Drug Administration. FDA approves Elyxyb (celecoxib) oral solution for acute treatment of migraine with or without aura In adults. Drugs.com; 2020. Available from: https://www.drugs.com/newdrugs/fda-approves-elyxyb-celecoxib-oral-solution-acute-migraine-without-aura-adults-5223.html.
34. U.S. Food and Drug Administration. Migraine: developing drugs for acute treatment. Guidance for Industry; 2018. Available from: https://www.fda.gov/media/89829/download.
35. Kimel M, Hsieh R, McCormack J, Burch SP, Revicki DA. Validation of the revised Patient Perception of Migraine Questionnaire (PPMQ-R): measuring satisfaction with acute migraine treatment in clinical trials. Cephalalgia. 2008;28(5):510–523.
36. Imitrex (Sumatriptan Succinate Tablet) [Prescribing Information]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
37. Schabenberger O. Mixed Model Influence Diagnostics. SAS® Institute Inc; 2005. Available from: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.597.609&rep=rep1&type=pdf.
38. Van der Meer T, Grotenhuis MT, Pelzer B. Influential cases in multilevel modeling: a methodological comment. ASR. 2010;75(1):173–178.
39. Speciali JG, Peres M, Bigal ME. Migraine treatment and placebo effect. Expert Rev Neurother. 2010;10(3):413–419.
40. Loder E, Goldstein R, Biondi D. Placebo effects in oral triptan trials: the scientific and ethical rationale for continued use of placebo controls. Cephalalgia. 2005;25(2):124–131.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.