")
Back to Journals » The Application of Clinical Genetics » Volume 13
A Homozygous Truncating Mutation in NALCN Causing IHPRF1: Detailed Clinical Manifestations and a Review of Literature
Authors Karimi AH, Karimi MR, Farnia P, Parvini F , Foroutan M
Received 12 May 2020
Accepted for publication 8 August 2020
Published 27 August 2020 Volume 2020:13 Pages 151—157
DOI https://doi.org/10.2147/TACG.S261781
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Amir Hossein Karimi,1 Mohammad Reza Karimi,1 Poopak Farnia,2,3 Farshid Parvini,1 Majid Foroutan4
1Department of Biology, Faculty of Basic Sciences, Semnan University, Semnan, Iran; 2Mycobacteriology Research Centre (MRC), National Research Institute of Tuberculosis and Lung Disease (NRITLD), Shahid Beheshti University of Medical Sciences, Tehran, Iran; 3Department of Biotechnology, School of Advanced Technologies in Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 4Department of Internal Medicine, Semnan University of Medical Sciences, Semnan, Iran
Correspondence: Farshid Parvini; Majid Foroutan Tel +98-2331533197
Fax +98-2333321005
Email [email protected]; [email protected]
Abstract: Infantile hypotonia, with psychomotor retardation and characteristic facies 1 (IHPRF1), is a rare disorder characterized by global developmental delay and dysmorphic features. This syndrome is caused by genetic anomalies within the NALCN gene. The current report examines a 9-year-old female IHPRF1 patient. Our objective was to contribute to the delineation of the underlying factors influencing this rare condition. Whole exome sequencing (WES) was utilized to identify the disease-causing mutation in the affected individual. Subsequently, Sanger sequencing was performed for the patient, her parents, and two close relatives in order to confirm the detected mutation. Moreover, detailed clinical examinations including EEG, echocardiography, and biochemical/physical tests were carried out to elucidate the effects of the mutation. WES identified a homozygous nonsense mutation in the NALCN gene (c.2563C>T p.R855X). This mutation was confirmed by Sanger sequencing in the patient and her family members and segregated with the autosomal recessive inheritance pattern of IHPRF1. Moreover, genotype-phenotype correlation analysis confirmed the disease-causing nature of this mutation. The current report provides the first detailed description of a patient with this homozygous nonsense mutation (c.2563C>T p.R855X) and expands the clinical spectrum of IHPRF1 disease. Possible influences of sex and other factors on this disease are discussed and a review of the literature is also provided.
Keywords: global developmental delay, dysmorphism, intellectual disability, motor retardation, cognitive delay
Plain Language Summary
Infantile hypotonia with psychomotor retardation and characteristic facies 1 (IHPRF1), is a rare neurological disorder caused by mutations in the NALCN gene. The product of this gene is the main component of a conserved ion transporting channel involved in determining the resting membrane potential of different cell types including neurons and is involved in many physiological processes such as respiration, intestinal pacemaking, and regulation of circadian cycle. Individuals affected by IHPRF1 are most prominently characterized by global developmental delay and dysmorphic features. Due to our poor understanding of the NALCN ion transporting channel and the limited number of reported IHPRF1 patients so far, there is no developed comprehensive perspective on the underlying factors affecting this disorder and the variability of the affected systems and processes. Herein, we provide a detailed description of an IHPRF1 patient with a homozygous truncating mutation, manifesting a wide range of severe abnormal symptoms. We also provide a thorough review of the literature and propose sex and other elements as possible factors influencing this rare syndrome. Moreover, we draw the attention of the future investigators to a described activator of the NALCN ion transporting channel known as substance P, as a potential participant in many of the anomalies observed in the diseases associated with NALCN ion transporting channel.
Introduction
Sodium leak channel, non-selective (NALCN) is the sole non-selective member of the 24-TM ion channel superfamily.1 This channel is an essential component in the regulation of the resting membrane potential of several cell types including neurons by conducting a sodium background conductance.2,3 NALCN along with UNC80 and UNC79 and NLF-1/FAM155A are the four main subunits of a channelosome which is known to be involved in rhythmic behaviors including circadian cycle, respiration, and intestinal pacemaking.3
The activity of the NALCN channelosome is regulated by different types of mediators. This includes its activation via neurotensin, acetylcholine, and substance P (SP),4,5 and its inhibition by GABA, dopamine, and extracellular calcium.6,7
Several diseases have been shown to be associated with the NALCN channelosome. While no disease-causing mutation has been reported for the UNC79 gene, mutations in the NALCN and UNC80 genes can cause infantile hypotonia with psychomotor retardation and characteristic facies 1 (IHPRF1, OMIM #615,419) and IHPRF2 (OMIM #616,801), respectively. Both IHPRF1 and IHPRF2 are shown to follow the autosomal recessive inheritance pattern of Mendelian disorders.8,9 It is worth mentioning that mutations in NALCN could also lead to autosomal dominant congenital contractures of the limbs and face, hypotonia, and developmental delay syndrome (CLIFAHDD, OMIM #616,266). A recent study indicates that IHPRF1 and CLIFAHDD are caused by loss and gain of function mutations, respectively.10
Here, we report a 9-year-old female IHPRF1 patient from a highly consanguineous Afghan family residing in Iran. The patient manifests a wide range of symptoms including severe hypotonia, severe global developmental delay, lack of speech, severe muscle atrophy, hypersalivation, and severe constipation (Table 1). The patient has also had an older male sibling with similar abnormalities who has died undiagnosed at the age of 3.5 months. Parents sought genetic counseling during the mother’s third pregnancy after their gynecologist suspected genetic defects in the proband. Our examinations revealed a homozygous nonsense mutation in the NALCN gene (NM_052867:c.2563C>T) as the cause of the defects in the proband and the heterozygosity for this mutation was confirmed prenatally for the third sibling. The present study further expands the clinical symptoms associated with IHPRF1 by describing the wide range of abnormal phenotypes manifested in the affected individual. A review of the literature is provided and a number of issues that might prove beneficial to be taken into account in future reports and studies are also discussed.
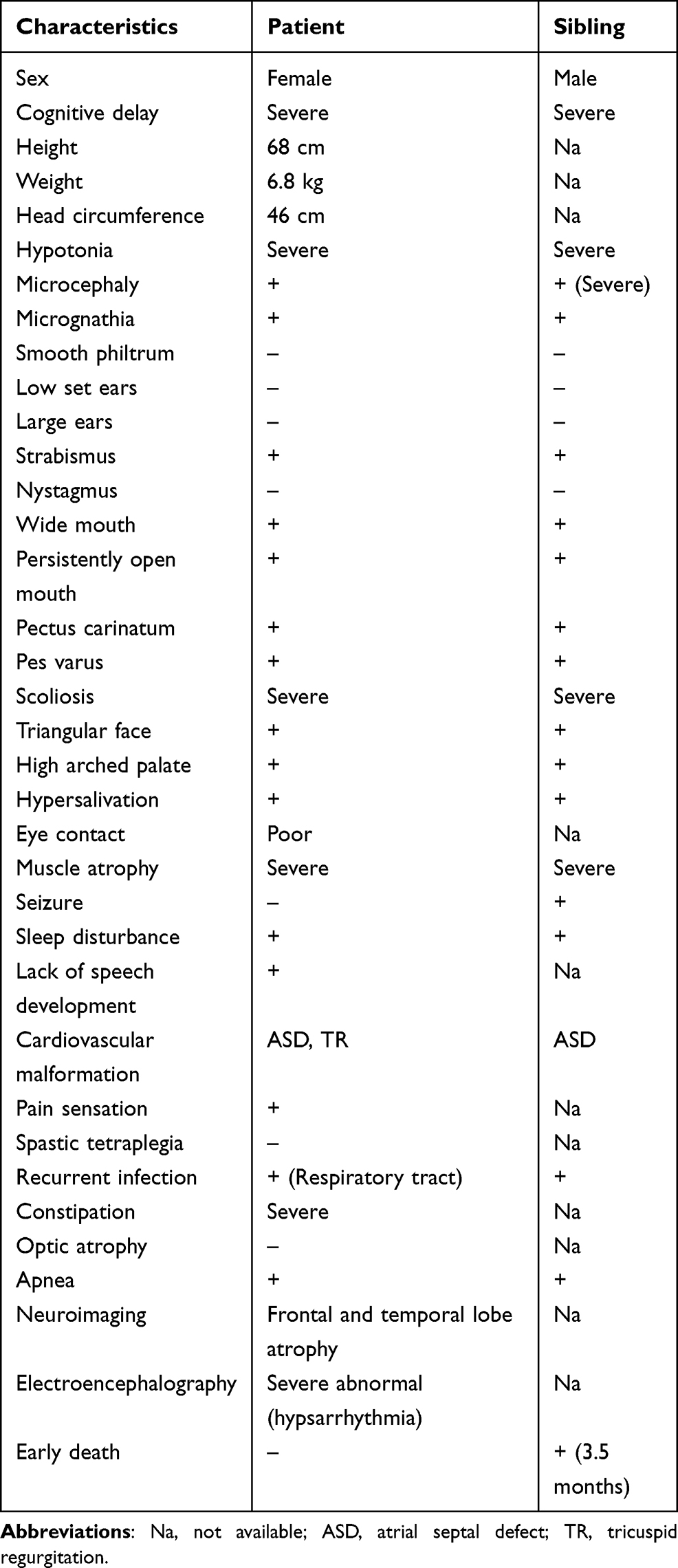 |
Table 1 Clinical Characteristics of the Proband and Her Deceased Sibling |
Materials and Methods
This study was approved by the ethics committee of Semnan University of medical sciences, Semnan, Iran. Informed consent was obtained from the legal guardians and Karyotype test was performed for the patient. Since no chromosomal abnormalities were detected, genomic DNA samples were prepared with the QIAamp DNA Blood Mini Kit (Germany) and whole exome sequencing (WES) was carried out for about 100 million reads, using the Illumina Hiseq2000 sequencer platform. Obtained results were analyzed by open-access bioinformatics tools, namely BWA aligner,11 GATK,12 and annovar.13 In order to inquire about the novelty of the mutation, public resources including ClinVar,14 gnomAD,15 Kaviar,16 and GME were utilized. Moreover, the local population database with more than 3000 unrelated individuals (BayanGene) was examined in order to determine the frequency of the mutation in the local population.
Online bioinformatics tools such as MutationTaster17 and CADD_phred18 were used in order to predict the probable effects of the mutation. Subsequently, Sanger sequencing was performed for the patient, her parents, and two close relatives to confirm the presence of the identified mutation and analyze its segregation. The primers used for this test were: F-5′ AGCAAAGAGGAGCAGCATGAT 3′ and R-5′ CTCTCATTCGGGAATGGCAAAAT 3′. Chromas software was applied to analyze the resulting data from Sanger sequencing. To determine the status of the mutation in the fetus, genomic DNA isolated from the amniotic fluid cells was sequenced. Furthermore, quantitative fluorescence polymerase chain reaction (QF-PCR) was carried out to investigate short tandem repeat (STR) markers on chromosomes X, Y, 13, 18, and 21 of the fetus, using “3130 Genetic Analyzer” and fragment analysis software.
Clinical examinations including echocardiography and electroencephalography (EEG) as well as physical examinations were performed for the patient. Since any further invasive examination was recognized to be life-threatening for the patient, we were confined to rely on the patient’s clinical history including brain CT scan, complete blood count and biochemistry profile, as well as abdominopelvic ultrasound at 8, 9, and 11 months of age, respectively.
Results
The WES analysis identified a stop gain mutation in the NALCN gene on chromosome13 (NM_052867); chr13:101107503G/A c.2563C>T p.R855X (rs376152742). The mutation was predicted to be pathogenic by MutationTaster and CADD_phred. Our review of the public resources, local population database, and the literature revealed that there is no previous publication describing this mutation in homozygous form. However, a single ClinVar submission confirmed the pathogenic effects of this genetic anomaly but merely provided an overview of the resulting manifestations with no detailed information. Furthermore, it failed to distinguish between IHPRF1 and CLIFAHDD and resulted in confusion by referring to several publications on CLIFAHDD as supporting evidence (SCV000742166.1). Sanger sequencing confirmed the presence of the mutation and its homozygosity in the proband and segregated with the autosomal recessive inheritance pattern of IHPRF1 in all tested individuals (Figure 1).
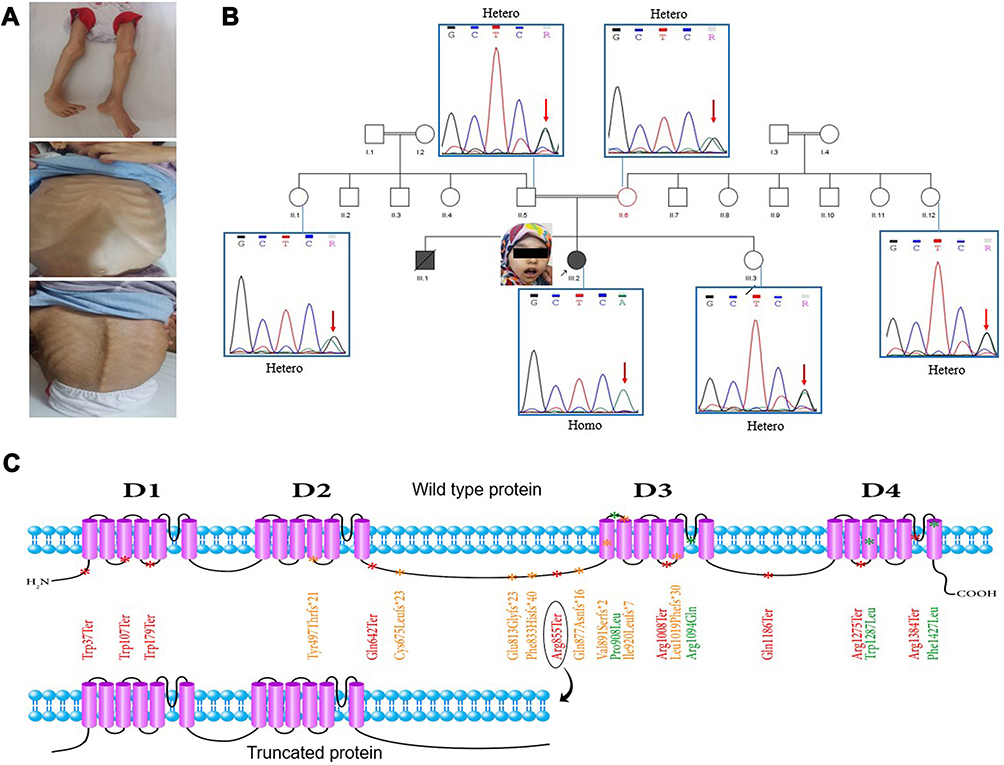 |
Figure 1 (A) Patient’s dysmorphic phenotypes. Pectus carinatum, severe muscle atrophy, pes varus, and scoliosis are evident. (B) Patient’s pedigree along with the results of Sanger sequencing for the respective individuals. All tested individuals, except for the proband, were revealed to be heterozygote for the detected mutation in the NALCN gene. (C) The NALCN protein structure. This protein consists of four transmembrane domains (shown by D1 to D4), each of which contains six transmembrane segments. Twenty-one of the described missense, frameshift, and nonsense mutations are marked with green, orange, and red, respectively. The homozygous nonsense mutation detected in our patient (Arg855Ter) (distinguished by an ellipse) is located on the cytoplasmic loop, between the second and the third transmembrane domains. The hypothetical truncating effect of this mutation on the protein is illustrated. |
Clinical examination results of the proband included severe muscle atrophy, severe scoliosis (improved by bracing), constipation, poor eye contact, and dysmorphic features such as a triangular face, long thin fingers, strabismus, pectus carinatum, and pes varus. Echocardiography results diagnosed the patient with atrial septal defect (ASD) and tricuspid regurgitation (TR). Additionally, our examination of the patient’s clinical history revealed that severe global developmental delay was evident from the first months of life. She never gained the ability to roll over or sit without support. Neuroimaging showed frontal and temporal lobe atrophy, particularly around the Sylvian fissure region, and cortical perisylvian polymicrogyria was suspected. EEG was performed for the patient during sleep and included generalized slow activity and attenuation with superimposed beta range activity. Asymmetry was present and the results were compatible with hypsarrhythmia. The complete blood count and biochemistry profile included a high level of white blood cells, low hematocrit, low levels of creatinine and blood ammonia, a very high level of aspartate aminotransferase (AST), and a normal level of alanine aminotransferase (ALT). Plasma amino acid profile included low levels of glycine, alanine, alpha-amino butyric acid, tryptophan, valine, and leucine. Abdominopelvic ultrasound was unremarkable. The TSH and T4 levels were within the normal range. The Patient also manifested recurrent and prolonged respiratory tract infections (Table 1).
The results of the QF-PCR performed prenatally for the third sibling showed a female fetus with no chromosomal abnormalities. Sanger sequencing revealed that the fetus was heterozygote for the identified mutation in the NALCN. Echocardiography at 26 weeks of gestation diagnosed the fetus with ASD. She passed away at 3 months of age due to an unknown cause.
Discussion
Here, we report an IHPRF1 patient with a homozygous mutation c.2563C>T p.R855X in the NALCN gene. The wild type NALCN protein contains four homologous repeats, each of which comprises six transmembrane segments with the pore-forming region of each domain located between the fifth and the sixth transmembrane segments.3 Due to the truncating effect of the mutation, loss of 884 out of 1738 amino acids of the wild type protein is predicted. Hence, the resulting protein would contain only two out of the four above-mentioned domains (Figure 1).
Although the same mutation was previously described in a compound heterozygote individual (pat: p.R855X, mat: p.R1094Q),19 there are no previous publications of this mutation in the homozygous form. Hence, the current report provides the first description of the direct effects of this truncating homozygous mutation. Interestingly, the patient described by Campbell et al (2018) is probably the least affected individual among all the reported IHPRF1 patients, so far. They suggested that the milder manifestation of the disease phenotypes might be due to the fact that the missense mutation of the compound heterozygote patient is located on the third transmembrane domain of the NALCN protein. However, it is noteworthy that a missense mutation in the fourth transmembrane domain of the NALCN also resulted in a milder manifestation of the disease.20 Moreover, there are reports of several truncating mutations located both in and after the third transmembrane domain of the NALCN protein, which resulted in much more severe manifestations of the disease.20–22 Considering these, the exhibition of milder clinical symptoms in the patients reported by Campbell et al (2018), the patients in the family 2 described by Al-Sayed et al (2013), and individuals 16 and 17 described by Bramswig et al (2018) might also be attributed to the missense nature of the mutation. Three points, however, should be taken into account: First, hypothetically, mRNAs produced from the mutated genes with stop gain mutations can undergo nonsense-mediated decay (NMD), thus, the more severe exhibition of the disease phenotypes in patients with stop gain mutations in or after the third transmembrane domain of the NALCN might be due to the general downregulation of the truncated protein by this mechanism. Second, to the best of our knowledge, there are only four reported missense mutations in the NALCN gene which are all located in the third and the fourth transmembrane domains of NALCN protein (Figure 1).19,20,23 Third, in a recent study, no sodium background current was detected in cells expressing p.Trp1287Leu missense mutation which might suggest underlying factors other than residual channel activity for the milder exhibition of the symptoms in the individuals affected by this mutation.10 With that being said, further accumulation of the IHPRF1 reports can help in the delineation of this matter. Beyond any doubt, rapid progress in the field of next-generation sequencing allows for a more accurate and less expensive diagnosis and study of genetic defects.24–26
Considering the extremely high rate of constipation in all the IHPRF1 patients reported so far (present in 31 out of 34 examined individuals), constipation can be considered as one of the most prominent symptoms of this disease, along with global developmental delay and dysmorphic features (for a summary of the symptoms manifested in all the 36 patients reported so far, please see the Supplementary table). Cardiovascular malformation, however, is seldom associated with IHPRF1. To the best of our knowledge, other than the current case, there is only one other reported individual diagnosed with ASD,27 and there is no previous report of TR in IHPRF1 patients.
The patient described in this report had an older male sibling with similar phenotypes who passed away genetically undiagnosed at the age of 3.5 months. Interestingly, the male sibling also exhibited seizures which is a common phenotype in IHPRF1 patients (present in more than 47% of the described individuals). This made us suspect that sex might play a role in the extent of the disease severity. We specifically compared the disease status in all the seven opposite-sex IHPRF1 siblings described in the previous reports.19,20,22,28,29 Our interpretation of the information provided by the authors in their respective manuscripts is that there seems to be a bias towards the more severe exhibition of the IHPRF1 phenotypes in the male individuals. In two of the described siblings (individuals 2 and 3, and individuals 11 and 12 described by Bramswig et al (2018)), we were not able to identify any major difference between the individuals. In all the other siblings described, the male individuals seem to present a more severe phenotype compared to their female siblings. Although the number of reported siblings is not enough to draw conclusions, we suggest that the sex of the affected individual might have an influence on the extent to which IHPRF1 phenotypes manifest. In agreement with this hypothesis, the study of the effects of estrogen and progesterone hormones on the regulation of the NALCN expression in human myometrial smooth muscle cells suggests a sex-dependent biology for the NALCN.30
NALCN has been identified as a cation channel involved in the conductance of substance P-activated cation channel current.4 Considering that it has been shown that SP plays important roles in a wide variety of physiological processes including cardiovascular system activity, cell growth and proliferation, inflammation, pain sensation, respiration, and pacemaking activity in the gastrointestinal tract,31–33 some of the phenotypes associated with IHPRF1 disease can be attributed to the disability of the mutated NALCN in the conductance of the SP-activated cation channel current. Indeed, it has been shown that NALCN is an important component in the SP-mediated modulation of gastrointestinal motility through interstitial cells of Cajal and also is essential in respiratory system response to SP.34,35 Furthermore, the study of IHPRF1 from the perspective of the other known regulators of the NALCN channelosome, namely neurotensin, acetylcholine, GABA, extracellular calcium, and dopamine, would be essential for a better understanding of this disorder.5–7
Conclusion
We utilized WES and Sanger sequencing to identify a disease-causing mutation in the NALCN gene in a patient with dysmorphic features and severe global developmental delay. Our main limitation was the patient’s intolerance for further clinical examinations. Our results further expand the clinical spectrum of IHPRF1. We propose sex-based differences as possible influencers of this disease. Further study of the biology of NALCN is essential for a better understanding of this disease. Regarding the importance of SP in a wide variety of physiological activities, the future study of the role of NALCN in these processes might prove beneficial.
Abbreviations
ALT, alanine aminotransferase; AST, aspartate aminotransferase; ASD, atrial septal defect; CLIFAHDD, congenital contractures of the limbs and face, hypotonia, and developmental delay syndrome; EEG, electroencephalography; IHPRF, infantile hypotonia with psychomotor retardation and characteristic facies; NALCN, sodium leak channel, non-selective; NMD, nonsense-mediated decay; QF-PCR, quantitative fluorescence polymerase chain reaction; STR, short tandem repeats; SP, substance P; TR, tricuspid regurgitation; WES, whole exome sequencing.
Ethics Approval
This study was conducted under the approval of the ethics committee of Semnan University of medical sciences, Semnan, Iran. Written informed consent was obtained from the legal guardians prior to all the relevant clinical tests and the preparation and submission of the manuscript. Consent for publication of patient’s data and any identifiable features is also available.
Acknowledgments
The authors express their sincere gratitude to the patient and her respected family who kindly consented to join the present study. We would like to thank Dr. Nooshin Masoudian (Department of Internal Medicine, School of Medicine, Semnan University of Medical Sciences, Semnan, Iran) and Dr. Samira Mehralizade (Semnan University of Medical Sciences, Semnan, Iran) for their technical collaborations, as well.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Snutch TP, Monteil A. The sodium “leak” has finally been plugged. Neuron. 2007;54(4):505–507. doi:10.1016/j.neuron.2007.05.005
2. Ren D. Sodium leak channels in neuronal excitability and rhythmic behaviors. Neuron. 2011;72(6):899–911. doi:10.1016/j.neuron.2011.12.007
3. Cochet-Bissuel M, Lory P, Monteil A. The sodium leak channel, NALCN, in health and disease. Front Cell Neurosci. 2014;8(MAY):1–17. doi:10.3389/fncel.2014.00132
4. Lu B, Su Y, Das S, et al. Peptide neurotransmitters activate a cation channel complex of NALCN and UNC-80. Nature. 2009;457(7230):741–744. doi:10.1038/nature07579
5. Swayne LA, Mezghrani A, Varrault A, et al. The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic β-cell line. EMBO Rep. 2009;10(8):873–880. doi:10.1038/embor.2009.125
6. Lu B, Zhang Q, Wang H, Wang Y, Nakayama M, Ren D. Extracellular calcium controls background current and neuronal excitability via an UNC79-UNC80-NALCN cation channel complex. Neuron. 2010;68(3):488–499. doi:10.1016/j.neuron.2010.09.014
7. Philippart F, Khaliq ZM. G i/o protein-coupled receptors in dopamine neurons inhibit the sodium leak channel NALCN. Elife. 2018;7:1–19. doi:10.7554/eLife.40984
8. Perez Y, Kadir R, Volodarsky M, et al. UNC80 mutation causes a syndrome of hypotonia, severe intellectual disability, dyskinesia and dysmorphism, similar to that caused by mutations in its interacting cation channel NALCN. J Med Genet. 2016;53(6):397–402. doi:10.1136/jmedgenet-2015-103352
9. Karakaya M, Heller R, Kunde V, et al. Novel mutations in the nonselective sodium leak channel (NALCN) lead to distal arthrogryposis with increased muscle tone. Neuropediatrics. 2016;47(4):273–277. doi:10.1055/s-0036-1584084
10. Bouasse M, Impheng H, Servant Z, Lory P, Monteil A. Functional expression of CLIFAHDD and IHPRF pathogenic variants of the NALCN channel in neuronal cells reveals both gain- and loss-of-function properties. Sci Rep. 2019;9(1):1–14. doi:10.1038/s41598-019-48071-x
11. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi:10.1093/bioinformatics/btp698
12. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a map reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi:10.1101/gr.107524.110
13. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):1–7. doi:10.1093/nar/gkq603
14. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(D1):980–985. doi:10.1093/nar/gkt1113
15. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv. 2019;531210. doi:10.1101/531210.
16. Glusman G, Caballero J, Mauldin DE, Hood L, Roach JC. Kaviar: an accessible system for testing SNV novelty. Bioinformatics. 2011;27(22):3216–3217. doi:10.1093/bioinformatics/btr540
17. Schwarz JM, Cooper DN, Schuelke M, Seelow D. Mutationtaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi:10.1038/nmeth.2890
18. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D894. doi:10.1093/nar/gky1016
19. Campbell J, FitzPatrick DR, Azam T, et al. NALCN dysfunction as a cause of disordered respiratory rhythm with central apnea. Pediatrics. 2018;141(Supplement 5):S485–S490. doi:10.1542/peds.2017-0026
20. Bramswig NC, Bertoli-Avella AM, Albrecht B, et al. Genetic variants in components of the NALCN–UNC80–UNC79 ion channel complex cause a broad clinical phenotype (NALCN channelopathies). Hum Genet. 2018;137(9):753–768. doi:10.1007/s00439-018-1929-5
21. Bourque DK, Dyment DA, MacLusky I, Kernohan KD, McMillan HJ. Periodic breathing in patients with NALCN mutations. J Hum Genet. 2018;63(10):1093–1096. doi:10.1038/s10038-018-0484-1
22. Angius A, Cossu S, Uva P, et al. Novel NALCN biallelic truncating mutations in siblings with IHPRF1 syndrome. Clin Genet. 2018;93(6):1245–1247. doi:10.1111/cge.13162
23. Al-Sayed MD, Al-Zaidan H, Albakheet A, et al. Mutations in NALCN cause an autosomal-recessive syndrome with severe hypotonia, speech impairment, and cognitive delay. Am J Hum Genet. 2013;93(4):721–726. doi:10.1016/j.ajhg.2013.08.001
24. Mantere T, Kersten S, Hoischen A. Long-read sequencing emerging in medical genetics. Front Genet. 2019;10(MAY):1–14. doi:10.3389/fgene.2019.00426
25. Carneiro TNR, Krepischi ACV, Costa SS, et al. Utility of trio-based exome sequencing in the elucidation of the genetic basis of isolated syndromic intellectual disability: illustrative cases. Appl Clin Genet. 2018;11:93–98. doi:10.2147/TACG.S165799
26. Noavar S, Behroozi S, Tatarcheh T, Parvini F, Foroutan M, Fahimi H. A novel homozygous frame-shift mutation in the SLC29A3 gene: a new case report and review of literature. BMC Med Genet. 2019;20(1):4–10. doi:10.1186/s12881-019-0879-7
27. Takenouchi T, Inaba M, Uehara T, Takahashi T, Kosaki K, Mizuno S. Biallelic mutations in NALCN: expanding the genotypic and phenotypic spectra of IHPRF1. Am J Med Genet Part A. 2018;176(2):431–437. doi:10.1002/ajmg.a.38543
28. Köroǧlu Ç, Seven M, Tolun A. Recessive truncating NALCN mutation in infantile neuroaxonal dystrophy with facial dysmorphism. J Med Genet. 2013;50(8):515–520. doi:10.1136/jmedgenet-2013-101634
29. Gal M, Magen D, Zahran Y, et al. A novel homozygous splice site mutation in NALCN identified in siblings with cachexia, strabismus, severe intellectual disability, epilepsy and abnormal respiratory rhythm. Eur J Med Genet. 2016;59(4):204–209. doi:10.1016/j.ejmg.2016.02.007
30. 1,2,3. 2020;(314):1–32.
31. Mistrova E, Kruzliak P, Chottova Dvorakova M. Role of substance P in the cardiovascular system. Neuropeptides. 2016;58:41–51. doi:10.1016/j.npep.2015.12.005
32. Mashaghi A, Marmalidou A, Tehrani M, Grace PM, Pothoulakis C, Dana R. Neuropeptide substance P and the immune response. Cell Mol Life Sci. 2016;73(22):4249–4264. doi:10.1007/s00018-016-2293-z
33. Zhang N, Gao D, Liu Y, Ji S, Sha L. Effects of Neuropeptide substance p on proliferation and β-cell differentiation of adult pancreatic ductal cells. Front Neurosci. 2018;12(NOV):1–11. doi:10.3389/fnins.2018.00806
34. Yeh SY, Huang WH, Wang W, et al. Respiratory network stability and modulatory response to substance P require NALCN. Neuron. 2017;94(2):294–303.e4. doi:10.1016/j.neuron.2017.03.024
35. Kim BJ, Chang IY, Choi S, et al. Involvement of Na + -leak channel in substance P-induced depolarization of pacemaking activity in interstitial cells of Cajal. Cell Physiol Biochem. 2012;29(3–4):501–510. doi:10.1159/000338504
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.