Back to Journals » Journal of Inflammation Research » Volume 14
A Gender-Dependent Molecular Switch of Inflammation via MyD88/Estrogen Receptor-Alpha Interaction
Authors El Sabeh R, Bonnet M, Le Corf K, Lang K ![]() , Kfoury A, Badran B, Hussein N, Virard F, Treilleux I, Le Romancer M
, Kfoury A, Badran B, Hussein N, Virard F, Treilleux I, Le Romancer M ![]() , Lebecque S, Manie S, Coste I, Renno T
, Lebecque S, Manie S, Coste I, Renno T
Received 16 February 2021
Accepted for publication 29 April 2021
Published 21 May 2021 Volume 2021:14 Pages 2149—2156
DOI https://doi.org/10.2147/JIR.S306805
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Rana El Sabeh,1,2 Mélanie Bonnet,1 Katy Le Corf,1 Kevin Lang,1 Alain Kfoury,1 Bassam Badran,2 Nader Hussein,2 Francois Virard,1 Isabelle Treilleux,3 Muriel Le Romancer,1 Serge Lebecque,1 Serge Manie,1 Isabelle Coste,1,* Toufic Renno1,*
1Université de Lyon, Université Claude Bernard Lyon 1, INSERM 1052, CNRS 5286, Centre Léon Bérard, Centre de Recherche en Cancérologie de Lyon, Lyon, France; 2Université Libanaise, PRASE, Hadath, Lebanon; 3Pathology Department, Centre Léon Bérard, Lyon, France
*These authors contributed equally to this work
Correspondence: Toufic Renno; Isabelle Coste
Centre de Recherche en Cancétologie de Lyon, 28 Rue Laennec, Lyon, 69373, France
Tel +33 4 691 666 29; +33 4 691 666 30
Email [email protected] ; [email protected]
Introduction: Most Toll-like receptors and IL-1/IL-18 receptors activate a signaling cascade via the adaptor molecule MyD88, resulting in NF-κB activation and inflammatory cytokine and chemokine production. Females are less susceptible than males to inflammatory conditions, presumably due to protection by estrogen. The exact mechanism underlying this protection is unknown.
Methods: MCF7 cells expressing wild-type or mutated LXXLL motif were used to determine MyD88/estrogen receptor (ER)-a interaction by immunoprecipitation and cell activation by ELISA and luciferase reporter assay. IL-1b and/or E2 were used to activate MCF7 cells expressing normal or knocked down levels of PRMT1. Finally, in situ proximity ligation assay with anti-MyD88 and anti-methylated ER-a (methER-a) antibodies was used to evaluate MyD88/methylated ER-a interaction in THP1 cells and histological sections.
Results: We show that MyD88 interacts with a methylated, cytoplasmic form of estrogen receptor-alpha (methER-α). This interaction is required for NF-κB transcriptional activity and pro-inflammatory cytokine production, and is dissociated by estrogen. Importantly, we show a strong gender segregation in gametogenic reproductive organs, with MyD88/methER-α interactions found in testicular tissues and in ovarian tissues from menopausal women, but not in ovaries from women age 49 and less – suggesting a role for estrogen in disrupting this complex in situ.
Discussion: Collectively, our results indicate that the formation of MyD88/methER-α complexes during inflammatory signaling and their disruption by estrogen may represent a mechanism that contributes to gender bias in inflammatory responses.
Keywords: nuclear receptor, post-translational modification, inflammation, protein-protein-interaction
Introduction
Females are relatively protected from certain diseases with an underlying inflammatory component, such as atherosclerosis,1 ischemic stroke,2 adipocyte atrophy,3 blood-brain barrier disruption,4 LPS-induced endotoxic shock,5 colitis,6 and cancers that develop on an inflammatory background, such as hepatocellular carcinoma (HCC).7 The relative protection from inflammation is thought to be mainly due to the high levels of estrogen in pre-menopaused women.8,9 Menopaused or ovariectomized females have increased inflammation, whereas estrogen-treated males had milder inflammatory responses.10 Estrogen is produced both in males and females, but at much higher levels in the latter until menopause. Estrogen has been shown to be an inhibitor of inflammation, mainly via decreasing NF-kB signaling, leading to decreased cytokine and chemokine production.11 It was reported that MyD88-dependent activation of interleukin-6 is negatively regulated by estrogen via the estrogen receptor alpha (ER-α).12 MyD88 is the main adaptor protein in TLR and IL-1R signaling that induces the activation of NF-kB.13
Here we describe a novel mechanism of inflammatory signaling regulation via the interaction between MyD88 and a cytoplasmic form of ER-α, with implications for gender disparity in inflammatory responses.
Materials and Methods
Cell Lines
The MCF7 and THP1cell lines were purchased from ATCC.
Immunoprecipitation Assay and Western Blotting
After treatment, cells were lysed using NP-40 lysis buffer supplemented with protease and phosphatase inhibitors. Protein extracts (1mg) were incubated with primary antibody G20 from Santa Cruz (overexpressed ER-α) or antibody specific to endogenous methER-α.14,15 Protein G-sepharose (Sigma Aldrich®) or L-agarose (Thermo Fisher Scientific®) beads for ER-a and metER-a were added and incubated 1h at 4°C. The immunoprecipitated products were denatured 5 min at 95°C in Laemmli buffer. After separation on SDS–PAGE, the antibodies listed below were used for Western Blot: antiFlag (Sigma), anti-MyD88 (Assay Design), anti- ER-a (60C, Millipore), anti-PRMT1 and anti-p85 (Upstate Biotechnology), anti-Src and anti-phospho-NF-κB p65 (Ser468) (Cell Signaling), anti-IκBa (BD) and anti-actin (MP Biomedicals).
NF-κB Luciferase Assay
pNF-κB-Luc and pRL-TK plasmids were transfected with Lipofectamine 2000 (Life Technologies®) according to the manufacturer’s instructions. Twenty-four hours after transfection, luciferase activity was measured using the DualLuciferase Reporter Assay system (Promega®) according to the manufacturer’s instructions.
Small Interfering RNA (siRNA) Sequences and Transfection
MCF-7 cells were transfected using Lipofectamine 2000 (Life Technologies®) with the appropriate siRNA concentration according to the manufacturer’s protocol. The sequences of siRNA are as follows: Control siRNA: 5ʹ-GGU GUU CAU ACG CAU GAA GTT −3ʹ (100nM, Dharmacon®), PRMT1 siRNA: 5ʹ-CCA UCG ACC UGG ACU UCA A-3ʹ (100nM, Eurogentec®).
ELISA Assay
IL-8 production in culture supernatants was measured by using the Human IL-8 ELISA MAX™ Standard, Biolegend® kit.
Proximity Ligation Assay
Cells were fixed with 4% PFA, permeabilized and blocked with 0.3% saponin, 10% BSA in PBS. Cells were then incubated with primary antibodies to MyD88 (rabbit polyclonal from Assay Designs) and ER-α (Santa Cruz, clone D12) overnight at 4°C and the next steps were performed according to the manufacturer’s instructions (Duolink®, Sigma Aldrich). For tissue microarray sections, PLA was performed according the manufacturer’s instructions with primary antibodies to MyD88 (Assay Designs) and methylated ER-α.14,15 Tissue microarrays of normal multiple organs (BN1002B and BN00011A), normal ovaries (OV806), and normal testis (TEN601) were purchased from US Biomax (Euromedex). Additional histology slides of ovarian tissue from menopaused or non-menopaused women were kindly provided by Dr. Isabelle Treilleux (Pathology Dept, Centre Léon Bérard) according to strict ethical guidelines. Indeed, biological material collection and retention activity (n°BB-0033-00050) is declared to the Ministry of Research (DC-2008-99 and AC-2019-3426). Samples were used in the context of patient diagnosis. Unused parts of the samples might be used for research if patient is not opposed to it (information notice transmitted to each patient). This study was approved by the ethical review board of Centre Léon Bérard and data analyzed with respect to GDPR MR004. This BRC process is certified according to AFNOR NFS96900 (N° 2009/35,884.2) and ISO 9001 (Certification N° 2013/56,348.2).
Immunofluorescence
Cells were fixed with 4% PFA, permeabilized with cold methanol for 5 minutes at 4°C and then incubated with PBS 5% BSA for 1h. Cells were then incubated with p65 antibody recognizing both phosphorylated and unphosphorylated p65 (Cell Signaling®) in PBS 1% BSA for 2 hours at RT. After PBS washes, cells were incubated with Alexa 488-secondary goat anti-rabbit antibody (Life Technologies®) for 1 hour at RT.
Statistical Analysis
Statistical analysis on pooled experiments was performed using the software GraphPad Prism, and P-values were indicated by (*) when P<0.05, (**) when P<0.01, and (***) when P<0.001.
Results
MyD88, a key adaptor protein in TLR/IL-1R signaling, harbors an evolutionarily conserved LXXLL motif in its death domain between amino acids 90 to 94. This motif is necessary and sufficient for interaction with nuclear receptors.16 Since MyD88 and the nuclear receptor ER-α are functionally linked in inflammatory signaling,12 we asked whether these two proteins physically interact. As seen in Figure 1A, wild-type MyD88, but not forms of the protein truncated for the DD (ΔDD) or mutated in the LXXLL motif, immunoprecipitates with ER-α.
 |
Figure 1 MyD88 forms a functional complex with ER-α. (A) HEK293T cells were transfected with a plasmid encoding ER-α and/or Flag-MyD88 (WT), Flag-MyD88-LXXAA (LXXAA), Flag-MyD88-delta LXXLL (∆LXXLL), and Flag-MyD88-delta death domain (∆DD). Lysates were immunoprecipitated with anti-ER-α antibody and immunoblotted with anti-Flag antibody. The experiment was performed twice with similar results. (B) Luciferase activity from NF-κB luciferase in HEK293T cells overexpressing MyD88 WT, LXXAA, ∆LXXLL, or ∆DD. All luciferase values were normalized based on Renilla luminescence. (**), P<0.01. Data are representative of 3 independent experiments, and n=3 in each experiment (upper graph). IL-8 production by HEK293T cells was measured by ELISA in culture supernatants of HEK293T cells overexpressing MyD88 WT, LXXAA, ∆LXXLL, or ∆DD. (**), P<0.01. The experiment was performed at least three times with similar results, and n=3 in each experiment (lower graph). (C) Luciferase activity from NF-κB-luciferase reporter in MCF7 cells pretreated with cell-permeable peptides containing the LXXLL motif (WT) or a mutant form (mut), and stimulated with 10 ng/mL IL-1. The experiment was performed twice with similar results. (D) THP-1 monocytic cells were stimulated with 1 µg/mL LTA and/or 10 nM estrogen (E2), then stained for MyD88 and ER-α according to the Duolink protocol. Magnification: 40x. The experiment was performed twice with similar results. Abbreviation: NT, non-treated cells. |
To address whether this interaction plays a role in NF-κB activation, an NF-κBluciferase reporter assay was performed, in which HEK293T cells were transfected either with wild-type, or with ΔDD or LXXLL mutated forms of MyD88. As shown in Figure 1B, whereas wild-type MyD88 activates a NF-κB-luc repoter and induces high levels of
IL-8, the MyD88 mutant forms unable to interact with ER-α produced virtually no IL-8. To unequivocally ascertain the importance of MyD88/ER-α interaction itself to inflammatory signaling, we used a cell-permeable peptide to inhibit the endogenous. MyD88/ER-α interaction in IL-1b-treated cells. This peptide contains the sequence of the MyD88 death domain containing the LXXLL motif responsible for the ER-α binding. As shown in Figure 1C, the specific peptide (WT), but not peptides that were mutated in the LXXLL motif (mut1-3), reduced NF-κB transcriptional activity in response to IL-1b. Therefore, MyD88 interacts with ER-α via its LXXLL motif, and this interaction is required for NF-κB activation and cytokine production.
We then investigated the regulation of endogenous MyD88/ER-α interaction. To this end, we used the highly sensitive in situ proximity ligation assay17 on the acute monocytic leukemia THP-1 cell line that is known to be responsive to both estrogen and TLR ligands.18 THP-1 cells were treated with LTA (lipoteichoic acid, TLR2 agonist) and/or with estrogen, before performing PLA. As seen in Figure 1D, untreated THP-1 cells show some interaction between MyD88 and ER-α (a). However, upon treatment with LTA, there is an increase in the interaction between MyD88 and ER-α (b), which is strongly reduced in presence of estrogen (d). Therefore, MyD88 and ER-α interact at the endogenous level. This interaction is enhanced by TLR stimulation and is dissociated in the presence of estrogen.
ER-α signaling can be divided into two major pathways, the genomic and the nongenomic pathways. In the genomic pathway, ligand-bound ER-α translocates to the nucleus, where it activates target gene transcription. The non-genomic or “rapid signaling” pathway is essentially cytoplasmic and is mainly mediated by methylated ER-α. Upon rapid estrogen stimulation (less than 5 minutes), the methyltransferase PRMT1 methylates ER-α on arginine 260 present in its DNA binding domain and which also lies within one of the nuclear localization signals of ER-α, thus maintaining methylated ERα (methER-α) exclusively in the cytoplasm.14 Since MyD88 is also a cytosolic protein, we investigated whether it particularly interacts with methER-α. We showed in coimmunoprecipitation experiments that upon cell stimulation with IL-1β, ER-α is methylated and it interacts with MyD88. Treatment of cells with IL-1β in presence of estrogen, or with siRNA specific for PRMT1, inhibits ER-α methylation, and therefore prevents ER-α interaction with MyD88 (Figure 2A).
 |
Figure 2 IL-1 stimulation leads to the formation of MyD88/methER-α complexes, which are required for NF-κB transcriptional activity and IL8 production. (A) MCF7 cells were transfected with control siRNA (siControl) or PRMT1-specific siRNA (siPRMT1) and then treated with 10 nM E2, 10 ng/mL IL-1, or both. Methylated ER-α was then immunoprecipitated and blotted with anti-MyD88 and anti-ER-α antibodies. (B) MCF7 cells were transfected with siControl or siPRMT1 and were treated with IL-1. Methylated ER-α was then immunoprecipitated and blotted for MyD88, ER-α, p85, and Src. (C–E) MCF7 cells were transfected with siControl or siPRMT1, and treated with IL-1. After 24h, IL-8 production was measured in culture supernatants by ELISA (C); the amount of p-p65 (Ser468), IKBα, PRMT1 and actin was determined by Western blot (D); and luciferase activity from an NF-κB-luciferase reporter was measured. Values were normalized based on Renilla luminescence (E). (F) MCF7 cells were pretreated with cell-permeable WT or mutant (mut) peptides for 1h and then stimulated with IL-1 for the indicated times. The amount of p-p65 (Ser468), IKBα, PRMT1, and actin was determined by Western blot. In this figure, blots have been cropped to improve clarity (white line). All these experiments were performed three times with similar results. P-values are indicated by (**) when P<0.01, and (***) when P<0.001. |
It has been shown that upon its methylation, ER-α interacts with p85, the regulatory subunit of PI3K, and with Src.14 To investigate whether MyD88, methylated ER-α, and these proteins are in the same complex, we performed an endogenous immunoprecipitation of methylated ER-α from MCF7 cells upon IL-1b stimulation in the presence or absence of PRMT1. As shown in Figure 2B, we detected the presence of p85 and Src in the MyD88/methER-α complex under the same conditions. This is consistent with previous findings that showed that MyD88 and p85 can be found in the same complex.19 These interactions were abrogated upon PRMT1 knockdown. Taken together, these data suggest that inflammatory signals trigger methylation of ER-α and its interaction with MyD88 in a complex containing PI3K p85 and Src.
To investigate whether the disruption of the MyD88/metER-α complex by PRMT1 knockdown is associated with biological consequences, MCF7 cells were stimulated with IL-1b after non-silencing or PRMT1-specific siRNA transfection. We found a decrease in IL-8 production when PRMT1 is silenced (Figure 2C).
We then sought to decipher the mechanisms whereby PRMT1-methylated ER-α and its association with MyD88 are implicated in the inflammatory response induced by IL-1b. Since PRMT1 has been described to methylate mainly late downstream targets such as histones, transcription factors, and cell-cycle and repair proteins20 and given that ER-α methylation by PRMT1 occurs early in the response to IL-1b (15 min, Supplemental Figure 1), we reasoned that analyzing early consequences of PRMT1 silencing should reduce the contamination of the results with off-target, indirect effects.
With this in mind, we looked at NF-kB activation in presence or absence of PRMT1. We found that early cytoplasmic events after IL-1b stimulation such as IκB degradation were not affected by knocking down PRMT1 (Figure 2D). At this time, most of the MyD88-methER-α complexes have not yet been formed. We therefore asked whether p65 translocation into the nucleus also occurs in absence of PRMT1. We showed that the p65 subunit of NF-kB complex translocates into the nucleus between 15 and 30 minutes after IL-1b stimulation independently of PRMT1 expression levels (Supplemental Figure 2). We then asked whether the p65 that was translocated into the nucleus in absence of PRMT1 is capable of activating the transcription of NF-kB target genes. To address this question, a NF-κB-luciferase reporter assay was performed under PRMT1 inhibition. Results show that transcriptional NF-κB activity is reduced as early as 2 hours after stimulation in absence of PRMT1 (Figure 2E). A post-translational modification of p65, the serine 468 phosphorylation, had been described to occur in the nucleus 15 min after activation and to be essential for enabling NF-kB transcriptional activation of several target genes.21 We therefore assayed p65 S468 phosphorylation in response to IL-1b and found that this nuclear post-translational modification is reduced post-treatment with IL-1b when PRMT1 is knocked down (Figure 2D).
To ascertain whether lack of MyD88/ER-α interaction resulting from knocking down PRMT1 is responsible for the defect in p65 activation, we used the cell-permeable LXXLL peptide described above to inhibit the endogenous MyD88/ER-α interaction in IL-1b-treated cells. Similar to what was observed upon PRMT1 knockdown, the LXXLLcontaining peptide did not affect IkB degradation (Figure 2F). However, also as with siPRMT1, the specific peptide inhibited p65 phosphorylation on S468 in response to IL-1b in MCF7 cells (Figure 2F).
To examine whether MyD88/methER-a interaction also occurs in human tissue, we performed PLA on tissue microarrays with a wide panel of tissues. We found that overall, MyD88/methER-α interaction was detected in 66/182 of tissue sections (Table 1). However, when tissue samples were segregated according to sex, a male-to-female bias emerged (57/103 vs 9/79). This bias is consistent with our data showing that estrogen disrupts the MyD88/methER-α complex. We then reasoned that since differences in estrogen production are most pronounced in gametogenic reproductive tissues, this bias should be more obvious in these tissues. Indeed, we found that whereas none (0/29) of ovarian tissues from women at non-menopausal age (range: 18–49) displayed MyD88/methER-α interactions, all testicular tissues tested (35/35) exhibited strong PLA staining (Table 1). The cells that stained positive in the testes were histologically identified as Leydig cells (Figure 3). This could be explained by the fact that Leydig cells do not have functional estrogen since they express estrogen sulfotransferase (EST) in both mice and humans. EST is an enzyme that modifies estrogen, rendering it unable to bind to its receptors.22 We further reasoned that if non-menopaused ovarian tissues do not have MyD88/methER-α complexes because they express high levels of estrogen, ovarian tissues from women at menopausal age, which are known to produce reduced levels of estrogen,23 should have MyD88/methER-α complexes. Indeed, we found that 3/11 ovarian samples from women aged 50–69 were positive for MyD88/methER-α PLA staining (Table 1 and Figure 3). Collectively, these data show a strong segregation of MyD88/methER-α interaction according to gender in gametogenic reproductive organs and suggest a role for estrogen in disrupting this complex in situ.
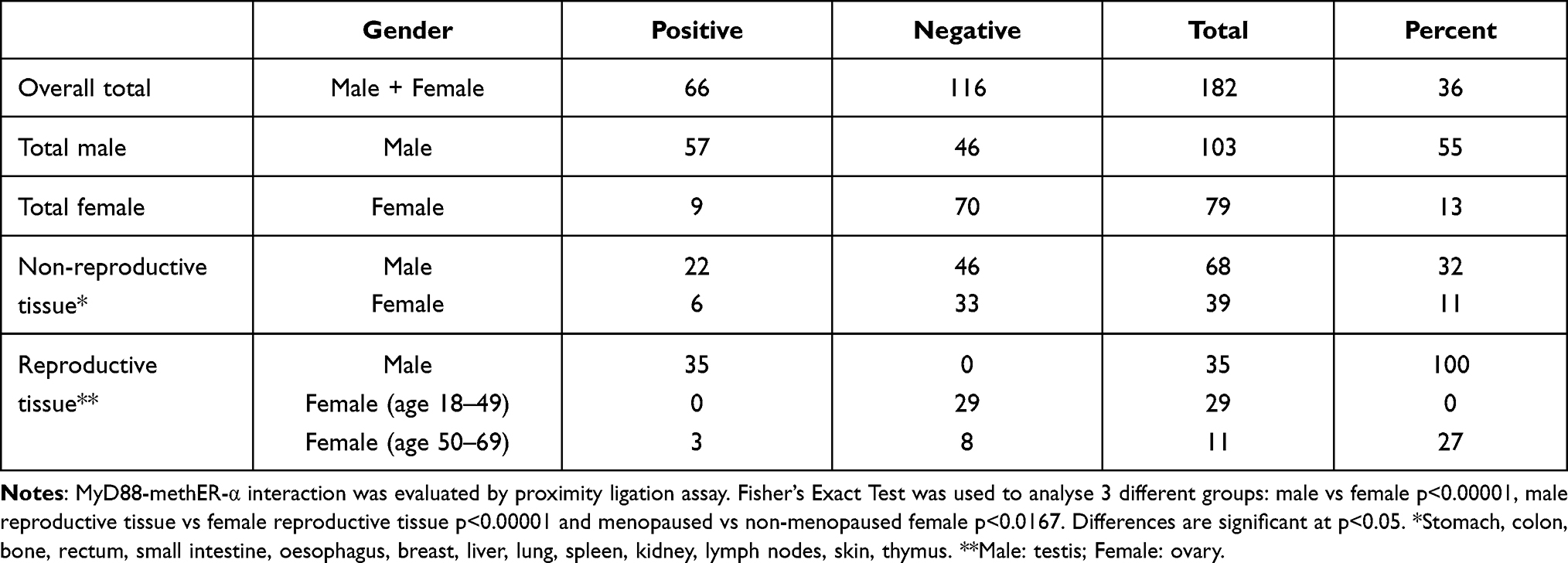 |
Table 1 MyD88-methER-α Interaction in Normal Human Tissue |
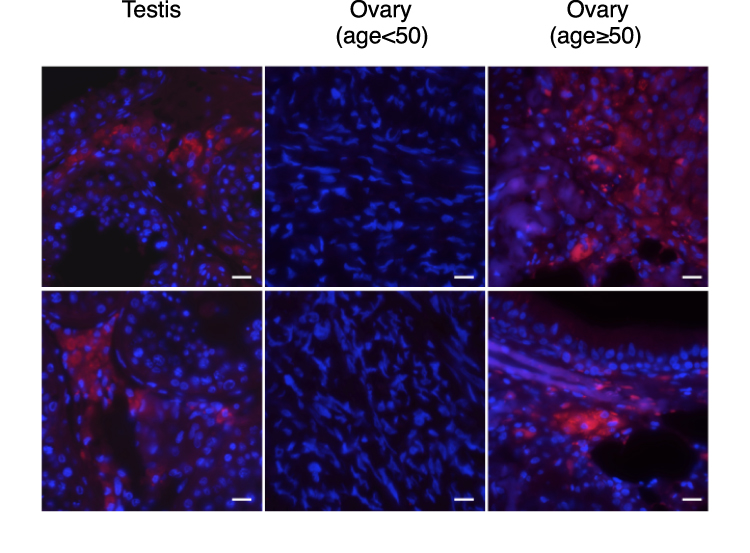 |
Figure 3 Segregation of MyD88/ER-α interaction according to gender. MyD88 and methERα interaction in human normal testis and ovary tissues revealed by proximity ligation assay (red dots). Magnification: 60x. |
Discussion
In this study, we describe a novel mechanism of inflammatory signaling regulation via the interaction between MyD88 and a cytoplasmic form of ER-α, with implications for gender disparity in inflammatory responses.
Sex-based differences in susceptibility to a wide variety of diseases are increasingly recognized as a confounding factor in the interpretation of clinical trials and responses to medication, with consequences on therapeutic decisions.24 Indeed, it has long been observed that females are relatively protected from certain diseases with an underlying inflammatory component, and estrogen has emerged as a major contributing factor to this relative protection from inflammatory diseases. Estrogen’s antiinflammatory activity is effected via one of its receptors, ER-α, which was described to exert a transcriptional inhibitory effect on NF-kB activation.11,25 Our data, on the other hand, implicate the methylated, cytoplasmic form of ER-α in gender disparity in response to inflammatory mediators. This pathway allows a rapid response to infection or tissue injury and thus may be useful in the early stages of the insult. Interestingly, in our hands, this response does not seem to be restricted to professional cells of innate immunity, but it also takes place in epithelial and interstitial cells. This is consistent with recent data indicating that non-hematopoietic cells respond to TLR and/or IL-1R ligands by producing cytokines and chemokines.26–28
In summary, our results indicate that gender bias in inflammatory responses may be at least partly due to the MyD88/methER-α complex formation and its regulation by estrogen.
Acknowledgment
This work was supported by the Ligue Nationale Contre le Cancer (R14009CC), the Institut National du Cancer (PLBIO11-071), and the SIRIC LYriCAN (INCa-DGOS-Inserm_12563).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fairweather D. Sex differences in inflammation during atherosclerosis. Clin Med Insights Cardiol. 2015;8:49–59. doi:10.4137/CMC.S17068
2. Koellhoffer EC, McCullough LD. The effects of estrogen in ischemic stroke. Transl Stroke Res. 2013;4(4):390–401. doi:10.1007/s12975-012-0230-5
3. Stubbins RE, Najjar K, Holcomb VB, Hong J, Núñez NP. Oestrogen alters adipocyte biology and protects female mice from adipocyte inflammation and insulin resistance. Diabetes Obes Metab. 2012;14(1):58–66. doi:10.1111/j.1463-1326.2011.01488.x
4. Maggioli ES, McArthur S, Mauro C, et al. Estrogen protects the blood-brain barrier from inflammation-induced disruption and increased lymphocyte trafficking. Brain Behav Immun. 2016;51:212–222. doi:10.1016/j.bbi.2015.08.020
5. Everhardt Queen A, Moerdyk-Schauwecker M, McKee LM, Leamy LJ, Huet YM. Differential expression of inflammatory cytokines in male and female mice in response to a lipopolysaccharide challenge. PLoS One. 2016;11(4):e0152289. doi:10.1371/journal.pone.0152289
6. Bábíčková J, Tóthová L, Lengyelová E, et al. Sex differences in experimentally induced colitis in mice: a role for estrogens. Inflammation. 2015;38(5):1996–2006. doi:10.1007/s10753-015-0180-7
7. Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127(5 Suppl 1):S5–S16. doi:10.1053/j.gastro.2004.09.011
8. Morale MC, Serra PA, L’episcopo F, et al. Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience. 2006;138(3):869–878. doi:10.1016/j.neuroscience.2005.07.060
9. Arnal JF, Douin-Echinard V, Brouchet L, et al. Understanding the oestrogen action in experimental and clinical atherosclerosis. Fundam Clin Pharmacol. 2006;20(6):539–548. doi:10.1111/j.1472-8206.2006.00445.x
10. Choudhry MA, Bland KI, Chaudry IH. Gender and susceptibility to sepsis following trauma. Endocr Metab Immune Disord Drug Targets. 2006;6(2):127–135. doi:10.2174/187153006777442422
11. Stein B, Yang MX. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Mol Cell Biol. 1995;15(9):4971–4979. doi:10.1128/MCB.15.9.4971
12. Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–124. doi:10.1126/science.1140485
13. Medzhitov R, Preston-Hurlburt P, Kopp E, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi:10.1016/S1097-2765(00)80136-7
14. Le Romancer M, Treilleux I, Leconte N, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. 2008;31(2):212–221. doi:10.1016/j.molcel.2008.05.025
15. Poulard C, Treilleux I, Lavergne E, et al. Activation of rapid oestrogen signalling in aggressive human breast cancers. EMBO Mol Med. 2012;4(11):1200–1213. doi:10.1002/emmm.201201615
16. Darimont BD, Wagner RL, Apriletti JW, et al. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12(21):3343–3356. doi:10.1101/gad.12.21.3343
17. Söderberg O, Gullberg M, Jarvius M, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3(12):9951000. doi:10.1038/nmeth947
18. Cutolo M, Villaggio B, Bisso A, Sulli A, Coviello D, Dayer JM. Presence of estrogen receptors in human myeloid monocytic cells (THP-1 cell line). Eur Cytokine Netw. 2001;12(2):368–372.
19. Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi:10.1002/eji.200323376
20. Wei H, Mundade R, Lange KC, Lu T. Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle. 2014;13(1):32–41. doi:10.4161/cc.27353
21. Moreno R, Sobotzik JM, Schultz C, Schmitz ML. Specification of the NF-kappaB transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKK epsilon. Nucleic Acids Res. 2010;38(18):6029–6044. doi:10.1093/nar/gkq439
22. Song WC, Qian Y, Sun X, Negishi M. Cellular localization and regulation of expression of testicular estrogen sulfotransferase. Endocrinology. 1997;138(11):5006–5012. doi:10.1210/endo.138.11.5512
23. Judd HL, Judd GE, Lucas WE, Yen SS. Endocrine function of the postmenopausal ovary: concentration of androgens and estrogens in ovarian and peripheral vein blood. J Clin Endocrinol Metab. 1974;39(6):1020–1024. doi:10.1210/jcem-39-6-1020
24. Simon V. Wanted: women in clinical trials. Science. 2005;308(5728):1517. doi:10.1126/science.1115616
25. Boyce BF, Xing L, Franzoso G, Siebenlist U. Required and nonessential functions of nuclear factor-kappa B in bone cells. Bone. 1999;25(1):137–139. doi:10.1016/S8756-3282(99)00105-2
26. Kiziltas S. Toll-like receptors in pathophysiology of liver diseases. World J Hepatol. 2016;8(32):1354–1369. doi:10.4254/wjh.v8.i32.1354
27. Coulter KR, Wewers MD, Lowe MP, Knoell LD. Extracellular regulation of interleukin (IL)-1beta through lung epithelial cells and defective IL-1 type II receptor expression. Am J Respir Cell Mol Biol. 1999;20(5):964–975. doi:10.1165/ajrcmb.20.5.3458
28. Diemer T, Hales DB, Weidner W. Immune-endocrine interactions and leydig cell function: the role of cytokines. Andrologia. 2003;35(1):55–63. doi:10.1046/j.1439-0272.2003.00537.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.