Back to Journals » OncoTargets and Therapy » Volume 8
A human leucyl-tRNA synthetase as an anticancer target
Authors Gao G, Yao Y, Li K, Mashausi D, Li D, Negi H, Kamle S, Chen H, Wu Z, Zhou H, Li D
Received 19 May 2015
Accepted for publication 11 August 2015
Published 15 October 2015 Volume 2015:8 Pages 2933—2942
DOI https://doi.org/10.2147/OTT.S88873
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Guangwei Gao,* Ying Yao,* Kun Li, Dhahiri Saidi Mashausi, Dongsheng Li, Hema Negi, Suchitra Kamle, Hao Chen, Zhenghua Wu, Huchen Zhou, Dawei Li
School of Pharmacy, Shanghai Jiao Tong University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Several aminoacyl-tRNA synthetases have been reported to be overexpressed for charging essential aminoacyl-tRNAs in many cancer types. In this study, we aimed to explore the potential role of leucyl-tRNA synthetase (LARS) as an anticancer target. MTT assay was performed to screen inhibitors to human LARS (hsLARS) from compounds AN2690 and its derivatives, compounds 1–6, in U2OS and SKOV3 cells. The compound with the strongest inhibitory ability was further investigated for its inhibitory effect in cancer cell lines and in an animal tumor model. Additionally, a LARS-rescue experiment was performed to explore the potential target in U2OS using Western blot and flow cytometry. Luciferase reporter assay was designed to analyze the effect of of hsLARS inhibitor on p21 activation. We identified an hsLARS inhibitor (compound 2) that suppressed the proliferation of U2OS and SKOV3 cells in vitro. A LARS-rescue experiment demonstrated that the proliferation inhibition was induced by targeting intracellular LARS. In addition, the hsLARS inhibition was shown to activate the p21 early transcription and promote cell apoptosis, as well as reduce implanted EMT6 tumor progression in mice. Our results suggest that LARS might serve as a potential anticancer target through the p21 signaling pathway and that the nutritional signaling pathway may provide a valuable anticancer strategy for further investigation.
Keywords: leucyl-tRNA synthetase, apoptosis, mouse model, anticancer target
Introduction
The uptake of amino acids in cancer cells is significantly faster than that in normal cells. It has been reported that balanced amino acid nutrients support tumor proliferation, whereas unbalanced amino acid nutrients suppress tumor cell proliferation.1–3 Reduction of essential amino acids interferes with cellular metabolism and their functions, and inhibits tumor growth by inhibiting protein synthesis.4,5
Aminoacyl-tRNA synthetases (aaRSs) are highly conserved during the evolutionary process. In protein synthesis, aaRSs catalyze the assembly of amino acids to their respective tRNAs and play an important role in the maintenance of cell survival.6,7 These enzymes have also been shown to be promising targets in the development of antimicrobial agents8 as well as in therapeutics against cancers and other diseases.9,10 Leucine is the most important amino acid in nutrients sensing through uncharged leucine-specific tRNA,11 and leucyl-tRNA synthetase (LARS) is reported to be overexpressed in lung cancer cells.12 The gene of LARS is homozygously amplified in 17% of kidney renal cell carcinoma and mutated/amplified in approximately 4% of malignant peripheral nerve sheet tumor, melanoma, and small cell lung carcinoma.13 Also, this homozygously expressed gene was deleted in 3% of acute myeloid leukemia.14 In addition, overexpressed mitochondrial LARS suppresses the A3243G mutation, which causes a number of human diseases, including tumorigenesis.15,16 LARS-targeted reagents, for example AN2690 (5-fluoro-1-hydroxy-1,3-dihydro-2,1-benzoxaborole), have been shown to be novel molecules possessing the antifungal ability.17 Moreover, a series of AN2690 derivatives have shown different degrees of LARS inhibition in vitro.18 However, whether LARS can serve as a target for anticancer drugs has not been thoroughly studied.
In this study, we have screened a small molecule inhibitor of LARS from a series of AN2690 derivatives, and this LARS inhibitor served as a probe to study the effects of LARS inhibition on cancer cell growth, cell cycle, signal pathway, and animal tumor model. Our study aimed to explore whether LARS may play a role as a potential target in oncotherapy.
Materials and methods
Cell culture
HOSE (human ovarian surface epithelial cells) were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA). U2OS (human osteosarcoma cells), SKOV3 (human ovarian cancer cells), EMT6 (mouse mammary carcinoma cells), and 3T3 (mouse embryonic fibroblast cells) were purchased from the American Type Culture Collection (Manassas, VA, USA). The cell lines were cultured under conditions recommended by their respective depositors. Briefly, these five cell lines were all grown in Dulbecco’s Modified Eagle’s Medium (GIBCO, New York, NY, USA) and kept in a 5% CO2 incubator at 37°C. Specifically, HOSE, U2OS, and SKOV3 cell lines were supplemented with 10% fetal bovine serum (Biowest, Nuaille, France) and 1% penicillin–streptomycin (Solarbio, Beijing, People’s Republic of China), while EMT6 and 3T3 cell lines were supplemented with 0.5% fetal bovine serum and 1% penicillin–streptomycin. This study was approved by the Ethics Committee of Shanghai Jiao Tong University.
hsLARS and tbLARS expression
To obtain the Homo sapiens LARS (hsLARS) and Trypanosoma brucei LARS (tbLARS), total RNA was extracted from HOSE cells and T. brucei cells using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Entire coding cDNA fragments of hsLARS and tbLARS were amplified using AMV Reverse transcriptase XL (TaKaRa, Tokyo, Japan) and then subcloned into the multicloning site of pET21a (Novagen, Madison, WI, USA) and pcDNA3.1 (Invitrogen), respectively. The primer sets for amplification were as follows: forward, 5′-ATCGATATCATGGCGGAAAGAAAAG-3′ and reverse, 5′-ATGGATATCATGAACCAGATAGATTATTG-3′ (pET21a-hsLARS); forward, 5′-GGCTGGCATATGTCGACTGTACGAC-3′ and reverse, 5′-TTCGGATCCGCTGCCTTCTTGTC-3′ (pET21a tbLARS); forward, 5′-CGCGGATCCATGGCGGAAAGAAAAG-3′ and reverse, 5′-CCGCTCGAGAGCCGGATCTCAGTGG-3′ (pcDNA3.1-hsLARS). The expression of tbLARS and hsLARS was induced with 0.1 mM IPTG (Sigma, St Louis, MO, USA), and the proteins were purified using a His-Bind kit (Novagen).
Western blot and fluorescent microscopy analysis
U2OS cells were cotransfected with pcDNA3.1-hsLARS and pcDNA3.1-enhanced green fluorescent protein (EGFP) in the ration of 0, 0.25, 0.5, 1, 2, 4, 8 and were transfected with pcDNA3-EGFP only. Transient transfection was carried out using a 25 kDa polyethylenimine reagent (Polysciences, Inc., Warrington, PA, USA) following the manufacturer’s instructions. After 24 hours, both groups of transfected cells were mixed in a 1:1 ratio and treated with 100 μM hsLARS-specific or control inhibitors. Cells transfected with pDsRed-Monomer served as an internal control. After another 24 hours of incubation, the green to red fluorescent cell ratios in each group were quantified by fluorescent microscopy with four random fields and compared with their own initial green/red ratio. At least 200 cells per slide were counted.
Meanwhile, U2OS cells were then lysed in NP-40 buffer (1% Np-40, 150 mM NaCl, 50 mM Tris–HCl, pH 8.0) containing 1 mM phenylmethanesulfonyl fluoride. Whole cell lysates were resolved by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membrane (Pall, Port Washington, NY, USA). Blots were subsequently probed with primary antibodies including mouse anti-His monoclonal antibody (1:1,000, Biotech, Shanghai, People’s Republic of China) and mouse anti-β-actin (1:2,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight. Horseradish peroxidase-conjugated goat anti-mouse IgG antibody (1:10,000, MultiSciences Biotech, Hangzhou, People’s Republic of China) was used as a secondary antibody and incubated for 40 minutes at 37°C. Visualization of blots was carried out using Ultra ECL kit (MultiSciences Biotech), and β-actin was used as a loading control.
Aminoacylation assay
All compounds (AN2690 and its derivatives, compounds 1–6) were dissolved in dimethyl sulfoxide (DMSO), and the aminoacylation assay was performed as previously described.19 Briefly, the experiments were performed in 70 μL reaction mixtures containing 50 mM HEPES–KOH (pH 7.8), 5 mM MgCl2, 45 mM KCl, 1 mM DTT, 0.02% BSA (W/V), 0.4 mg/mL of brewer’s yeast tRNA (Roche, Basel, Switzerland), 10 μM Leu (PerkinElmer, Waltham, MA, USA), and 2 nM tbLARS (or hsLARS). The reaction mixture was incubated at 37°C for 20 minutes and then initiated by adding 4 mM adenosine triphosphate. After 20 minutes, three aliquots of 20 μL reaction solution were quenched on a Whatman qualitative filter with 5% trichloroacetic acid and washed three times with 95% ethanol. The filter was then dried under an infrared heat lamp, and the radioactivity of the precipitate was quantified using a scintillation counter (Beckman Coulter, Irvine, CA, USA).
MTT assay
Growth inhibition by compounds AN2690 and 1–6 were evaluated in vitro using MTT assays,20 and in all our experiments whenever treated with these compounds, the same volume of DMSO was used as a blank control. The testing compounds were first dissolved in DMSO to a stock concentration of 100 mM. Compound stocks were further diluted with cell culture medium to the final concentration before cell treatment, and the maximum concentration of DMSO was 0.1%. U2OS and SKOV3 cells were plated onto 96-well plate at a density of 3×103 cells in 100 μL medium per well. After 24 hours of incubation, the medium was changed and supplemented with 100 mM compounds in DMSO, and the cells were incubated for another 48 hours. Then, 10 μL of 5 mg/mL MTT solution (pH 7.4, Sigma) was added to the plates. Formazan, as the product during the reaction, is liposoluble and cannot be dissolved in water. Therefore, after additional incubation for 4 hours, the MTT solution was removed and replaced with 150 μL DMSO for absorbance measurement. The 96-well plate was further incubated for 10 minutes at room temperature using a shaker at a low speed. The absorbance at 550 nm was recorded using a Microplate Reader 450 (Thermo, Menlo Park, CA, USA). Moreover, the compound concentration that caused 50% inhibition (IC50) of U2OS cells was obtained by adding the compound with concentrations of 10, 25, 40, 55, 70, 85, and 100 μM in the culture medium. The compound with the strongest inhibitory ability was chosen as the LARS inhibitor.
Furthermore, the LARS inhibitor with concentrations of 3.2, 10, 25, 40, 55, and 70 μM was also added to the 96-well plate with EMT6 cells and 3T3 cells to analyze the inhibitory effect using MTT assay. Meanwhile, IC50 values of the LARS inhibitor for EMT6 cells and 3T3 cells were calculated.
Detection of cell apoptosis
To further investigate the inhibitory mechanism of the LARS inhibitor identified, the effect of the LARS inhibitor was compared with cycloheximide (CHX) and actinomycin D (ActD), both of which are known to inhibit transcription and induce apoptosis. In detail, U2OS cells were treated with the indicated concentration of discovered inhibitor (40, 80 μM), ActD (0.1, 0.5 μM; Sigma), and CHX (0.35, 3.5 μM; Sigma) for 24 hours, respectively. Then, U2OS cells were stained with DAPI (Sigma) or propidium iodide (Solarbio) according to the manufacturer’s instructions. Apoptosis of cells stained with DAPI was visualized under a fluorescent photometer (Olympus, Tokyo, Japan) at an excitation wavelength of 340–380 nm, while cells stained with propidium iodide were analyzed by a FACSCalibur (EPICS, BD Biosciences, San Jose, CA, USA) at a wavelength of 488 nm. ActD-treated cells served as an apoptosis control.
Luciferase reporter assay
The luciferase reporter assay was further designed to explore the signaling pathway related to LARS inhibition. U2OS cells were cotransfected with pGL3-Basic-p21-promoter reporter and an internal control pcDNA3.1-β-galactosidase reporter, and the two reporters were in a 1:1 (w/w) ratio. The Con-transfected cells were subsequently treated with discovered indicator and CHX for 12 and 48 hours, respectively. Luciferase activity was detected with d-luciferin free acid, and β-galactosidase activity was measured by ortho-nitrophenyl-d-galactopyranoside hydrolysis using the β-Gal assay kit (Invitrogen) following the manufacturer’s instructions. The cells cotransfected with empty vectors pGL3-Basic and pcDNA3.1 were taken as a blank control.
Animal models
Female BALB/c mice (3–4 weeks old, ~25 g) were purchased from Shanghai Slac Laboratory Animal Co. LTD (Shanghai, People’s Republic of China) and maintained in a sterile animal facility. Totally, 5×105 EMT6 cells were resuspended in 0.5 mL of phosphate-buffered saline and injected subcutaneously per animal with a syringe (n=20/experiment). After 5 days, the inoculated mice were assigned to two treatment groups (six mice per group) according to the tumor size (diameter =5 mm). The LARS inhibitor was dissolved in DMSO, diluted in Poloxamer188 (Sigma), and administrated for nine consecutive days with 100 mg/kg or 0.5 mL volume by intraperitoneal injection. The tumor size was calculated according to the formula: tumor volume (mm3) =0.5× L × W2, where L is the length and W is the width. Finally, the mice were euthanized when the tumor growth reached 15 mm.
All animal care and experimental protocols were approved by the Animal Care and Use Committee of Shanghai Jiao Tong University-School of Pharmacy (SYXK-2007-0025). All efforts were made to minimize animal suffering and reduce the number of animals used.
Statistical analysis
Statistical analysis was performed with SPSS software (version 12.0.1; SPSS, Chicago, IL, USA). Data are represented as mean ± standard deviation from three independent experiments. The statistical comparisons were made by analysis of variance (ANOVA), and Newman–Keuls post hoc tests were applied when ANOVA values were significant. P<0.05 was defined as statistically significant.
Results
Identification of human LARS inhibitors
To identify the inhibitory ability of the selected compounds, MTT assays were performed on U2OS and SKOV3 cells. As shown in Figure 1A, compounds 2 and 5 had inhibition rates above 50% against hsLARS enzymatic activity among the 7 compounds. Additionally, compound 2 could inhibit >50% cell growth for both U2OS and SKOV3 cells, and compound 5 showed weaker inhibited function than that of compound 2 (Figure 1B). Interestingly, compound 1 neither targeted hsLARS nor inhibited SKOV3 cells, while the compound had an off-target effect to kill U2OS cells. Further tests showed that compound 2 inhibited the proliferation of U2OS cells in a dose-dependent manner with IC50 =66.8 μM (Figure 1C). Compound 2, a derivative of AN2690, was selected as the hsLARS inhibitor used in our study. Chemical structures of compound AN2690 and compound 2 are shown in Figure 1D.
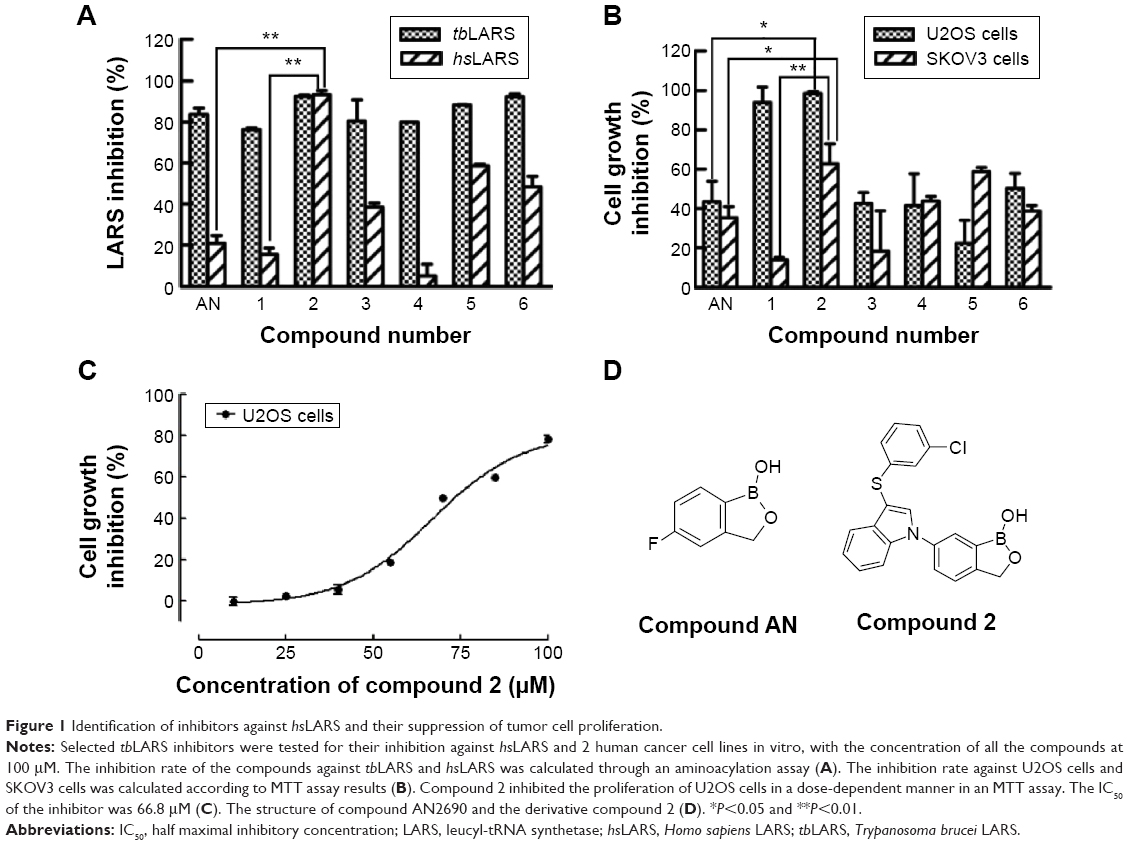 | Figure 1 Identification of inhibitors against hsLARS and their suppression of tumor cell proliferation. |
The effect of compound 2 against intracellular LARS on tumor cells
To confirm that the intracellular LARS was the major target of compound 2 for the inhibition of proliferation, the LARS-rescue experiment was performed. The expression of hsLARS showed that the protein expression level of hsLARS was increased in a dose-responsive manner with an increase in transfected pcDNA3-hsLARS (Figure 2A).
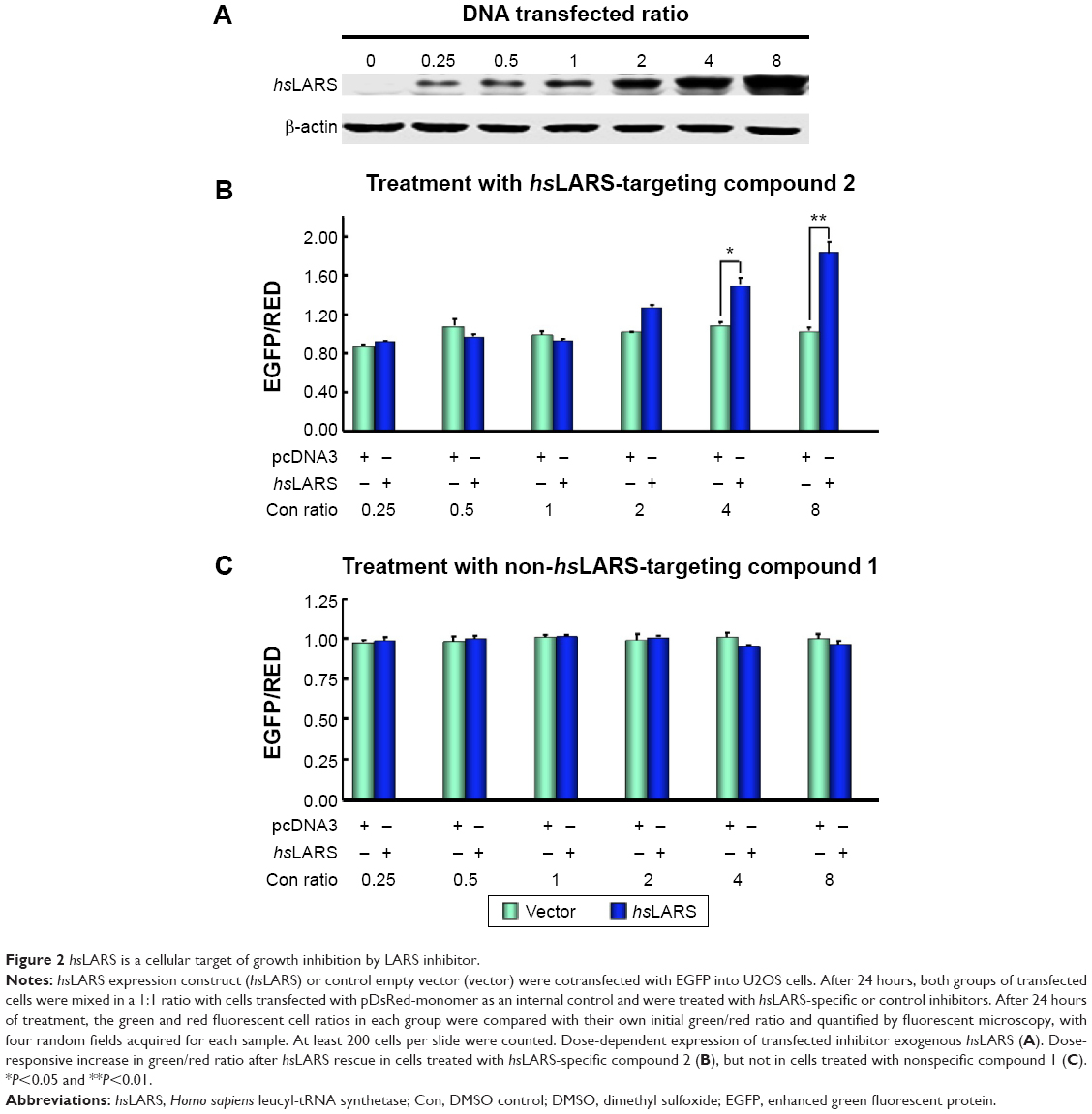 | Figure 2 hsLARS is a cellular target of growth inhibition by LARS inhibitor. |
As shown in Figure 2B and C, a dose-responsive increase in the green/red ratio indicated that the increase of exogenously expressed hsLARS rescued the inhibited endogenous hsLARS, with an increased cell survival. Evidently, a dose-responsive increase in the green/red ratio after hsLARS rescue existed only in cells treated with hsLARS-specific compound 2, but not in cells treated with nonspecific compound 1, indicating that compound 2 targets to hsLARS, but compound 1 does not.
Inhibition of LARS-induced apoptosis and p21 activation in U2OS cells
To explore the potential mechanism for growth inhibition, the inhibitory effect of the LARS inhibitor was compared with that of ActD and CHX. After incubation for 24 hours, U2OS cells treated with compound 2 and ActD showed remarkable suppression in cell division. However, cells treated with CHX showed no evident sign of apoptosis. As shown in Figure 3A, cells treated with compound 2 showed condensed and fragmented nuclei, which is characteristic of the morphology of cells undergoing apoptosis and is similar to cells treated with ActD. A statistical comparison of the apoptotic rate is presented in Figure 3B. The apoptotic rate of cells treated with DMSO, as a negative control, was found to be 0.86%. Notably, the apoptotic rates were 4.77% and 7.16% in cells treated with 40 and 80 μM of compound 2, respectively, which indicated a dose-dependent response. ActD induced apoptosis in a similar manner as compound 2, while CHX did not significantly induce apoptosis compared with DMSO control. Flow cytometry results (Figure 3C and D) also showed that the cell apoptotic rate significantly increased in the LARS inhibitor-treated group, while CHX treatment had little influence on apoptosis.
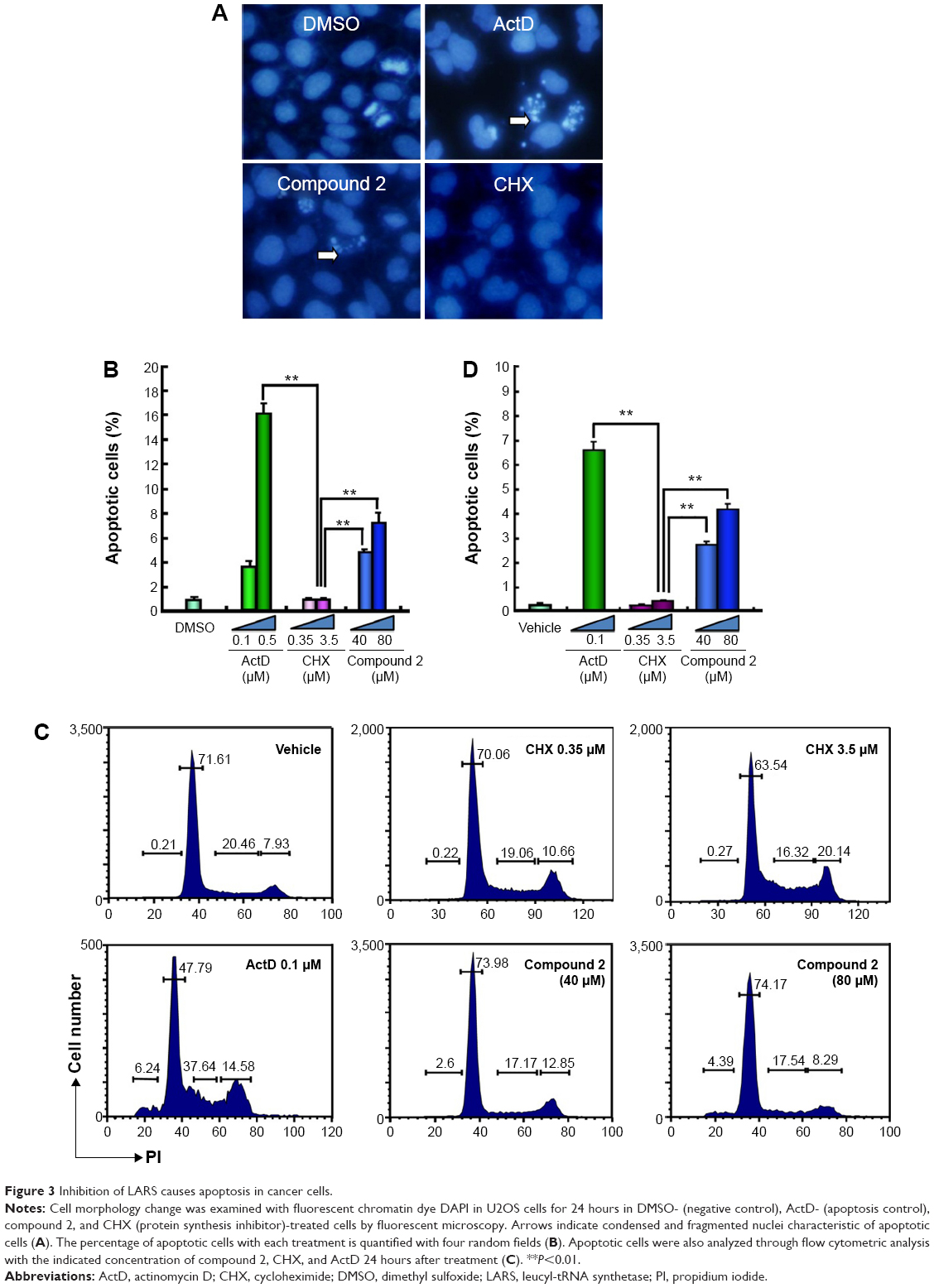 | Figure 3 Inhibition of LARS causes apoptosis in cancer cells. |
To elucidate the cell signaling pathway of LARS inhibition, we investigated the change in p21 promoter activity using p21 promoter-driven luciferase assay. Figure 4 shows that the relative p21 promoter activity increased within 12 hours after being treated with compound 2 in a dose-dependent manner. In contrast, CHX repressed p21 promoter activity in a dose-dependent manner. Nevertheless, both compounds 2 and CHX repressed p21 promoter activity in a dose-dependent manner after 48 hours of treatment.
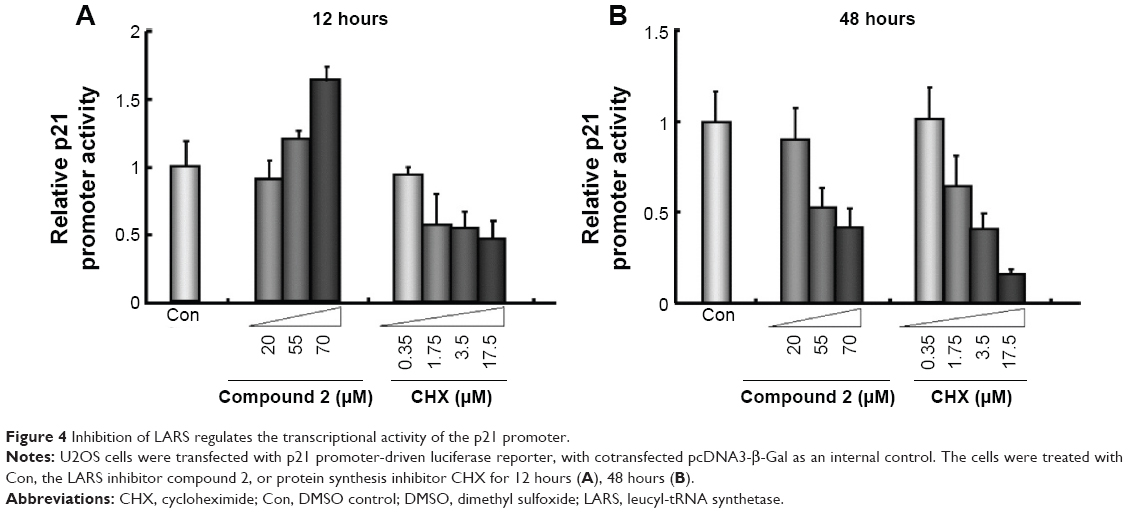 | Figure 4 Inhibition of LARS regulates the transcriptional activity of the p21 promoter. |
LARS inhibitor selectively inhibits fast growing tumor cells in vitro and in vivo
To assess whether the LARS-targeting inhibitor compound 2 can selectively inhibit tumor cell proliferation, we selected the EMT6 cells and 3T3 cells for further investigation. As shown in Figure 5A, compound 2 inhibited cell proliferation in a concentration-dependent manner, with significant differences between EMT6 and 3T3 cells. The IC50 of compound 2 was 15 μM for EMT6 cells and 30 μM for 3T3 cells, suggesting that cancer cells (EMT6) were more sensitive than normal cells (3T3).
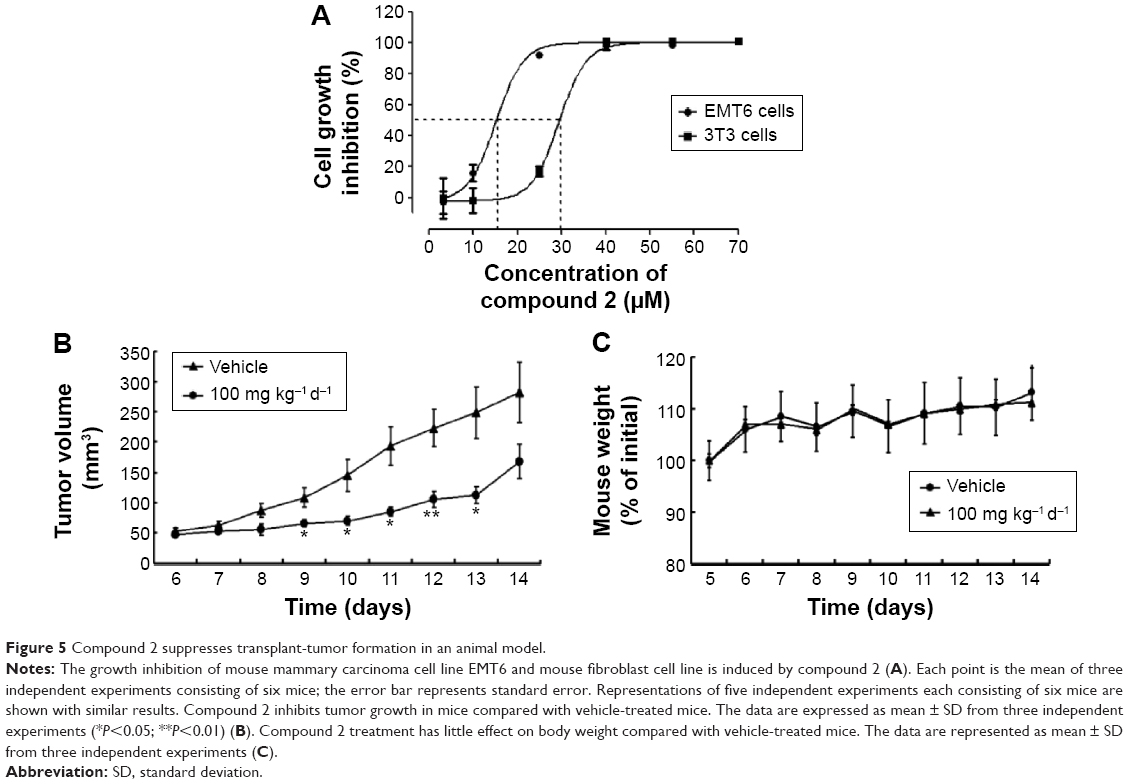 | Figure 5 Compound 2 suppresses transplant-tumor formation in an animal model. |
To determine the antitumor effect of LARS inhibitor in vivo, we have examined the effect of compound 2 in EMT6 transplant mouse model. As shown in Figure 5B, tumor growth was significantly suppressed from the ninth day in compound 2-treated mice compared with that in the control group. Nevertheless, no significant difference was observed in body weight loss between mice in the two groups within 14 days (Figure 5C), which indicates that compound 2 might has less side effects on the organisms.
Discussion
LARS has been successfully targeted in designing antimicrobial drugs. Currently, most of the drugs targeting aaRS concentrate on the anti-infective drugs, with antibacterial drugs being most prominent and followed by antifungal drugs.21 In this study, LARS in HOSE cells has been successfully expressed to serve as a target for screening new hsLARS inhibitor in vitro. The cell proliferation was found to be suppressed by targeting intracellular LARS. In addition, hsLARS inhibition was demonstrated to activate p21 early transcription and promote cell apoptosis. Finally, hsLARS inhibition was shown to reduce the progression of implanted EMT6 tumor in mice. The aforementioned results indicated that LARS may be a potential target at the center of a crucial signaling pathway controlling cell metabolism and cell proliferation.
Previous studies have reported that lack of leucine is the most important nutrition-sensing mechanism through uncharged leucine-specific tRNA, signaling necessary for suppression of cell growth.11,22 Aminoacyl-tRNAs was the first product in protein biosynthesis, and inhibition of aminoacyl-tRNA synthesis is described to inhibit protein synthesis.23 A study by Shin et al12 demonstrated that knockdown LARS in lung cancer cells may reduce the cells’ ability to migrate and form colonies. LARS is reported to be overexpressed in cancer cells and is considered as a potential metabolic anticancer target.10,24,25 In addition, the wide use of natural or synthetic anticancer drugs against metabolic pathways, such as 5-fluorouracil and taxol derivatives, suggests the selectivity of these antimetabolic drugs based on the nature of cancer cells whose proliferation relies on unregulated signaling pathways. In this study, the LARS inhibitor could significantly inhibit tumor cell proliferation by targeting intracellular LARS in a dose-dependent manner, which suggests that LARS may be a potential anticancer target.
LARS is known to interact with AIMP1 and other multifunctional proteins.26 In genotoxic damage, these multifunctional proteins were found to activate the p53 activity27,28 and then regulate the expression of p21 protein. Protein p21 has been recognized as a critical mediator through which p53 inhibits human neoplasia. By observing p53-dependent G1 arrest, p53 expression was found to be completely abrogated in p21-deficient cells.29 Despite its profound role in promoting antiproliferation and p53 tumor suppressor pathway, p21 is also involved in promoting carcinogenesis and tumor progression.30 Consequently, p21 is often misregulated in human cancers, and the gene plays a role as a tumor suppressor or as an oncogene, depending on the cellular context and circumstances. On the other hand, as CHX inhibits protein synthesis by interfering with the translocation step in protein synthesis including p21 protein, p21 expression is predicted to be inhibited during treatment by LARS inhibitors. In this study, compound 2 was found to have an early induction of p21 expression, indicating the protective signaling of cell starvation at 12 hours treatment. The repression of p21 expression at 48 hours suggests general repression of protein synthesis at a later time. Moreover, compound 2 was shown to induce apoptosis in a dose-dependent manner within 24 hours. Meanwhile, the CHX, as a protein synthesis inhibitor, did not lead to activation of p21 signal transduction pathway, but inhibited protein synthesis in a dose-dependent manner. Therefore, we suggest that the performance of the LARS inhibitor is quite different from that of the general inhibitors of protein synthesis such as CHX. Moreover, the inhibition of LARS caused an increase in uncharged leucine-specific tRNA, which triggered a chain of malnutrition signaling despite a normal supply of leucine in cells. The performance of compound 2 in human cancers might be due to the misregulation of p21, while the precise way needs to be studied further.
Taken together, our results show that inhibition of LARS may reduce the proliferation of cancer cells through the p21 signaling pathway and cause apoptosis. In addition, treatment with the LARS inhibitor significantly reduces the implanted tumor growth in an animal model. Our findings suggest that LARS may serve as a potential anticancer target and that the nutritional signaling pathway may provide valuable anticancer targets, leading to novel anticancer therapeutics.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81201769 and 81373319) and Shanghai Government Science and Technology Funding (10431903900).
Disclosure
The authors report no conflicts of interest in this work.
References
Medina MA, Marquez J, de Castro IN. Interchange of amino acids between tumor and host. Biochem Med Metab Biol. 1992;48(1):1–7. | ||
Giese C, Lepthien S, Metzner L, Brandsch M, Budisa N, Lilie H. Intracellular uptake and inhibitory activity of aromatic fluorinated amino acids in human breast cancer cells. Chem Med Chem. 2008;3(9): 1449–1456. | ||
Shikano N, Ogura M, Okudaira H, et al. Uptake of 3-[125I]iodo-alpha-methyl-L-tyrosine into colon cancer DLD-1 cells: characterization and inhibitory effect of natural amino acids and amino acid-like drugs. Nucl Med Biol. 2010;37(2):197–204. | ||
Cascino A, Muscaritoli M, Cangiano C, et al. Plasma amino acid imbalance in patients with lung and breast cancer. Anticancer Res. 1995;15(2):507–510. | ||
Nishihira T, Takagi T, Kawarabayashi Y, et al. Anti-cancer therapy with valine-depleted amino acid imbalance solution. Tohoku J Exp Med.1988;156(3):259–270. | ||
Lamb RF. Amino acid sensing mechanisms: an Achilles heel in cancer? FEBS J. 2012;279(15):2624–2631. | ||
Goberdhan DC. Intracellular amino acid sensing and mTORC1-regulated growth: new ways to block an old target? Curr Opin Investig Drugs. 2010;11(12):1360–1367. | ||
Hurdle JG, O’Neill AJ, Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob Agents Chemother. 2005;49(12):4821–4833. | ||
Lee SW, Kang YS, Kim S. Multifunctional proteins in tumorigenesis: aminoacyl-tRNA synthetases and translational components. Curr Proteom. 2006;3(4):233–247. | ||
Park SG, Schimmel P, Kim S. Aminoacyl tRNA synthetases and their connections to disease. Proc Natl Acad Sci U S A. 2008;105(32): 11043–11049. | ||
Gallinetti J, Harputlugil E, Mitchell JR. Amino acid sensing in dietary-restriction-mediated longevity: roles of signal-transducing kinases GCN2 and TOR. Biochem J. 2013;449(1):1–10. | ||
Shin SH, Kim HS, Jung SH, Xu HD, Jeong YB, Chung YJ. Implication of leucyl-tRNA synthetase 1 (LARS1) over-expression in growth and migration of lung cancer cells detected by siRNA targeted knock-down analysis. Exp Mol Med. 2008;40(2):229–236. | ||
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. | ||
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi:10.1158/2159-8290.CD-12-0095. | ||
Park H, Davidson E, King MP. Overexpressed mitochondrial leucyl-tRNA synthetase suppresses the A3243G mutation in the mitochondrial tRNA(Leu(UUR)) gene. RNA. 2008;14(11):2407–2416. | ||
Mayr JA, Meierhofer D, Zimmermann F, et al. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin Cancer Res. 2008;14(8):2270–2275. | ||
Baker SJ, Zhang YK, Akama T, et al. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2690), for the potential treatment of onychomycosis. J Med Chem. 2006;49:4447–4450. | ||
Zhao Y, Wang Q, Meng Q, et al. Identification of Trypanosoma brucei leucyl-tRNA synthetase inhibitors by pharmacophore- and docking-based virtual screening and synthesis. Bioorg Med Chem. 2012;20(3): 1240–1250. | ||
Gao G, Yao Y, Ding D, Ye L, Zhou H, Li D. The establishement of an anti-Trypanosoma drug screening system with leucyl-tRNA synthetase as an inhibition target. China Biotechnol. 2009;29(12):13–17. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Tao J, Schimmel P. Inhibitors of aminoacyl-tRNA synthetases as novel anti-infectives. Expert Opin Investig Drugs. 2000;9(8):1767–1775. | ||
Schwartz GJ. Central leucine sensing in the control of energy homeostasis. Endocrinol Metab Clin North Am. 2013;42(1):81–87. | ||
Kawamura T, Liu D, Towle MJ, et al. Anti-angiogenesis effects of borrelidin are mediated through distinct pathways: threonyl-tRNA synthetase and caspases are independently involved in suppression of proliferation and induction of apoptosis in endothelial cells. J Antibiot (Tokyo). 2003;56(8):709–715. | ||
Antonellis A, Green ED. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet. 2008;9:87–107. | ||
Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316(5832):1759–1761. | ||
Kim S, You S, Hwang D. Aminoacyl-tRNA synthetases and tumorigenesis: more than housekeeping. Nat Rev Cancer. 2011;11(10): 708–718. | ||
Han JM, Park BJ, Park SG, et al. AIMP2/p38, the scaffold for the multi-tRNA synthetase complex, responds to genotoxic stress via p53. Proc Natl Acad Sci U S A. 2008;105(32):11206–11211. | ||
Park BJ, Kang JW, Lee SW, et al. The haploinsufficient tumor suppressor p18 upregulates p53 via interactions with ATM/ATR. Cell. 2005;120(2):209–221. | ||
Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55(22): 5187–5190. | ||
Roninson IB. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 2002;179(1):1–14. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.