Back to Journals » Cancer Management and Research » Volume 11
A Molecular Epidemiological Analysis Of Programmed Cell Death Ligand-1 (PD-L1) Protein Expression, Mutations And Survival In Non-Small Cell Lung Cancer
Authors Schabath MB ![]() , Dalvi TB, Dai HA, Crim AL
, Dalvi TB, Dai HA, Crim AL ![]() , Midha A, Shire N, Gimbrone NT, Walker J, Greenawalt DM, Lawrence D, Rigas JR
, Midha A, Shire N, Gimbrone NT, Walker J, Greenawalt DM, Lawrence D, Rigas JR ![]() , Brody R, Potter D, Kumar NS, Huntsman SA, Gray JE
, Brody R, Potter D, Kumar NS, Huntsman SA, Gray JE
Received 7 June 2019
Accepted for publication 18 October 2019
Published 7 November 2019 Volume 2019:11 Pages 9469—9481
DOI https://doi.org/10.2147/CMAR.S218635
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Matthew B Schabath,1,2 Tapashi B Dalvi,3 Hongyue A Dai,4 Alan L Crim,4 Anita Midha,5 Norah Shire,3 Nicholas T Gimbrone,1 Jill Walker,6 Danielle M Greenawalt,7 David Lawrence,8 James R Rigas,9 Robert Brody,9 Danielle Potter,9 Naveen S Kumar,4 Shane A Huntsman,4 Jhanelle E Gray2
1Department of Cancer Epidemiology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, USA; 2Department of Thoracic Oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, USA; 3Oncology R&D, AstraZeneca, Gaithersburg, MD, USA; 4M2Gen, Tampa, FL, USA; 5Department of Personalised Healthcare and Biomarkers, AstraZeneca, Cambridge, UK; 6Department of Precision Medicine Oncology, AstraZeneca, Cambridge, UK; 7Department of iMED Oncology Informatics, AstraZeneca, Waltham, MA, USA; 8Department of Global Medicines Development, AstraZeneca, Cambridge, UK; 9Department of Global Medical Affairs Oncology, AstraZeneca, Gaithersburg, MD, USA
Correspondence: Matthew B Schabath
H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL 33612, USA
Tel +1 813 745 4150
Fax +1 813 745 6525
Email [email protected]
Purpose: To characterize programmed cell death ligand-1 (PD-L1) expression in relation to survival and gene mutation status in patients with advanced NSCLC. The study also explored the influence of tumor mutational burden (TMB) on PD-L1 expression and patient characteristics.
Patients and methods: Adult patients with histologically or cytologically documented Stage IIIB/Stage IV/recurrent/progressive NSCLC, Eastern Cooperative Oncology Group performance status 0 to 3, and >2 lines of prior systemic treatment regimens were included in this retrospective analysis. Patients were treated from 1997 to 2015 at H. Lee Moffitt Cancer Center and Research Institute, Tampa, or at 7 community centers across the United States. PD-L1 expression level was determined using the VENTANA PD-L1 (SP263) Assay. EGFR and KRAS mutation status and ALK rearrangements were determined by targeted DNA sequencing; these were obtained from clinical records where targeted DNA sequencing was not performed. TMB was calculated as the total number of somatic mutations per sample.
Results: From a total of 136 patients included in the study, 23.5% had tumors with high PD-L1 expression (≥25%). There were no significant differences in patient characteristics, overall survival (OS), and progression-free survival (PFS) between patients with high PD-L1 expression (median OS: 39.5 months; median PFS: 15.8 months) vs low PD-L1 expression (<25%; median OS: 38.1 months; median PFS: 18.6 months). PD-L1 expression level correlated (P=0.05) with TMB and was consistent with The Cancer Genome Atlas data.
Conclusion: In this retrospective analysis, survival outcomes of patients with advanced NSCLC were comparable by PD-L1 expression level. EGFR and KRAS mutation status were not found to be significantly associated with PD-L1 expression level, while TMB was weakly associated with PD-L1 expression level. Overall, PD-L1 expression level was not observed to be an independent prognostic biomarker in this cohort of patients with advanced NSCLC treated with chemotherapy.
Keywords: non-small cell lung cancer, patient outcomes, tumor mutational burden, prognostic biomarker
Introduction
In the United States (US), lung cancer is the second most common cancer and the leading cause of cancer-related deaths in both sexes.1 Despite considerable improvement in patient survival for many cancers over the last decades, there has been little improvement in the 5-year survival rates for lung cancer. The 5-year relative survival rate for all lung cancers (ie, non-small cell lung cancer [NSCLC] and small cell lung cancer combined) is 18%.1 Among NSCLCs, the 5-year relative survival rates are 26%, 10%, and 1% for Stages IIIB, IVA, and IVB, respectively.2
Immuno-oncology (IO), especially immune checkpoint inhibition therapy (immunotherapy), an emerging therapeutic area, may hold the key to improving patient survival in the early and/or advanced stages of NSCLC in patients with metastatic disease.3–5 Immunotherapy involves recognition and eradication of tumors by restoration of the host’s immune system capacity.6 The programmed cell death-1/programmed cell death ligand-1 (PD-1/PD-L1) pathway is an important checkpoint used by tumor cells to inhibit antitumor responses.3–9 Blocking the PD-1/PD-L1 pathway has been shown to reverse the formation of immunosuppressive tumor microenvironment and enhance endogenous antitumor immune responses.7 Several anti–PD-1/PD-L1 antibodies are approved for the treatment of NSCLC, including nivolumab, pembrolizumab, durvalumab, and atezolizumab.8–10
In addition to PD-L1 expression, NSCLCs are also characterized by genetic alterations in oncogenes and tumor suppressor genes critical to tumor growth and survival that can be exploited using specific targeted therapy.11 As many lung cancers harbor somatic mutations and/or alterations, targeted therapy is important to improve outcomes of this disease.10 Pro-oncogenic mutations in NSCLC can involve components of the mitogen-activated protein (MAP) kinase pathway, such as Kirsten rat sarcoma viral oncogene (KRAS) and B-Raf proto-oncogene (BRAF) mutations.11 Presently, there are US Food and Drug Administration (FDA)-approved therapies targeting genetic mutations, such as epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) gene rearrangements.11–18 High tumor mutational burden (TMB) in diverse cancers, including NSCLC, has been associated with a high probability of response to immunotherapy, particularly with PD-1/PD-L1/cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) blockade.19–21 Based on these observations, future clinical trials will incorporate TMB as a biomarker for single-agent immunotherapies, including checkpoint inhibitors.19
Limited retrospective, noninterventional studies have previously assessed the co-occurrence of PD-L1 expression with somatic mutations and gene alterations.22 These data could provide greater understanding of how genomic aberrations affect predictive ability for patient prognosis. The aim of this retrospective study was to characterize PD-L1 expression and its relationship to common somatic mutations and alterations associated with NSCLC (EGFR, KRAS, and ALK), and to correlate clinical outcomes to PD-L1 expression levels in patients with advanced NSCLC treated with ≥2 lines of chemotherapy.
Materials And Methods
Patients
This study included 136 patients with NSCLC treated between 1997 and 2015 at the H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida, or from 7 community centers throughout the state of Florida and across the United States as part of Moffitt’s Total Cancer Care (TCC®) protocol.23 TCC is a multi-institutional observational study of patients diagnosed with cancer that prospectively collects patient data and tissue samples for research purposes. The study included adult patients aged ≥18 years with histologically or cytologically documented Stage IIIB/Stage IV/recurrent/progressive NSCLC and an Eastern Cooperative Oncology Group (ECOG) performance status 0 to 3. All patients had formalin-fixed paraffin-embedded tissue (FFPET) in sufficient amounts to complete ≥1 test for PD-L1 and gene mutations. All tumors collected under the TCC protocol are microscopically evaluated by a qualified Anatomic Pathologist and subsequently macro-dissected to enrich for percent tumor nuclei. Specifically, the TCC protocol aims to enrich percent tumor nuclei to ≥60% by removing stromal and normal tissue and reduce necrotic tissue to <20%.
Patients who received ≥2 prior systemic treatment regimens for NSCLC were included in this study. Prior platinum-containing adjuvant, neoadjuvant, or definitive chemoradiation therapy administered for locally advanced disease was considered first-line therapy only if recurrent (local or metastatic) disease developed ≤6 months of completing therapy. Additionally, patients who experienced disease progression or recurrence after a platinum-based chemotherapeutic regimen and patients who received ≥1 additional systemic therapy (maintenance therapy following platinum doublet-based chemotherapy was not considered a separate regimen of therapy) were included for survival analysis. All patients with recurrent disease had disease progression ≥6 months after a subsequent platinum-based chemotherapeutic regimen was administered to treat the recurrence. All patients were immunotherapy-naïve. The inclusion/exclusion criteria for the survival analysis cohort closely matched the ATLANTIC clinical trial.24
The protocol of this study was approved by the Chesapeake Institutional Review Board (Columbia, MD). The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and was consistent with Good Clinical Practice guidelines and applicable regulatory requirements. Written informed consent was obtained from all patients for their participation in the study.
PD-L1 Testing
The PD-L1 expression in FFPET samples was assessed by immunohistochemistry (IHC) performed using the VENTANA PD-L1 (SP263) Assay (Ventana Medical Systems, Inc., Tucson, AZ, USA) per the manufacturer’s guidelines.25,26 High expression of PD-L1 was defined as ≥25% of the tumor cells with membrane positivity for PD-L1 at any intensity. Low expression of PD-L1 was defined as <25% of the tumor cells with membrane positivity for PD-L1 at any intensity.
Targeted DNA Sequencing
Targeted DNA sequencing was performed to assess EGFR and KRAS mutation status and ALK fusion from FFPET samples of 88 patients. Where DNA sequencing was not performed, EGFR, KRAS, and ALK status were obtained from electronic clinical records where available.
DNA was extracted from FFPET using Qiagen DNeasy® Blood and Tissue Kits (QIAGEN, Valencia, CA, USA) per manufacturer’s instructions, quantified using Nanodrop 2000 (Thermo Fisher Scientific, Wilmington, DE, USA), and its integrity was assessed by electrophoresis. Extracted tumor genomic DNA was fragmented into 200 to 300 base pairs (bp) using a Covaris® M220 focused-ultrasonicator (Covaris, Woburn, MA, USA). In brief, cell-free DNA (cfDNA) or sheared tissue DNA was enriched with end-repairing, A-tailing, adapter ligation, and size selection using Agencourt® AMPure® XP beads (Beckman Coulter, Indianapolis, IN, USA). Libraries were then subjected to ligation-mediated polymerase chain reaction (LM-PCR) amplification and purification and hybridized to the Roche NimbleGen SeqCap® EZ Exome probe (Roche NimbleGen, Madison, WI, USA). Targeted DNA profiling was performed using the TumorCare panel designed by BGI Genomics (Cambridge, MA, USA) and manufactured by Roche NimbleGen (Roche NimbleGen, Madison, WI, USA). The panel detects genomic alterations at extremely high coverage in 1053 cancer-related genes spanning a 4.6 Mb region of the genome, including base substitutions, insertions and deletions, copy number alterations, and rearrangements (Table S1). Both noncaptured and captured LM-PCR products were subjected to quantitative polymerase chain reaction (qPCR) to estimate the magnitude of enrichment.
The enriched libraries were sequenced on Illumina HiSeq 4000 (Illumina, San Diego, CA, USA) next-generation sequencing platforms independently to ensure that each sample achieved the desired average fold-coverage. The delivered targeted DNA sequencing had an average coverage of 512X across all samples. Raw image files were processed by Illumina base-calling software 1.7 for base calling with default parameters and the sequences for all patients were generated as 100 bp paired-end reads. FASTQ files were aligned to build 37 genes to reference sequence Human Genome version 19 (hg19) using Burrows-Wheeler Aligner (BWA) with optimized parameters.27 All data processing and secondary analysis were performed using Bcbio-Nextgen best-practice pipeline (https://github.com/chapmanb/bcbio-nextgen). Variant calling was performed using VarDict,28 and variant effects were annotated using SnpEff.29 Variants were filtered at an allele frequency threshold of 5%, a minimum sequencing depth of 5X, and a minimum variant depth of 3X. Putative sequencing artifacts were removed based on a cohort frequency <75%, and germline variants were filtered by removing variants identified in the Single Nucleotide Polymorphism Database (dbSNP; version138), 1000 Genome project, while keeping variants if they were found in ClinVar or COSMIC.30–32
Using the resulting processed variant information from the aforementioned pipeline above, TMB was calculated as the total number of somatic mutations per sample, including all nonsynonymous mutations and indels.
Statistical Analysis
The primary covariates were PD-L1 expression and mutation status of EGFR, KRAS, and ALK determined by targeted DNA sequencing. The prevalence of PD-L1 expression ≥25% and <25% was compared in subgroups based on age, sex, smoking history, race, ethnicity, histology, tumor stage, EGFR and KRAS mutation status, and ALK rearrangements using the χ2/Fisher’s exact test and Monte-Carlo estimation method. The Wilcoxon 2-sample test was used for testing differences in median age and the Student’s t-test was used for testing differences in mean age. Analyses were also conducted comparing demographic subgroups by TMB that was dichotomized at the median value. Overall survival (OS) was defined as the time from index date (time of diagnosis or time of therapy initiation) to death due to any cause or last follow-up. Progression-free survival (PFS) was defined as the time from index date to the date the patient changed therapy due to first documented disease progression or death due to any cause. Survival analyses were performed using Kaplan-Meier survival curves and log-rank test statistics. PD-L1 expression level ≥25% was used as the reference group.
Results
Patient Characteristics
A total of 136 patients with NSCLC were included in this study (Table 1); the mean (standard deviation [SD]) age of patients at diagnosis was 61.9 (9.3) years, and 52% were male (n=70). The most frequent (>70% incidence) histological diagnosis was adenocarcinoma of the lung (71.3%). Overall, 113 (83.1%) patients were smokers and 17 (12.5%) patients were nonsmokers. Information on smoking history was not available for 6 (4.4%) patients. Overall, 53 (39.0%) patients were diagnosed with Stage IV disease, 29 (21.3%) patients did not have metastatic disease, and 39 (28.7%) patients had received 4 lines of therapy. No patients were treated with immunotherapy. The ECOG performance status was 0 in 72 (52.9%) patients, 1 in 45 (33.1%) patients, 2 in 9 (6.6%) patients, and 3 in 2 (1.5%) patients. Information on performance status was missing for 8 (5.9%) patients.
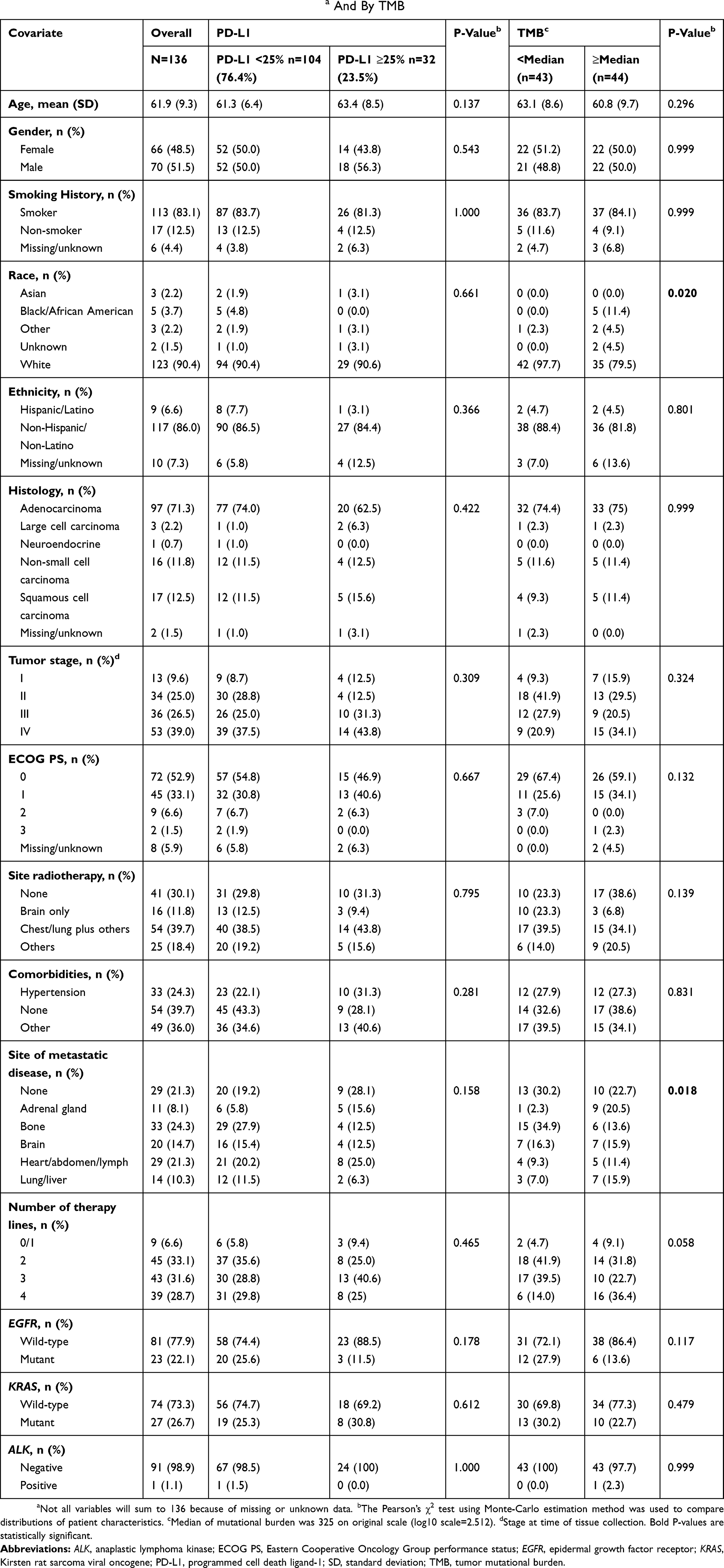 |
Table 1 Patient Characteristics By PD-L1 Expression Levela And By TMB |
PD-L1 And Tissue Status
The mean (SD) sample age (number of years of sample storage) was 7.2 (2.8) years. Overall, 84.2% of the tumor samples that were used for PD-L1 testing were collected by surgical resection and 15.8% of the tumor samples were biopsy samples. Moreover, 63.3% of the tumor samples were collected (biopsy or resection) prior to any treatment (ie, chemotherapy-naïve) and 36.7% of the tumor samples were collected (biopsy or resection) post-any treatment (defined as any chemotherapy following surgical resection or biopsy); 59% of the tumor samples tested involved primary tissue and 41% of the tumor samples were from metastatic tissue. There was no significant difference in PD-L1 expression level by timing of sample collection (prior to any treatment vs post-any treatment; P=0.532) and no significant difference in PD-L1 expression level by tissue type (primary vs metastatic; P=0.836).
PD-L1 Expression And Survival
High expression of PD-L1 (≥25%) was observed in 23.5% of the patients. Overall, there were no statistically significant differences in patient characteristics by PD-L1 expression level (Table 1). There was no significant association between PD-L1 expression level and OS (log-rank test, P=0.968; Figure 1A) or PFS (log-rank test, P=0.714; Figure 1B). The percentiles of survival times (third quartile, median, and first quartile) and survival rates by PD-L1 expression level at 6, 12, 18, and 24 months were largely similar (Table 2). Median OS was 38.1 months from diagnosis for the PD-L1 <25% group and 39.5 months for the PD-L1 ≥25% group (Table 2). Median PFS was 18.6 months for the PD-L1 <25% group and 15.8 months for the PD-L1 ≥25% group (Table 2). Similar OS results by PD-L1 expression level were observed for patients receiving second-line (log-rank test, P=0.523; Figure 1C) and third-line systemic therapies (log-rank test, P=0.607; Figure 1D). We also utilized a PD-L1 ≥90% cut-off to examine OS, including OS from the initiation of second- and third-line therapy, and PFS by PD-L1 expression level (Figures S1–S4).
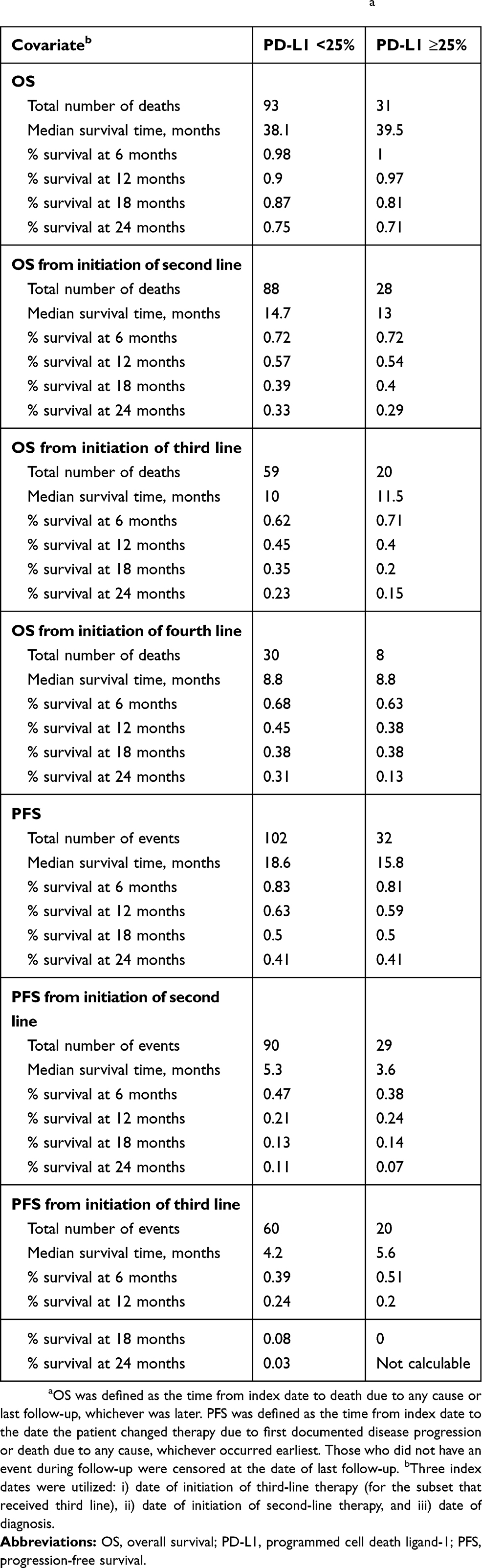 |
Table 2 OS And PFS By PD-L1 Expression Levela |
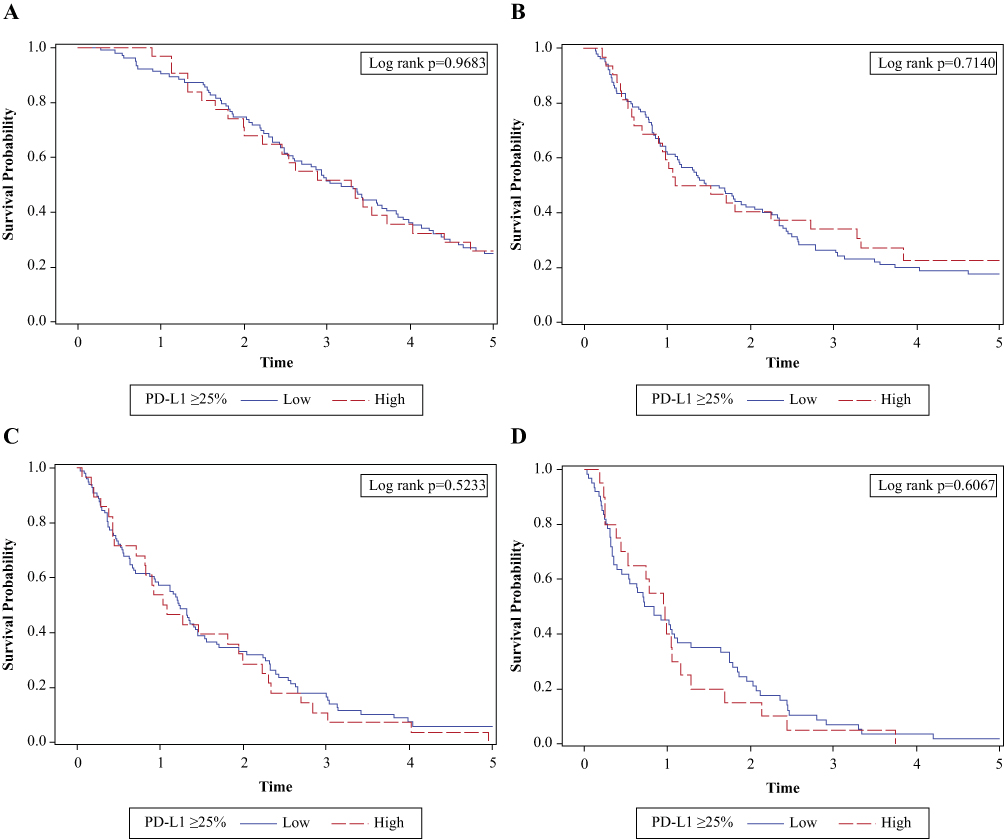 |
Figure 1 OS and PFS by PD-L1 expression level. (A) OS by PD-L1 expression ≥25% showing no significant association between PD-L1 expression level and OS (log-rank test, P=0.968). (B) PFS by PD-L1 expression ≥25% showing no significant association between PD-L1 expression level and PFS (log-rank test, P=0.714). (C) OS by PD-L1 expression ≥25% showing no significant association between PD-L1 expression level and OS for patients receiving second-line therapies (log-rank test, P=0.523). (D) OS from third-line therapy by PD-L1 expression ≥25% showing no significant association between PD-L1 expression level and OS for patients receiving third-line systemic therapies (log-rank test, P=0.607). X-axis is in years. The analyses are censored at 5 years. Abbreviations: OS, overall survival; PD-L1, programmed cell death ligand-1; PFS, progression-free survival. |
Mutation Analysis And TMB
Targeted DNA sequencing was performed on a 4 Mb region of the genome capturing 1,053 cancer-associated genes, including known fusion events (Table S1). On average, 70 million reads were mapped per sample, with 73% of reads on target; 98% of the targeted regions achieved 50X coverage and 94% achieved 100X coverage. Our primary analysis focused on EGFR and KRAS mutation status and ALK fusion. We identified 18 patient samples with EGFR mutations; 6 patient samples with L858R mutations, 7 samples with exon 19 deletions, and 1 tumor sample with both L858R and T790M mutations. KRAS mutations were found in 23 tumor samples and were mutually exclusive of EGFR mutations. An ALK-echinoderm microtubule-associated protein-like 4 (EML4) fusion was only identified in 1 sequenced tumor sample and was mutually exclusive of EGFR and KRAS mutations. There were no significant differences in PD-L1 expression for the distributions of EGFR and KRAS mutations. Given that only 1 ALK fusion was identified, further statistical analysis was not performed for this gene.
Figure 2A shows the scatter plot of log TMB and the percentage of tumor cells with PD-L1 expression. PD-L1 expression, as a continuous covariate, was weakly but positively associated with the number of somatic mutations (Pearson’s correlation coefficient=0.22, P=0.05). This finding was replicated (Figure 2B) using available data on lung cancer samples in The Cancer Genome Atlas (TCGA).33 However, no significant difference was observed when TMB was analyzed by PD-L1 expression ≥25% (Figure 2C). When TMB was dichotomized at the median value, it was significantly associated with race (P=0.020) and site of metastatic disease (P=0.018; Table 1). With an H-score positivity of >5%, 71.9% (64/89) of the patients with EGFR mutations were found to have high PD-L1 expression compared with EGFR wild-type (P=0.067). In patients with EGFR mutations treated with an EGFR–tyrosine kinase inhibitor (TKI), high PD-L1 expression did not have an impact on OS compared with patients with low PD-L1 expression (P=0.932).
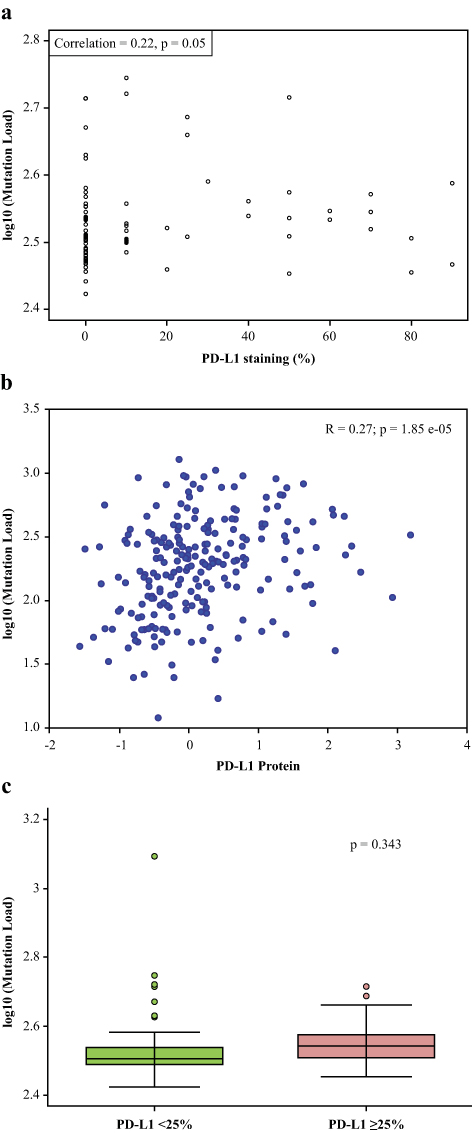 |
Figure 2 Mutational load vs PD-L1 expression level. (A) Scatter plot of log TMB and the percentage of tumor cells with PD-L1 expression demonstrating correlation between mutational load and % PD-L1 staining. PD-L1 expression, as a continuous covariate, was weakly but positively associated with the number of somatic mutations (Pearson’s correlation coefficient=0.22, P=0.05). (B) Correlation between mutational load and % PD-L1 staining in the TCGA demonstrating similar results as in Figure 2A. (C) Mutational load by high vs low PD-L1 expression level demonstrating no significant difference observed when TMB was analyzed by PD-L1 expression ≥25%. Abbreviations: PD-L1, programmed cell death ligand-1; TCGA, The Cancer Genome Atlas; TMB, tumor mutational burden. |
For genes with a mutation frequency >20%, the distribution of PD-L1 expression by mutation status (wild-type vs mutation) was assessed and survival analyses were performed. There were 8 genes with somatic mutation/variants at a frequency ≥20% (Table 3). Among these 8 genes, nuclear receptor corepressor 2 (NCOR2) was mutated in 86 out of 87 samples, which may be an artifact due to a reference genome issue. None of these genes were significantly associated with PD-L1 expression level. Tumor protein p53 (TP53) mutations were associated with significantly poorer OS from primary diagnosis (P=0.002), OS from second-line therapy (P=0.014), PFS from primary diagnosis (P=0.021) and PFS from second-line therapy (P=0.041) (Table 3). RNA-binding motif protein encoded on the X chromosome (RBMX) mutations was associated with significantly worse OS from second-line therapy (P=0.024) and KRAS mutations were associated with PFS from second-line therapy (P=0.028) (Table 3).
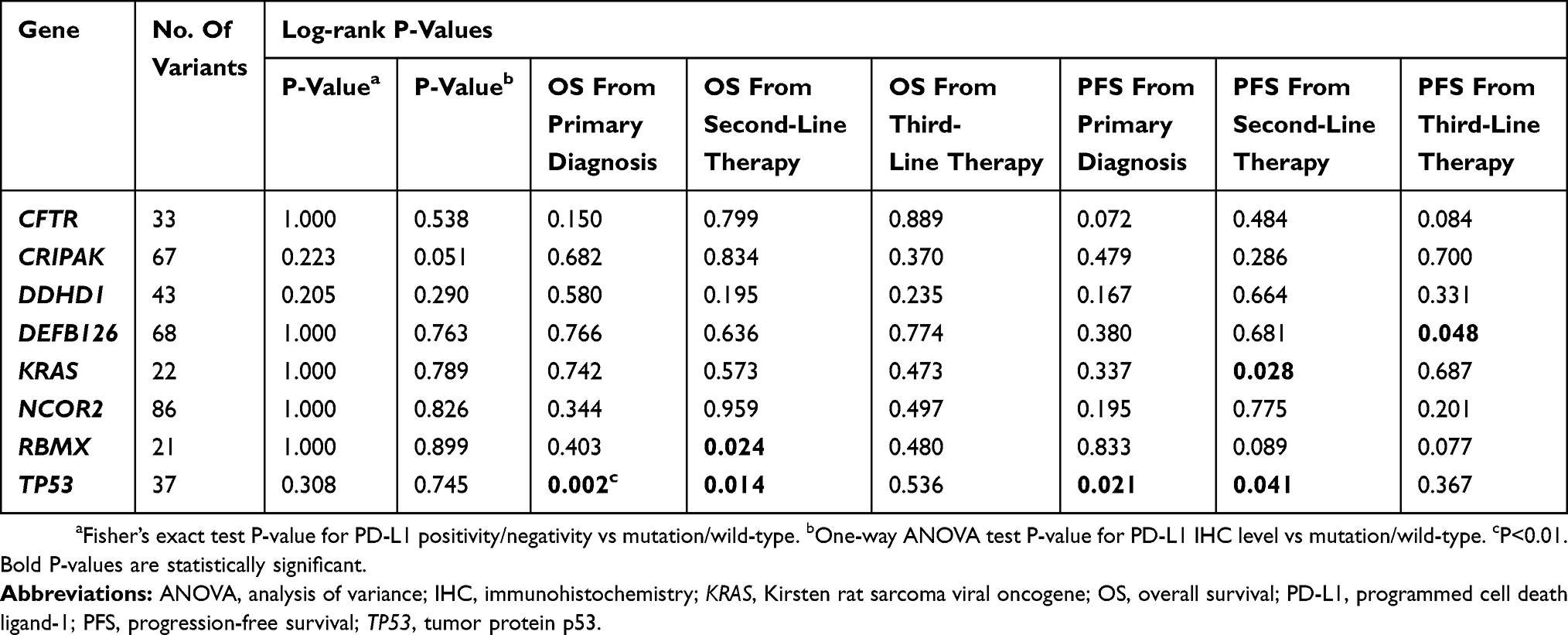 |
Table 3 High-Frequency Mutations And PD-L1 Expression |
Combinatorial And Stratified Analyses By Mutation Status And Histology
There were no significant associations for the combinatorial analyses of PD-L1 and mutation status on OS and PFS (Figures S5–S8). Further analyses were performed stratified by histology (nonsquamous NSCLC only) and mutation status (EGFR wild-type/ALK non-rearranged, EGFR mutant/ALK non-rearranged, KRAS wild-type/ALK non-rearranged, and KRAS mutant/ALK non-rearranged), but there were no statistically significant differences in demographics, clinical characteristics, and survival by PD-L1 expression level (data not shown).
Discussion
The current retrospective study revealed no significant differences between patient characteristics or survival outcomes with low vs high PD-L1 expression level (<25% vs ≥25%). Moreover, EGFR, KRAS, and ALK status were not significantly associated with PD-L1 expression level; however, TMB was weakly but positively associated with PD-L1 expression level. As described by Shukuya and Carbone,34 the response rates (RRs) of patients treated with anti-PD-1/PD-L1 antibodies were low and averaged approximately 20% in unselected patients with NSCLC. Since patients in the current analysis were not treated with anti-PD-1/PD-L1 antibodies, conclusions about the predictive capabilities of the assay could not be substantiated.
To date, studies of various anti-PD-1/PD-L1 antibodies have utilized different companion diagnostic kits and algorithms to determine PD-L1 expression in tumor samples of patients with NSCLC. Durvalumab studies have utilized the VENTANA PD-L1 (SP263) Assay for PD-L1 detection (25% cut-off), nivolumab studies have utilized the Dako PD-L1 IHC 28–8 pharmDx to quantitate PD-L1 expression on tumor cell surface (1% cut-off), pembrolizumab studies have utilized the Dako PD-L1 IHC 22C3 pharmDx (1% cut-off for nonsquamous NSCLC, 50% cut-off for NSCLC), and atezolizumab studies have utilized the VENTANA PD-L1 (SP142) Assay (1% cut-off for urothelial carcinoma and 50% for metastatic NSCLC).35 Furthermore, the Blueprint PD-L1 assay comparison project demonstrated that the VENTANA PD-L1 (SP263) Assay, Dako PD-L1 IHC 22C3 pharmDx, and Dako PD-L1 IHC 28–8 pharmDx stained tumor cells in a highly consistent manner, while the VENTANA PD-L1 (SP142) Assay identified fewer tumor cells with PD-L1 expression.36 The current study utilized the VENTANA PD-L1 (SP263) Assay and demonstrated that PD-L1 expression level was not associated with survival in patients with advanced NSCLC. A recent systematic literature review assessed the association of PD-L1 expression level and disease characteristics in patients with advanced NSCLC to determine its prognostic significance as a predictive biomarker of response to anti-PD-1/PD-L1 antibodies.22 It demonstrated no association between PD-L1 expression level and demographic characteristics such as gender, age, smoking history, tumor histology (adenocarcinoma vs squamous cell carcinoma), performance status, pathologic tumor grade, or EGFR/KRAS/ALK status.22 The study also suggested that considerable research links PD-L1 expression level in tumors to shorter survival in advanced/metastatic NSCLC; however, its use as a prognostic factor requires more research.22
From a clinical decision-making standpoint, it is interesting to note that trials evaluating the prognostic capabilities of the 22C3 PD-L1 antibody have reported disparate results.37,38 Sorensen et al evaluated the tissue specimens of 204 patients with advanced NSCLC treated in Denmark with a cut-off traceable to the “clinical trial version of the assay” and found no significant association between PD-L1 expression and survival.37 In a multivariate analysis of 678 Australian patients with Stage I-III NSCLC treated with chemotherapy, Cooper et al found that high expression of PD-L1 (≥50%) was associated with significantly longer OS (P<0.05). There were no associations with sex, stage, and EGFR or KRAS mutation status.38 The retrospective study design, different demographic characteristics of patients, antibodies, and cut-offs used make cross-study comparisons challenging. Nevertheless, there is clearly variability in the prognostic potential of PD-L1 expression level testing.
The relationship between PD-L1 expression level and response to immunotherapy has been demonstrated in multiple clinical studies, with increased enrichment of response with higher levels of PD-L1 expression; however, responses are also observed (to a lesser degree) in PD-L1 low/negative patient subgroups.25,39,40 While tumor heterogeneity may play a role in these observations, other mechanisms are likely involved in response to immunotherapy/evasion of immune response.
Smoking (ie, current and former smoker status)39 and TMB21 have previously been associated with response to nivolumab. In the current analysis, smoking history was not associated with PD-L1 expression level. Interestingly, a weak positive association between TMB and PD-L1 expression level was observed in this study and this was validated using data on lung cancer from TCGA.33 The positive correlation between PD-L1 expression level and TMB is consistent with the expectation that more somatic mutations lead to a higher probability of neo-antigens, which is likely to attract more infiltrating immune cells to the tumor and drive increased PD-L1 expression in tumor cells for survival advantage.41 The weak correlation between PD-L1 expression level and TMB may also indicate that there are many other factors influencing PD-L1 levels. TMB has been further explored in anti-PD-1/PD-L1/CTLA-4–treated cohorts of patients with NSCLC for predictive abilities, with promising results.19–21 Whether TMB is a superior predictor of response to anti-PD-1/PD-L1 therapy is yet to be established prospectively. Currently, its most promising clinical applicability may be in PD-L1 low/negative patients (<25%). Given the complexities of intra-tumoral heterogeneity and the interplay with host immune response, it is likely that multiple biomarkers may be required to accurately predict response to immunotherapy. Such analyses are beyond the scope of this retrospective study.
Molecular testing is a standard aspect to the work-up of patients with NSCLC, especially nonsquamous NSCLC.42 Key molecular markers of NSCLC include EGFR and KRAS mutations, and ALK rearrangements, which are expressed in several patients with NSCLC and are mutually exclusive. In this study, in addition to TMB, we also evaluated these individual mutations. While KRAS mutations were not significantly associated with PD-L1 expression levels, patients with EGFR mutations demonstrated high PD-L1 expression compared with EGFR wild-type. In a subset analysis of the KEYNOTE 001 trial (Phase I trial of single-agent pembrolizumab for treatment of advanced NSCLC; Dako PD-L1 IHC 22C3 pharmDx), 35% of the patients with EGFR mutations had high PD-L1 expression (tumor proportion score [TPS] ≥50%) and 43% of the patients with KRAS mutations had a TPS ≥50%. Interestingly, those with EGFR mutations and a TPS <1% had a 0% RR, while those with KRAS mutations and a TPS ranging from 1% to 49% had a 0% RR.43 Due to the small sample size of this study, the results must be interpreted with caution.44
Limitations of this study include its retrospective design. Furthermore, approximately 75% of the patients with NSCLC in this analysis were derived from a single institution and patients were mostly white. Hence, the results may not be generalizable to community practices or other cancer centers that treat patients with different ethnicities. Additionally, the significant association observed between TMB and race should be interpreted with caution as it may be an artifact of the analysis method, as opposed to a true measure of biological differences in TMB between ethnicities. Further, the covariates were largely limited to cancer registry data, which do not include a systematic assessment of lung cancer risk factors such as detailed smoking history, family history of cancer, comorbidities, and medical history. Another potential limitation is the sample size of this patient cohort. Regarding the 3 co-primary endpoints, if we assume approximately 40% of the patients have high PD-L1 expression, with different number of samples tested, the 95% confidence intervals (CIs) are as follows: 300 samples, 95% CI (32%–49%); 170 samples, 95% CI (30–53%); 100 samples, 95% CI (28–58%). However, the actual sample size was very low, which may limit our power to detect statistically significant results in sub-strata analyses. As the analyses were performed in an immunotherapy-naïve population with NSCLC, the predictive value of the IHC assay is beyond the scope of this study but can be found in the ATLANTIC trial publication.45 The final limitation is that details such as testing procedure and location of testing were not available for the patients where EGFR, KRAS, and ALK status was abstracted from medical records.
Conclusion
In this retrospective analysis conducted in immunotherapy-naïve patients with advanced NSCLC treated with ≥2 lines of chemotherapy, survival outcomes based on standard of care chemotherapy were comparable in patients with high and low levels of PD-L1 expression. EGFR and KRAS mutation status was not found to be significantly associated with PD-L1 expression level, while TMB was weakly associated with PD-L1 expression level. Overall, PD-L1 expression level was not observed to be an independent prognostic biomarker in patients with advanced NSCLC treated with chemotherapy.
Ethics Approval And Informed Consent
The protocol of this study was approved by the Chesapeake Institutional Review Board (Columbia, MD). The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and was consistent with Good Clinical Practice guidelines and applicable regulatory requirements. Written informed consent was obtained from all patients for their participation in the study.
Data Availability
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Acknowledgments
The authors thank Liam Gillies PhD, of Cactus Communications Pvt. Ltd. for editorial and submission support, which was in accordance with Good Publication Practice (GPP3) guidelines and funded by AstraZeneca. The authors also thank Suzanne Booth and Noolie Gregory for their work on the study. M2Gen received funding from AstraZeneca to manage this study and to perform data analyses.
Disclosure
Dr. Gray receives research grants and personal fees from AstraZeneca; research grants from Merck, Bristol Myers Squibb, and Genentech, during the conduct of the study. She is an advisor for AstraZeneca. She also reports research grants and personal fees from AstraZeneca and Genentech; research grants from Array, Merck, Epic Sciences, Bristol Myers Squibb, Boehringer Ingelheim, Trovagene, and Novartis; personal fees from Takeda, Eli Lilly, Celgene, and Janssen, outside of the submitted work. Drs. Dalvi, Midha, Shire, Walker, and Rigas are employees of AstraZeneca and hold stock in the company. Dr Brody is now a retiree and was an employee of AstraZeneca at the time of the study. Dr Potter was an employee of AstraZeneca at the time of the study and currently an AstraZeneca stockholder. Drs. Greenawalt and Lawrence were employees of AstraZeneca at the time of the study and are currently employed with Bristol-Myers Squibb, USA, and Novartis, Switzerland, respectively. Drs Dai and Crim are employees of M2Gen. Drs Kumar and Huntsman were employees of M2Gen at the time of the study. The authors report no other conflicts of interest in this work.
References
1. American Cancer Society. Cancer Facts & Figures 2018. Atlanta: American Cancer Society; 2018.
2. American Cancer Society. Non-small cell lung cancer survival rates, by stage. 2018. Available from: https://www.cancer.org/cancer/non-small-cell-lung-cancer/detection-diagnosis-staging/survival-rates.html.
3. Santarpia M, Giovannetti E, Rolfo C, et al. Recent developments in the use of immunotherapy in non-small cell lung cancer. Expert Rev Respir Med. 2016;10(7):781–798. doi:10.1080/17476348.2016.1182866
4. Tchekmedyian N, Gray JE, Creelan BC, et al. Propelling immunotherapy combinations into the clinic. Oncology (Williston Park). 2015;29(12):990–1002.
5. Brahmer JR. Harnessing the immune system for the treatment of non-small-cell lung cancer. J Clin Oncol. 2013;31(8):1021–1028. doi:10.1200/JCO.2012.45.8703
6. Jing W, Li M, Zhang Y, et al. PD-1/PD-L1 blockades in non-small-cell lung cancer therapy. Onco Targets Ther. 2016;9:489–502. doi:10.2147/OTT
7. He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci Rep. 2015;5:13110. doi:10.1038/srep13110
8. Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377(20):1919–1929. doi:10.1056/NEJMoa1709937
9. Seetharamu N, Preeshagul IR, Sullivan KM. New PD-L1 inhibitors in non-small cell lung cancer - impact of atezolizumab. Lung Cancer (Auckl). 2017;8:67–78. doi:10.2147/LCTT.S113177
10. Politi K, Herbst RS. Lung cancer in the era of precision medicine. Clin Cancer Res. 2015;21(10):2213–2220. doi:10.1158/1078-0432.CCR-14-2748
11. Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12(2):175–180. doi:10.1016/S1470-2045(10)70087-5
12. Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–1197. doi:10.1056/NEJMoa1311107
13. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–125. doi:10.1056/NEJMoa1713137
14. Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742. doi:10.1016/S1470-2045(11)70184-X
15. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi:10.1016/S1470-2045(11)70393-X
16. Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. doi:10.1200/JCO.2012.44.2806
17. Lilenbaum RA, Horn LA. Management of EGFR mutation-positive non-small cell lung cancer. J Natl Compr Canc Netw. 2016;14(5 Suppl):S672–S674. doi:10.6004/jnccn.2016.0189
18. Horn L. Targeted/emerging therapies for metastatic non-small cell lung cancer. J Natl Compr Canc Netw. 2015;13(5 Suppl):S676–S678. doi:10.6004/jnccn.2015.0201
19. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi:10.1158/1535-7163.MCT-17-0386
20. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093–2104. doi:10.1056/NEJMoa1801946
21. Carbone DP, Reck M, Paz-Ares L, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376(25):2415–2426. doi:10.1056/NEJMoa1613493
22. Brody R, Zhang Y, Ballas M, et al. PD-L1 expression in advanced NSCLC: insights into risk stratification and treatment selection from a systematic literature review. Lung Cancer. 2017;112:200–215. doi:10.1016/j.lungcan.2017.08.005
23. Fenstermacher DA, Wenham RM, Rollison DE, Dalton WS. Implementing personalized medicine in a cancer center. Cancer J. 2011;17(6):528–536. doi:10.1097/PPO.0b013e318238216e
24. AstraZeneca. A global study to assess the effects of MEDI4736 in patients with locally advanced or metastatic non small cell lung cancer (ATLANTIC). NLM identifier: NCT02087423. Available from: https://clinicaltrials.gov/ct2/show/NCT02087423.
25. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi:10.1056/NEJMoa1501824
26. Rebelatto MC, Mistry A, Sabalos C, et al. An immunohistochemical PD-L1 companion diagnostic assay for treatment with durvalumab (MEDI4736) in NSCLC patients [abstract].
27. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
28. Lai Z, Markovets A, Ahdesmaki M, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44(11):e108. doi:10.1093/nar/gkw227
29. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: sNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. doi:10.4161/fly.19695
30. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Databaseissue):D805–D811. doi:10.1093/nar/gku1075
31. Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868. doi:10.1093/nar/gkv1222
32. Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi:10.1093/nar/29.1.308
33. The Cancer Genome Atlas (TCGA) - cancer genome. Available from: https://cancergenome.nih.gov/.
34. Shukuya T, Carbone DP. Predictive markers for the efficacy of anti-PD-1/PD-L1 antibodies in lung cancer. J Thorac Oncol. 2016;11(7):976–988. doi:10.1016/j.jtho.2016.02.015
35. Diggs LP, Hsueh EC. Utility of PD-L1 immunohistochemistry assays for predicting PD-1/PD-L1 inhibitor response. Biomarker Res. 2017;5:12. doi:10.1186/s40364-017-0093-8
36. Hirsch FR, McElhinny A, Stanforth D, et al. PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J Thorac Oncol. 2018;12(2):208–222. doi:10.1016/j.jtho.2016.11.2228
37. Sorensen SF, Zhou W, Dolled-Filhart M, et al. PD-L1 expression and survival among patients with advanced non-small cell lung cancer treated with chemotherapy. Transl Oncol. 2016;9(1):64–69. doi:10.1016/j.tranon.2016.01.003
38. Cooper WA, Tran T, Vilain RE, et al. PD-L1 expression is a favorable prognostic factor in early stage non-small cell carcinoma. Lung Cancer. 2015;89(2):181–188. doi:10.1016/j.lungcan.2015.05.007
39. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643
40. Gettinger S, Rizvi NA, Chow LQ, et al. Nivolumab monotherapy for first-line treatment of advanced non–small-cell lung cancer. J Clin Oncol. 2016;34:2980–2987. doi:10.1200/JCO.2016.66.9929
41. Wirth TC, Kühnel F. Neoantigen targeting-dawn of a new era in cancer immunotherapy? Front Immunol. 2017;8:1848. doi:10.3389/fimmu.2017.01848
42. Lindeman NI, Cagle PT, Aisner DL, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch Pathol Lab Med. 2018;142(3):321–346. doi:10.5858/arpa.2017-0388-CP
43. Leighl NB, Hellmann MD, Hui R, et al. KEYNOTE-001: 3-year overall survival for patients with advanced NSCLC treated with pembrolizumab. J Clin Oncol. 2017;35(15 Suppl):9011. doi:10.1200/JCO.2017.35.15_suppl.9011
44. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi:10.1126/science.aaa1348
45. Garassino MC, Cho B-C, Kim J-H, et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): an open-label, single-arm, phase 2 study. Lancet Oncol. 2018;19(4):521–536. doi:10.1016/S1470-2045(18)30144-X
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.